

Abstract
Improgan is a congener of the H2 antagonist cimetidine which produces potent antinociception. Because a) the mechanism of action of improgan remains unknown and b) this drug may indirectly activate cannabinoid CB1 receptors, the effects of the CB1 antagonist/inverse agonist rimonabant (SR141716A) and three congeners with varying CB1 potencies were studied on improgan antinociception after intracerebroventricular (icv) dosing in rats. Consistent with blockade of brain CB1 receptors, rimonabant ( Kd=0.23 nM) and O-1691 (Kd=0.22 nM) inhibited improgan antinociception by 48% and 70% after icv doses of 43 nmol and 25 nmol, respectively. However, two other derivatives with much lower CB1 affinity (O-1876, Kd=139 nM and O-848, Kd=352 nM) unexpectedly blocked improgan antinociception by 65% and 50% after icv doses of 300 nmol and 30 nmol, respectively. These derivatives have 600-fold to 1,500-fold lower CB1 potencies than that of rimonabant, yet they retained improgan antagonist activity in vivo. In vitro dose-response curves with 35S-GTPγS on CB1 receptor-containing membranes confirmed the approximate relative potency of the derivatives at the CB1 receptor. Although antagonism of improgan antinociception by rimonabant has previously implicated a mechanistic role for the CB1 receptor, current findings with rimonabant congeners suggest that receptors other than or in addition to CB1 may participate in the pain-relieving mechanisms activated by this drug. The use of congeners such as O-848, which lack relevant CB1-blocking properties, will help to identify these cannabinoid-like, non-CB1 mechanisms.
Keywords: cannabinoid, CB1 receptor, antinociception, analgesia, improgan, rimonabant
Introduction
Improgan, a congener of the histamine H2 antagonist cimetidine (Fig. 1), shows promise as a novel analgesic with an unknown target in the brain10. Cimetidine and improgan both act in the brain to inhibit supraspinal and intraspinal nociceptive responses in rats, suggesting a morphine-like analgesic profile10. However, the antinociceptive mechanism is not opioid receptor-mediated and cannot be attributed to any known histamine receptor, possibly suggesting that it is new. In fact, over 100 brain receptors have been screened with improgan and all have shown low affinity10, 13. Improgan also shows no activity on GTPγS binding, adenylate cyclase, inositol phosphate, or MAP kinase assays in brain preparations (unpublished results). Extensive testing, including tail-pinch, tail flick, hot plate, tail-immersion, and rotarod methods suggests that improgan produces profound analgesia. Improgan has shown antinociceptive activity after intracerebral injection into the periaqueductal gray and raphe magnus21, both of which participate in endogenous descending analgesic mechanisms. Repeated administration of improgan did not result in tolerance10. These findings in rats suggest that improgan produces non-opioid analgesia with fewer side effects than morphine10.
Figure 1.

Chemical structures of improgan and rimonabant.
The analgesic mechanisms of cannabinoids may be the key to understanding improgan actions. Both cannabinoids and improgan activate non-opioid descending pathways leading to analgesia12. CB1, the principal cannabinoid target in the brain, is found in the periaqueductal gray and the rostral ventral medulla17,19, both of which are sites of improgan action21 . Improgan and CB1 agonists, such as WIN 55, 212−2 (WIN), produce antinociception that exhibits dose-dependent inhibition by rimonabant (SR141716A), a CB1 antagonist/inverse agonist12. Recently, improgan antinociception was found to be reduced by chronic cannabinoid administration, also supporting an improgan-cannabinoid (possibly CB1) interaction22. However, radioligand binding assays have confirmed that improgan, unlike WIN, does not directly bind to CB1 or CB2 sites12. Although WIN can activate both of these receptors, the existence of the latter in the CNS is very limited, and CB2-mediated analgesia occurs outside of the brain15. Spinal CB2 receptors may contribute to antihyperalgesic actions in inflammatory pain states, but not under control conditions30. These findings imply that improgan indirectly activates CB1 along its analgesic circuit. Improgan studies in CB1 null mice gave equivocal results which neither support nor refute the involvement of CB1 receptors12 (also see discussion).
Presently, the effects of several chemical congeners of rimonabant (Table 1) with varying CB1 affinities were studied on cannabinoid and improgan antinociception. These studies are needed to further test the cannabinoid hypothesis of improgan action and to search for novel ligands relevant to the mechanism of improgan antinociception.
Table 1.
Structures and CB1 activity of selected congeners of rimonabant.

| Drug | CB1 Ki (nM) | R1 | R2 | R3 | R4 |
|---|---|---|---|---|---|
| Rimonabant | 6.2 | 2,4-dichloro | 4-chloro | methyl | 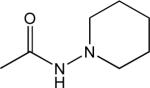 |
| O-1691 | 1.5 | 2,4-dichloro | 4-(n-pentyl) | bromo |  |
| O-1876 | 657 | 2,4-dichloro | 4-chloro | methyl | 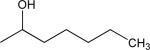 |
| O-848 | 2450 | 2,4-dichloro | 4-chloro | methyl | 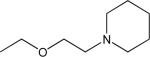 |
Literature Ki values are shown34 for drugs synthesized and tested presently.
Methods
Animals
Male Sprague-Dawley rats (175 − 200 g) from Taconic Farms, (Germantown, NY) were maintained on a 12-hr light/ dark cycle (lights on from 7:00am to 7:00pm) and provided with food and water. Rats were housed in groups of three or four until the time of surgery and individually thereafter. All animal experiments were approved by the Institutional Animal Care and Use Committee of Albany Medical College.
Surgery
For intracerebroventricular (icv) injections in rats, animals were anesthetized with pentobarbital sodium and supplemented with isoflurane. Cannulas were stereotaxically implanted into the left lateral ventricle and anchored to the skull with three stainless steel screws and cranioplast cement. The coordinates for the cannula (in mm from bregma) were: anterior-posterior −0.8, medial-lateral + 1.5, dorso-ventral −3.3. After surgery, the animals were individually housed with food and water available and were allowed to recover for at least 5 to 7 days before testing. Each animal was only studied once.
Rat ICV Injections and Nociceptive Testing
Rats were tested with the tail flick test3. A randomly selected location 2−5 cm from the tip of the ventral surface of the tail was exposed to radiant heat and the latency for tail movement was recorded. The heat source was set so that baseline latencies are generally between 3 and 4 sec with a 15-sec cutoff. The heat source was not adjusted for individual animals. The animals were tested with three tail flick tests performed at one-min intervals, and the third test was used as the baseline score. Animals were then gently secured by wrapping with a laboratory pad, the stylet was removed, and the icv injection cannula inserted. This cannula extends 1 mm beyond the guide to penetrate the lateral ventricle. Icv injections were performed manually over a one-min period with 2 μl of an antagonist solution or DMSO vehicle control. One min after the end of the infusion, the injection cannula was clipped approximately 2 mm above the juncture with the guide cannula. After a five min interval, a single tail flick test was performed, followed by a second icv injection of 10 μl improgan, WIN, or vehicle (60% DMSO). The second injection was performed by removing the first clipped injection cannula, and repeating the process above. The second cannula was clipped as before and single tail flick latencies were recorded five, ten, and thirty min later. Successful icv injections were assured by following the movement of an air bubble in the tubing between the syringe and the cannula and by the absence of leakage. After testing, animals received pentobarbital sodium (100 mg/kg, i.p.) and India Ink (5 μl, icv). Proper distribution of the ink in the cerebroventricular system indicated successful icv injections. Data from animals with poor placements or unsuccessful injections were excluded.
Drugs and Solutions
Small amounts of rimonabant and three of its chemical congeners (Table 1) were available from a previous study34. Additional amounts of O-848 were synthesized as given below. Additional rimonabant was provided by the National Institute on Drug Abuse Drug Supply Program. Improgan base (kindly provided by Prof. R. Leurs, Vrije University, Amsterdam11) and WIN (dosed as mesylate salt; RBI/Sigma, Natick, MA) were dissolved in 60% DMSO and 40% saline and all antagonists were dissolved in 100% DMSO. This icv vehicle has been widely used for cannabinoids and antagonists and has no adverse effects on motor behavior, nociceptive responses, or the ability to detect antagonism of antinociception14 . In all cases, both the identity (experimental group) of the subject, and the identity of the solutions injected were blinded to the experimenters.
Synthesis of O-848
Although O-848 has been described in the literature34, synthesis of this compound has not been previously published (Fig. 2). Ester 1 was prepared following a literature procedure16. This compound was then reduced to alcohol 2 using LiBH4 in THF according to another literature procedure27. A mixture of alcohol 2 (93 mg, 0.25 mmol) and PBr3 (4 mL) was stirred for 24 h. The reaction mixture was poured onto crushed ice and extracted with CH2Cl2. The organic layer was washed with water and brine, dried over sodium sulfate and concentrated to give intermediate 3, which was dissolved in 5 mL of DMF and added to a mixture of NaH (61 mg, 2.5 mmol), 1-(2-hydroxyethyl) piperidine (49 mg, 0.38 mmol) and DMF (5 mL). The resulting mixture was stirred for 24 h. The reaction was poured onto crushed ice and extracted with ethyl acetate. The organic layer was washed with water and brine, dried over sodium sulfate and concentrated to give a brown oil, which was purified by combiflash (CH2Cl2:MeOH:NH4OH 40:1:0.1 to 10:1:0.1) to give final product 4 (O-848, 90 mg, 74% from 2): (CDCl3) δ 7.39 (m, 1H), 7.28 (d, 2H, J = 8.6 Hz), 7.26 (m, 2H), 7.05 (d, 2H, J =8.6 Hz), 4.63 (s, 2H), 3.67 (t, 2H, J = 6.1 Hz), 2.60 (t, 2H, J = 6.1 Hz), 2.42 (m, 4H), 2.14 (s, 3H), 1.58 (m, 4H), 1.42 (m, 2H); MS (ESI, MH+).
Figure 2.

Chemical synthesis of O-848.
Analysis of Antinociceptive Data
Results are expressed as latencies (sec, mean ± SEM). Analysis of variance (between groups: drug treatment, within groups [repeated measures]: time) yielded highly significant (p<0.001) drug by time interactions from all nociception studies performed (Fig. 3-6). Bonferoni post-hoc tests were performed to determine significant differences between groups (Graphpad Prism 4.0, San Diego, CA).
Figure 3.

Effects of rimonabant (SR) on improgan (Imp, A) or WIN 55,212−2 (WIN, B) antinociception in rats. Animals were tested (ordinate, mean ± S.E.M. tail-flick latencies for the number of subjects in parentheses) for baseline responses (BL) then received an icv injection of 100% DMSO or rimonabant (doses in brackets are in nmol). Latencies were reassessed 5 min later (Post), followed by a second injection of 60% DMSO, improgan (388 nmol, 80 μg), or WIN (38.3 nmol, 20 μg). Latencies were then determined 5, 10, and 30 min after the second injection (abscissa). The same DMSO/DMSO and rimonabant [108]/DMSO groups are shown in Fig. 3A and 3B. Data were combined with some results which were previously published12. +, ++ p<0.05, <0.01 vs. DMSO/DMSO group at the same time point, respectively. *, ** P<0.05, p<0.01 vs. DMSO/Imp group at the same time point, respectively. #, ## P<0.05, P<0.01 vs. DMSO/WIN group at the same time point, respectively.
Fig. 6.

Effects of O-848 on improgan and WIN 55,212−2 antinociception in rats. Subjects were treated and tested as in Fig. 3, except that several doses of O-848 were given instead of rimonabant, and multiple doses of WIN were studied. Nociceptive testing (ordinate, tail-flick latencies in sec, mean ± S.E.M., number of subjects given in parentheses) was performed 5 (top), 10 (middle) and 30 (bottom) min after injections. Treatment groups consisted of improgan (388 nmol, 80 μg, left panels), WIN (20 μg [38.3 nmol] or 30 μg [57.5 nmol ]as labeled, right panels), or DMSO vehicles (Veh, all panels), along with the specified dose of O-848 (abscissa, nmol, all panels). The “vehicle” group (Veh) in the O-848/DMSO curve are pooled baseline latencies from 30, 100 and 300 nmol-treated groups. This does not affect the statistical analyses, which included baseline values from all groups. **P < 0.01 for O-848 by time interaction term among improgan-treated subjects by ANOVA.
CB1 receptor-stimulated [35S]GTPγS binding assay
Membrane fractions from a CB1-HEK 293 stable cell line were used to assess the activity at CB1 receptors in a manner similar to previously described studies1,18. Cells were suspended in phosphate buffered saline containing 1 mM EDTA and centrifuged at 500 × g for 5 min. The pellet was homogenized in homogenate buffer (50 mM Tris-HCl, 1 mM EDTA, 3 mM MgCl2, pH 7.4) and centrifuged (42,000 × g, 15 min, 4°C). The resulting pellet was resuspended in homogenate buffer and aliquots stored at −80°C. On the day of assay, aliquots were thawed on ice, centrifuged, and the pellet resuspended in assay buffer (50 mM Tris-HCl, 100 mM NaCl, 3 mM MgCl2, 0.2 mM EGTA, 0.1% bovine serum albumin, pH 7.4). Briefly, binding was initiated with addition of 20 μg of membrane protein into glass tubes containing 0.1 nM [35S]GTPγS (Perkin Elmer/NEN, Boston, MA) and 10 μM GDP in binding buffer. Nonspecific binding was assessed in the presence of 20 μM unlabeled GTPγS. Samples were incubated for 90 min at 30°C with various concentrations of WIN, along with candidate antagonists in a total volume of 500 μl, followed by rapid filtration through Whatman GF/B filters. Filters were mixed with scintillation cocktail, remained at room temperature for 3 hr, and were counted in a Beckman scintillation counter.
Results
As observed previously, icv improgan (388 nmol, 80 μg) produced a large increase in tail flick latency in rats at 5 and 10 min after injection, which returned to baseline 30 min later. This dose was chosen to produce near-maximal, but not supramaximal responses, based on recent dose-response studies14. The CB1 antagonist/inverse agonist rimonabant produced dose-dependent inhibition of improgan antinociception (Fig. 3A). The 43 nmol dose of rimonabant partially blocked, while the 108 nmol dose completely blocked improgan antinociception. The CB1 agonist WIN showed activity identical to that of improgan when injected alone. This effect was also blocked by the 43 nmol and the 108 nmol pretreatment with rimonabant (Fig. 3B). Neither dose of rimonabant had antinociceptive properties when injected alone.
O-1691, another potent CB1 antagonist, also inhibited improgan antinociception (Fig. 4A). Two doses of this drug (10 and 25 nmol) produced dose-related inhibition of antinociception. A higher dose (50 nmol) did not produce complete antagonism. A 25 nmol dose of O-1691 completely blocked WIN antinociception (Fig. 4B). This dose of O-1691 did not show antinociceptive properties when injected alone.
Figure 4.

Effects of O-1691 on improgan (A) or WIN 55,212−2 (B) antinociception in rats. Subjects were treated and tested as in Fig. 3, except O-1691 was injected instead of rimonabant. The same DMSO/DMSO, DMSO/Imp, and DMSO/WIN data are shown in Fig. 3. Footnotes indicating statistical significances are the same as in Fig. 3.
O-1876, a congener with 100-fold lower CB1 potency than rimonabant, also reduced improgan antinociception (Fig. 5A). The pattern of inhibition with this drug was remarkably similar to that seen after O-1691, except in this case maximal (but incomplete) antagonism was observed after 300 nmol of O-1876. This antagonist treatment also blocked WIN activity with similar potency (Fig. 5B). A 300 nmol dose of O-1876 did not show antinociceptive properties when injected alone.
Figure. 5.

Effects of O-1876 on improgan (A) or WIN 55,212−2 (B) antinociception in rats. Subjects were treated and tested as in Fig. 3, except O-1876 was injected instead of rimonabant. The same DMSO/DMSO, DMSO/Imp, and DMSO/WIN data are shown in Fig. 3. Footnotes indicating statistical significances are the same as in Fig. 3.
When given alone, O-848 tended to show a dose-related increase in tail flick latencies 5 and 10 min after injection (Fig. 6). However, repeated measures ANOVA of data from O-848-treated subjects in the absence of improgan or WIN failed to substantiate a statistical significance to this trend (O-848 treatment: P=0.11, time: P < 0.01, interaction: P=0.53). When combined with improgan, O-848 reduced improgan antinociception at 5 and 10 min after injection, but a slight enhancement was observed in the 30 min group (left panels, Fig. 6). ANOVA of data from improgan-treated subjects showed that O-848 significantly modified tail flick latencies (O-848 treatment: P=0.38, time: P<0.001, interaction: P<0.01). Since this significant interaction term could be due to either antagonism of improgan at 5 or 10 min, or due to enhancement at 30 min, a separate ANOVA was performed without the 30 min data (O-848 treatment: P=0.21, time: P<0.001, interaction: P=0.021). This second analysis confirms that O-848 inhibits improgan antinociception in either the 5 or 10 min groups (or both). Post-hoc analysis did not identify which groups show this inhibition. The magnitude of the inhibition (50%) was maximal in the 10 min data after 30 nmol of O-848. Latencies measured in these groups closely matched the effects of O-848 alone (especially at the higher doses), possibly consistent with a partial agonist profile.
In contrast to the effects of O-848 on improgan actions, this drug did not significantly inhibit WIN antinociception (Fig. 6, right panels). Because 20 μg (38.3 nmol) of WIN did not achieve maximal antinociception in this experiment, the effects of a larger dose (30 μg, 57.5 nmol) were also studied. When O-848 was combined with either dose of WIN, a tendency toward enhancement (not inhibition) of latencies was observed. ANOVA of WIN (20 μg)-treated subjects found significant main effects of O-848 (P<0.05), time (P<0.001) but no significant interaction (P=0.13). A similar analysis of the WIN (30 μg) treatment groups did not find significant changes.
35S-GTPγS-binding studies were used to quantify CB1-mediated responses and assess CB1 antagonist potencies (Fig. 7). The CB1 agonist WIN produced concentration-dependent increases in specific binding, which were competitively blocked by rimonabant and its congeners. All compounds produced concentration-dependent shifts in the WIN concentration-response curves, although antagonist concentrations needed to block the responses varied considerably. Thus, low concentrations (10−100 nM) of rimonabant and O-1691 were highly effective, yet larger concentrations of O-1876 (30−300 nM) produced only slight changes. Even larger concentrations of O-848 (1,000 – 3,000 nM) were needed to alter WIN responses. O-848 was also a very weak CB1 antagonist in membranes prepared from rat brain. Non-linear regression of all curves produced agonist dose ratios for each antagonist treatment, permitting Schild analyses, and estimates of CB1 antagonist Kd values (Fig. 7 and Table 2).
Fig. 7.

Effects of CB1 antagonists on CB1-mediated activation of GTPγS synthesis. Membranes from CB1-HEK 293 cells or rat brain were prepared and incubated as described in the presence of 35S-GTPγS, antagonists, and increasing concentrations of the CB1 agonist WIN55,212 (abscissa). Specific binding of GTPγS (ordinate, fmol/mg protein, or %baseline, mean ± S.E.M.) is shown. Each point is the mean ± S.E.M. of 3 separate experiments each performed in triplicate. Five panels show the effects of varying concentrations of each antagonist on agonist concentration-response curves. Non-linear regression was used estimate the apparent EC50 of each curve. Lower right: Schild plots are shown for each of the five antagonist experiments. For each plot, the log of antagonist concentration (abscissa) is plotted vs. log of dose ratio (DR) minus one (ordinate). The dose ratio was calculated as the ratio of the estimated agonist ED50 in the presence of antagonist divided by the agonist ED50 in the absence of antagonist. For each antagonist experiment, the slope of the Schild curve (S, expected to be 1.0) and the regression Kd (antilog of x-intercept) are given.
Table 2.
Anti-Improgan activity vs. CB1 activity.
| Drug | Active Dose (icv, nmol) | % Inhbition of Imp | Rel. Active Dose (/SR) | CB1 Kd (nM) | Rel. CB1 Kd (/SR) |
|---|---|---|---|---|---|
| Rimonabant | 43 | 48 | 1.0 | 0.23 | 1.00 |
| O-1691 | 25 | 70 | 0.6 | 0.22 | 0.96 |
| O-1876 | 300 | 65 | 7.0 | 139 | 604 |
| O-848 | 30 | 50 | 0.7 | 352 | 1,530 |
For each drug, the dose and degree of inhibition of improgan antinociception is given. Relative active dose refers to the quotient of the active dose of the specified drug divided by the active dose of rimonabant (43 nmol). CB1 Kd values were calculated from Schild plots (Fig. 7F). Relative CB1 Kd refers to the quotient of the calculated CB1 Kd of the drug divided by the calculated CB1 Kd of rimonabant (0.23 nM). All percent inhibition values were calculated using 10 minute post improgan injection data.
Discussion
Although improgan antinociception is reduced by WIN-blocking doses of rimonabant12 (confirmed in Fig. 3), radioligand experiments showed that improgan lacks affinity for CB1 and CB2 receptors from various biological sources assessed with several different radioligands12. In addition, improgan has virtually no effect on GTPγS activation in either the presence or absence of WIN in CB1-containing membranes14. Taken together, these studies show that CB1 is not a direct target for improgan. If CB1 receptors participate in improgan action, then an indirect mechanism must be involved. Improgan may act through an endocannabinoid mechanism, but this has not been explored. The compound has no effect on fatty acid amide hydrolase, an enzyme of endocannabinoid metabolism (unpublished).
Studies with improgan in CB1 knockout mice gave complex results. If the CB1 receptor mediates improgan effects, then improgan-induced antinociception should not have been observed in CB1 (−/−) animals. Surprisingly, improgan antinociception was observed in CB1 (−/−) mice12. One possible explanation for these data may be that rimonabant is blocking improgan at a receptor other than CB1. If so, then rimonabant should still have prevented improgan antinociception in CB1 (−/−) mice. However, improgan antinociception in CB1 (−/−) mice was not blocked by rimonabant, even though the same treatment was effective in wild-type control mice12. Thus, results with CB1 (−/−) mice failed to substrantiate the importance of CB1 mechanisms in improgan antinociception. However, these results also do not disprove the hypothesis. Thus these findings provide neither positive nor negative evidence on the role of CB1 mechanisms. Several studies suggest the up-regulation of a non-CB1, non-CB2 cannabinoid receptors in the CNS of germ line CB1 (−/−) mice (e.g. see20). Thus, improgan antinociception may utilize the CB1 receptor in normal mice, but the germ line knockout animal may acquire alternate circuits during development6. Clearly, additional studies were needed in order to learn the role of CB1 in improgan antinociception.
Rimonabant is the first potent and selective CB1 antagonist which is active in vivo29 . However, rimonabant can act at non-CB1sites as well8,24,26 . Thus, blockade of improgan by rimonabant (even at pharmacologically relevant doses) does not absolutely prove a CB1 mechanism. Therefore, the use of rimonabant congeners with varying potencies toward the CB1 receptor was designed to provide a powerful test of the CB1 cannabinoid hypothesis of improgan antinociception. If rimonabant acts at the CB1 receptor to block improgan antinociception, then various congeners of rimonabant should block improgan antinociception at doses commensurate with their CB1 potencies. Accordingly, drugs like O-1876 and O-848 should not have been blockers of improgan antinociception in vivo. If, on the other hand, rimonabant acts at a non-CB1 receptor to block improgan antinociception, there should have been no correlation between the potencies of congeners toward the CB1 receptor and the doses of these drugs needed to block improgan.
Previously several congeners of rimonabant were synthesized to determine structure-activity relationships at the CB1 receptor34. This study concluded that N(1)- and C(5) substituents in rimonabant which retain the central pyrazole structure (Fig. 1) are primarily involved in receptor antagonism, whereas replacements at the C(3) position (R4 in Table 1) can contribute to CB1 receptor recognition and agonism34. Of the many congeners made and tested, O-1691, O-1876, and O-848 were chosen for the present studies because of their close chemical similarities to rimonabant, and their widely divergent CB1 receptor affinities (Table 1).
Because O-1876 and O-848 were reported to be more than 100-fold less potent than rimonabant on CB1 receptors (Table 1), the in vivo inhibition of improgan action by these drugs was unexpected (Figs. 5, 6). Since only a limited amount of data on these compounds was available34, it seemed important to re-examine the CB1 antagonist affinity of these drugs. In addition, significant discrepancies in CB1 Ki values for compounds related to rimonabant have been noted, depending on the radioligand used28,32. For the present work, it seemed that the most unambiguous determination of CB1 potencies would be measurement of antagonism of cannabinoid responses in vitro (Fig. 7). The use of the same cannabinoid agonist (WIN) both in vivo and in vitro was thought to be an added strength of the experimental design. The Kd values of the four compounds obtained by this method (Fig. 7 & Table 2) confirm the earlier conclusions34 (Table 1) that rimonabant and O-1691 are both high potency CB1 antagonists, whereas O-1876 and O-848 are several hundred-fold lower in potency. However, the CB1 antagonist Kd values for all of the drugs (Table 2) were considerably lower (4−30 fold) than the previously-reported corresponding Ki values34 (Table 1). Notwithstanding these differences, both studies show that O-1876 and O-848 are 106-fold (Table 1) to 604-fold (Table 2) and 395-fold (Table 1) to 1,530-fold (Table 2) less potent, respectively, than rimonabant. Literature Ki or Kd values for rimonabant are 0.16 – 0.33 nM1,4,7.
In this study, two compounds with high CB1 potency (rimonabant and O-1691) blocked both improgan and WIN activity, thus yielding the hypothesized results (Figs 3-4, Table 2). The findings show that comparable doses of these two compounds have similar actions against both improgan and WIN antinociception, consistent with the CB1 hypothesis of improgan analgesia. Both present (Fig. 4) and previous34 data confirm that O-1691, which does not increase tail flick latency when injected alone, is a high potency CB1 antagonist that blocks cannabinoid antinociception.
Results with the remaining two compounds (O-1876 and O-848) are not consistent with the CB1 hypothesis. Both of these compounds have a very low CB1 affinity (Table 2) and they blocked improgan antinociception at doses not consistent with their ability to act on CB1. Table 2 shows that 604 times the active dose of rimonabant would be needed in order for O-1876 to produce comparable antagonism of improgan. However, a dose of O-1876 that is 6 times the dose of rimonabant was active. This discrepancy is even larger with O-848. A dose of O-848 that is 1,530 times the active dose of rimonabant should be needed to block improgan (Table 2). Surprisingly, nearly identical doses of O-848 and rimonabant reduced improgan antinociception by about 50% (Table 2). If rimonabant and O-848 block improgan antinociception by the same mechanism, this finding strongly suggests that the CB1 receptor does not mediate this response.
A number of possible non-CB1 sites of rimonabant action need to be considered when searching for the improgan mechanism. For example, rimonabant has activity in hippocampal slices of the CB1 −/− mouse brain9. This drug also inhibits anandamide-induced vasodilation in rat coronary and mesenteric arteries5,33,35. However, in these and other studies24, rimonabant's non-CB1 activity occurs at concentrations ranging from 0.5 to 10 μM, which is 1,000-fold larger than the concentrations needed to block CB1 (0.5 − 2 nM). The potency of rimonabant to block improgan activity is similar to its potency to block WIN antinociception (Fig. 3). Assuming that WIN antinociception is mediated through the CB1 receptor23, this argues that rimonabant must be acting in the nanomolar range to block both WIN-induced and improgan-induced antinociception (Fig. 7). Thus, targets for rimonabant acting in the micromolar range are not likely to be relevant to the antagonism of improgan.
One non-CB1 target of potential further interest for both WIN and improgan antinociception may be the TRPV1 receptor. Although best studied in the spinal cord, this site exists in brain, may have relevance for analgesia31, and may be a target for rimonabant at higher concentrations8. However, binding studies found that improgan (10 μM) has no affinity at these sites (unpublished).
Although many of the above studies show non-CB1 targets for rimonabant in the micromolar range, one report found evidence that rimonabant acts at a non-CB1, non-CB2 target in mouse vas deferens at a concentration in the nanomolar range24. Rimonabant's KB for antagonism at this novel receptor was reported to be 15.4 nM. In contrast, the KB of rimonabant for antagonism of WIN in the same assay (which is CB1-mediated) was 2.4 nM25, suggesting. T that rimonabant has a non-CB1 target in the relevant concentration range. It is possible that the improgan-blocking effect of rimonabant is due to its ability to block this novel receptor, and not its ability to block CB1. This could explain why O-1876 and O-848, two compounds with virtually no CB1 activity, are blockers of improgan antinociception.
Antagonism of improgan antinociception by rimonabant initially suggested a role for the CB1 receptor in the improgan analgesic circuit. However, the present data with rimonabant congeners argue against this hypothesis, suggesting the importance of non-CB1 receptors. It is possible that improgan may have both a CB1 and non-CB1 mediated component. In this scenario, drugs that could exclusively block either CB1 or a non-CB1 target would be able to partially block improgan antinociception. Only drugs with the ability to block both targets would be able to completely block improgan activity. The present findings are consistent with this prediction. O-1691, O-1876, and O-848 were able to partially block improgan antinociception at the maximum dose tested (Fig. 4-6), fitting the profile for a drug that blocks only one of the two targets. The action of O-1691 is most likely due to its ability to block CB1, because this drug has a high affinity towards this receptor. In contrast, O-1876 and O-848 most likely act on a non-CB1 target because both of these drugs have a very low affinity for the CB1 receptor. The complete inhibition of improgan activity by rimonabant (Fig. 3) could be explained by blockade of both CB1 and non-CB1 targets. A previous study has also suggested a dual CB1 and non-CB1 mechanism of action for a novel compound with cannabinoid properties24.
In the present study, the CB1 agonist WIN was used as a control to assess the ability of rimonabant and its congeners to block CB1 mediated antinociception. It was expected that drugs with a high CB1 potency (rimonabant and O-1691) would block WIN antinociception, and drugs with a low CB1 potency (O-1876 and O-848) would not. Three of the drugs (rimonabant, O-1691, and O-848) gave the predicted results. However, the unexpected antagonism of WIN antinociception by O-1876 (Fig. 5B) is not consistent with the CB1 receptor's role in mediating WIN responses. Even though WIN clearly acts at CB1 receptors, non-CB1 actions of this drug have also been reported2,9,20,26. Because some of the reported effects of WIN may be blocked by rimonabant, but not by all CB1 antagonists8, further studies are required to delineate this potential non-CB1 mechanism for WIN antinociception. O-848 will be a useful tool in searching for this mechanism and in the further characterization of improgan antinociception.
Acknowledgements
This work was supported by grants from the National Institute on Drug Abuse (DA-03816 and DA-15915 to LBH; DA-09978 and DA-05274 to MEA; DA 09789 to BRM) and the Samuel L. Powers, M.D. Fellowship. We thank Prof. Rob Leurs (Vrije University, Amsterdam) for kindly providing the sample of improgan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
PERSPECTIVE: This article describes new pharmacological characteristics of improgan, a pain-relieving drug which acts by an unknown mechanism. Improgan may use a marijuana-like (cannabinoid) pain-relieving mechanism, but it is shown presently that the principal cannabinoid receptor in the brain (CB1) is not solely responsible for improgan analgesia.
References
- 1.Abood ME, Ditto KE, Noel MA, Showalter VM, Tao Q. Isolation and expression of a mouse CB1 cannabinoid receptor gene. Comparison of binding properties with those of native CB1 receptors in mouse brain and N18TG2 neuroblastoma cells. Biochem Pharmacol. 1997;53:207–214. doi: 10.1016/s0006-2952(96)00727-7. [DOI] [PubMed] [Google Scholar]
- 2.Breivogel CS, Griffin G, Di M, V, Martin BR. Evidence for a new G protein-coupled cannabinoid receptor in mouse brain. Mol Pharmacol. 2001;60:155–163. [PubMed] [Google Scholar]
- 3.D' Amour F, Smith D. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- 4.DiMarzo V, Breivogel CS, Tao Q, Bridgen DT, Razdan RK, Zimmer AM, Zimmer A, Martin BR. Levels, metabolism, and pharmacological activity of anandamide in CB(1) cannabinoid receptor knockout mice: evidence for non-CB(1), non-CB(2) receptor-mediated actions of anandamide in mouse brain. J Neurochem. 2000;75:2434–2444. doi: 10.1046/j.1471-4159.2000.0752434.x. [DOI] [PubMed] [Google Scholar]
- 5.Ford WR, Honan SA, White R, Hiley CR. Evidence of a novel site mediating anandamide-induced negative inotropic and coronary vasodilatator responses in rat isolated hearts. Br J Pharmacol. 2002;135:1191–1198. doi: 10.1038/sj.bjp.0704565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fride E, Foox A, Rosenberg E, Faigenboim M, Cohen V, Barda L, Blau H, Mechoulam R. Milk intake and survival in newborn cannabinoid CB1 receptor knockout mice: evidence for a “CB3” receptor. Eur J Pharmacol. 2003;461:27–34. doi: 10.1016/s0014-2999(03)01295-0. [DOI] [PubMed] [Google Scholar]
- 7.Griffin G, Atkinson PJ, Showalter VM, Martin BR, Abood ME. Evaluation of cannabinoid receptor agonists and antagonists using the guanosine-5'-O-(3-[35S]thio)-triphosphate binding assay in rat cerebellar membranes. J Pharmacol Exp Ther. 1998;285:553–560. [PubMed] [Google Scholar]
- 8.Hajos N, Freund TF. Pharmacological separation of cannabinoid sensitive receptors on hippocampal excitatory and inhibitory fibers. Neuropharmacology. 2002;43:503–510. doi: 10.1016/s0028-3908(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 9.Hajos N, Ledent C, Freund TF. Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic synaptic transmission in the hippocampus. Neuroscience. 2001;106:1–4. doi: 10.1016/s0306-4522(01)00287-1. [DOI] [PubMed] [Google Scholar]
- 10.Hough LB, Nalwalk JW, Barnes WG, Leurs R, Menge WM, Timmerman H, Wentland M. A third life for burimamide. Discovery and characterization of a novel class of non-opioid analgesics derived from histamine antagonists. Ann N Y Acad Sci. 2000;909:25–40. doi: 10.1111/j.1749-6632.2000.tb06674.x. [DOI] [PubMed] [Google Scholar]
- 11.Hough LB, Nalwalk JW, Chen Y, Schuller A, Zhu Y, Zhang J, Menge WM, Leurs R, Timmerman H, Pintar JE. Improgan, a cimetidine analog, induces morphine-like antinociception in opioid receptor-knockout mice. Brain Res. 2000;880:102–108. doi: 10.1016/s0006-8993(00)02776-1. [DOI] [PubMed] [Google Scholar]
- 12.Hough LB, Nalwalk JW, Stadel R, Timmerman H, Leurs R, Paria BC, Wang X, Dey SK. Inhibition of improgan antinociception by the cannabinoid (CB)(1) antagonist N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-p yrazole-3-carboxamide (SR141716A): lack of obligatory role for endocannabinoids acting at CB(1) receptors. J Pharmacol Exp Ther. 2002;303:314–322. doi: 10.1124/jpet.102.036251. [DOI] [PubMed] [Google Scholar]
- 13.Hough LB, Nalwalk JW, Barnes WG, Leurs R, Menge WM, Timmerman H. A third legacy for burimamide: discovery and characterization of improgan and a new class of non-opioid analgesics derived from histamine antagonists. In: Watanabe T, Timmerman H, Yanai K, editors. Histamine Research in the New Millenium. Elsevier; Amsterdam: 2001. pp. 237–242. [Google Scholar]
- 14.Hough LB, de Esch IJP, Janssen E, Phillips J, Svokos K, Kern B, Trachler J, Abood ME, Leurs R, Nalwalk JW. Antinociceptive activity of chemical congeners of improgan: optimization of side chain length leads to the discovery of a new, potent, non-opioid analgesic. Neuropharmacology. 2006;51:447–456. doi: 10.1016/j.neuropharm.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, Davar G, Makriyannis F, Vanderah TW, Mata HP, Malan TP., Jr CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc.Natl.Acad.Sci.U.S.A. 2005;102:3093–3098. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katoch-Rouse R, Pavlova OA, Caulder T, Hoffman AF, Mukhin AG, Horti AG. Synthesis, structure-activity relationship, and evaluation of SR141716 analogues: development of central cannabinoid receptor ligands with lower lipophilicity. J Med Chem. 2003;46:642–645. doi: 10.1021/jm020157x. [DOI] [PubMed] [Google Scholar]
- 17.Lichtman AH, Cook SA, Martin BR. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: evidence supporting periaqueductal gray involvement. J Pharmacol Exp Ther. 1996;276:585–593. [PubMed] [Google Scholar]
- 18.McAllister SD, Hurst DP, Barnett-Norris J, Lynch D, Reggio PH, Abood ME. Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: the importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J Biol Chem. 2004;279:48024–48037. doi: 10.1074/jbc.M406648200. [DOI] [PubMed] [Google Scholar]
- 19.Meng ID, Manning BH, Martin WJ, Fields HL. An analgesia circuit activated by cannabinoids. Nature. 1998;395:381–383. doi: 10.1038/26481. [DOI] [PubMed] [Google Scholar]
- 20.Monory K, Tzavara ET, Lexime J, Ledent C, Parmentier M, Borsodi A, Hanoune J. Novel, not adenylyl cyclase-coupled cannabinoid binding site in cerebellum of mice. Biochem Biophys Res Commun. 2002;292:231–235. doi: 10.1006/bbrc.2002.6635. [DOI] [PubMed] [Google Scholar]
- 21.Nalwalk JW, Svokos K, Taraschenko O, Leurs R, Timmerman H, Hough LB. Activation of brain stem nuclei by improgan, a non-opioid analgesic. Brain Res. 2004;1021:248–255. doi: 10.1016/j.brainres.2004.06.066. [DOI] [PubMed] [Google Scholar]
- 22.Nalwalk JW, Svokos K, Hough LB. Cannabinoid-improgan cross-tolerance: Improgan is a cannabinomimetic analgesic lacking affinity at the cannabinoid CB(1) receptor. Neuropharmacol. 2006;549:79–83. doi: 10.1016/j.ejphar.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 23.Pertwee RG. Cannabinoid receptors and pain. Prog Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 24.Pertwee RG. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005;76:1307–1324. doi: 10.1016/j.lfs.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 25.Pertwee RG, Thomas A, Stevenson LA, Maor Y, Mechoulam R. Evidence that (−)-7-hydroxy-4'-dimethylheptyl-cannabidiol activates a non-CB(1), non-CB(2), non-TRPV1 target in the mouse vas deferens. Neuropharmacology. 2005;48:1139–1146. doi: 10.1016/j.neuropharm.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 26.Pistis M, Perra S, Pillolla G, Melis M, Gessa GL, Muntoni AL. Cannabinoids modulate neuronal firing in the rat basolateral amygdala: evidence for CB1- and non-CB1-mediated actions. Neuropharmacology. 2004;46:115–125. doi: 10.1016/j.neuropharm.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Pommery N, Taverne T, Telliez A, Goossens L, Charlier C, Pommery J, Goossens JF, Houssin R, Durant F, Henichart JP. New COX-2/5-LOX inhibitors: apoptosis-inducing agents potentially useful in prostate cancer chemotherapy. J Med Chem. 2004;47:6195–6206. doi: 10.1021/jm0407761. [DOI] [PubMed] [Google Scholar]
- 28.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, Westwood P, Marrs J, Thomson F, Cowley P, Christopoulos A, Pertwee RG, Ross RA. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 29.Rinaldi-Carmona M, Barth F, Heaulme M, Alonso R, Shire D, Congy C, Soubrie P, Breliere JC, Le FG. Biochemical and pharmacological characterisation of SR141716A, the first potent and selective brain cannabinoid receptor antagonist. Life Sci. 1995;56:1941–1947. doi: 10.1016/0024-3205(95)00174-5. [DOI] [PubMed] [Google Scholar]
- 30.Sagar DR, Kelly S, Millns PJ, O'Shaughnessey CT, Kendall DA, Chapman V. Inhibitory effects of CB1 and CB2 receptor agonists on responses of DRG neurons and dorsal horn neurons in neuropathic rats. Eur J Neurosci. 2005;22:371–379. doi: 10.1111/j.1460-9568.2005.04206.x. [DOI] [PubMed] [Google Scholar]
- 31.Steenland HW, Ko SW, Wu LJ, Zhuo M. Hot receptors in the brain.). Mol Pain. 2006;2:34. doi: 10.1186/1744-8069-2-34. 10.1186/1744-8069-2-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas BF, Francisco ME, Seltzman HH, Thomas JB, Fix SE, Schulz AK, Gilliam AF, Pertwee RG, Stevenson LA. Synthesis of long-chain amide analogs of the cannabinoid CB1 receptor antagonist N-(piperidinyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716) with unique binding selectivities and pharmacological activities. Bioorg Med Chem. 2005;13:5463–5474. doi: 10.1016/j.bmc.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Wagner JA, Varga K, Jarai Z, Kunos G. Mesenteric vasodilation mediated by endothelial anandamide receptors. Hypertension. 1999;33:429–434. doi: 10.1161/01.hyp.33.1.429. [DOI] [PubMed] [Google Scholar]
- 34.Wiley JL, Jefferson RG, Grier MC, Mahadevan A, Razdan RK, Martin BR. Novel pyrazole cannabinoids: insights into CB(1) receptor recognition and activation. J Pharmacol Exp Ther. 2001;296:1013–1022. [PubMed] [Google Scholar]
- 35.Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di M, V, Julius D, Hogestatt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]