

Summary
Cryptosporidium parvum attaches to intestinal and biliary epithelial cells via specific molecules on host-cell surface membranes including Gal/GalNAc-associated glycoproteins. Subsequent cellular entry of this parasite depends on host-cell membrane alterations to form a parasitophorous vacuole via activation of phosphatidylinositol 3-kinase (PI-3K)/Cdc42-associated actin remodelling. How C. parvum hijacks these host-cell processes to facilitate its infection of target epithelia is unclear. Using specific probes to known components of sphingolipid-enriched membrane microdomains (SEMs), we detected aggregation of host-cell SEM components at infection sites during C. parvum infection of cultured human biliary epithelial cells (i.e. cholangiocytes). Activation and membrane translocation of acid-sphingomyelinase (ASM), an enzyme involved in SEM membrane aggregation, were also observed in infected cells. Pharmacological disruption of SEMs and knockdown of ASM via a specific small interfering RNA (siRNA) significantly decreased C. parvum attachment (by ~ 84%) and cellular invasion (by ~ 88%). Importantly, knockdown of ASM and disruption of SEMs significantly blocked C. parvum-induced accumulation of Gal/GalNAc-associated glycoproteins at infection sites by ~ 90%. Disruption of SEMs and knockdown of ASM also significantly blocked C. parvum-induced activation of host-cell PI-3K and subsequent accumulation of Cdc42 and actin by up to 75%. Our results suggest an important role of SEMs for C. parvum attachment to and entry of host cells, likely via clustering of membrane-binding molecules and facilitating of C. parvum-induced actin remodelling at infection sites through activation of the PI-3K/Cdc42 signalling pathway.
Introduction
The heterogeneity of cellular membranes is characterized by the existence of functionally distinct membrane microdomains enriched in sphingolipids, cholesterol, ganglioside GM1 and glycosylphosphatidylinositol (GPI)-anchored proteins called sphingolipid-enriched membrane microdomains (SEMs) or rafts. Although not yet morphologically confirmed, SEMs are assumed to be very small ‘floating’ regions on the cellular membrane that have substantial lateral mobility. Whereas sphingolipids contain saturated acyl chains that enable each shingolipid molecule to pack tightly with each other in SEMs via hydrophilic and hydrophobic interactions, cholesterol integrates between these sphingolipids, in particular sphingomyelin, to give SEMs more stability and fluidity (Brown and London, 1998; Manes et al., 2003). Recent studies indicate that SEMs are involved in various membrane functions, including clustering of membrane receptors and activation of associated downstream intracellular signalling pathways.
A primary function of SEMs is thought to be to regulate protein interactions by their ability to selectively recruit or exclude proteins as well as their ability to cluster into larger platforms. It has been postulated that a single SEM is very small and only holds a few proteins, and therefore, clustering of multiple microdomains into larger platforms is important to bring signalling proteins closer together (Brown and London, 1998; Manes et al., 2003). Acidicsphingomyelinase (ASM) is a shingomyelin-specific phospholipase C that exists in lysosomal and secretory forms in many cell types. Recent data show that translocation of secretory ASM onto the outer leaflet of the plasma membrane plays a role in formation of larger SEM platforms (Grassme et al., 2003; Rotolo et al., 2005). This process has been shown to be stimulated by both membrane receptors (e.g. CD95, TNF receptors, CD40, Fcγ receptor II) and by pathogenic microbes (e.g. Neisseria gonorrhoeae, Pseudomonas aeruginosa and Sindbis virus) (Grassme et al., 2003; Kwiatkowska et al., 2003). Apparently activated ASM converts SEM-associated sphingomyelin into ceramide resulting in the clustering of smaller SEMs into larger platforms. These larger SEM platforms serve to re-organize molecules in the cell, presumably to cluster receptors, thus enhancing receptor signalling (Grassme et al., 2003). For example, SEMs are involved in the clathrin-independent endocytotic pathway mediated by caveolae, smooth invaginations of the plasma membrane that are enriched in sphingolipids and cholesterol which partition into SEM fractions and whose expression is associated with caveolin-1 (Duncan et al., 2002). Also, human growth factor induces membrane ruffling via SEM-associated activation of actin polymerization (Seveau et al., 2004). Aggregation of SEMs into larger platforms is also involved in the activation of downstream effectors of membrane-associated kinases such as SEM-associated Src family tyrosine kinases (Kwiatkowska et al., 2003).
Sphingolipid-enriched membrane microdomains have recently been recognized to be involved in cellular entry of pathogens and associated toxins. A well-known example is cholera toxin which enters cells via its binding to ganglioside GM1, a common component in SEMs (Lafont and van der Goot, 2005). Many intracellular pathogens, including viruses, bacteria and parasites, likely also utilize host-cell SEM machinery for their cellular entry. In many cases, these pathogens manipulate host-cell caveolae-dependent endocytosis via SEMs to enter host cells, a process hijacked by those pathogens to avoid the lysosomal endocytotic pathway that the host cell uses for defence (Duncan et al., 2002). Recently, studies indicate that SEMs are also involved in pathogen-induced actin cytoskeleton remodelling during cellular internalization (e.g. the involvement of SEMs in Shigella cellular internalization) (Duncan et al., 2002). Efficient binding of Shigella protein IpaB to its host-cell membrane receptor protein, CD44, requires SEM-associated platform formation, a process that triggers the ezrin–redixin–moesin network leading to actin cytoskeleton re-organization at the attachment sites resulting in bacterial entry into cells (Lafont et al., 2002).
Cryptosporidium parvum, an obligate intracellular parasite of the phylum Apicomplexia, primarily infects the gastrointestinal tract of humans and animals worldwide (Chen et al., 2002). The infection in humans is usually self-limiting except in cases involving immunocompromised patients, most notably AIDS patients, where severe diarrhoea can persist for weeks with no effective treatment available (Wuhib et al., 1994; Chen et al., 2002). C. parvum infects by the faecal-oral route and once oocysts are ingested, excysted sporozoites infect the epithelial cells of the intestinal tract (Zhu et al., 1998; Chen et al., 2002). Sporozoites may also travel up the biliary tree, infect cholangiocytes (i.e. epithelial cells lining the bile duct) and cause secondary sclerosing cholangitis in AIDS patients (Thompson et al., 2005). The infection process involves attachment of the sporozoite to the host-cell apical membrane via unknown surface-binding proteins including Gal/GalNAc epitopes of glycoproteins. Cellular entry of this parasite is also characterized by host-cell membrane protrusions that encapsulate the organism and form a parasitophorous vacuole (O’Donoghue, 1995; Tzipori and Griffiths, 1998; Clark, 1999). Actin remodelling is required for host-cell membrane protrusions and C. parvum initiates this via activation of the class IA phosphatidylinositol 3-kinase (PI-3K)/Cdc42 signalling pathway (Chen et al., 2004a).
Using an in vitro model of human biliary cryptosporidiosis, we demonstrate, for the first time, that C. parvum infection triggers the clustering of SEM components at infection sites. Disruption of SEM components or inhibition of SEM platform formation decreases C. parvum attachment to and entry of cultured cholangiocytes. Clustering of SEM components at infection sites appears also to be involved in aggregation of Gal/GalNAc-associated membrane receptors and C. parvum-induced activation of the PI-3K/Cdc42/actin signalling pathway, processes associated with parasite attachment to host cells and subsequent cellular entry. Thus, SEMs are required for C. parvum attachment to and entry of host cells, likely via clustering of membrane-binding proteins and facilitating of C. parvum-induced actin remodelling at infection sites via activation of the PI-3K/Cdc42 signalling pathway.
Results
Cryptosporidium parvum infection recruits components of SEMs to infection sites
Host-cell attachment and cellular entry by C. parvum is a process that involves direct binding of parasite ligands to host-cell membrane receptors followed by host-cell membrane protrusion to cover the parasite and form the parasitophorous vacuole. This process is limited to infection sites as the parasitophorous vacuole is formed at the attachment site which keeps the internalized organism intracellular but extracytoplasmic. To test whether SEMs are involved in C. parvum host-cell attachment and cellular entry, we first tested whether SEMs are recruited to and aggregated at infection sites. Various reagents and probes, known to specifically label various SEM components, were used in our study. Using cholera toxin B to label SEM-associated GM1 and Fillipin to label membrane cholesterol, we found a strong accumulation of both SEM-associated GM1 and cholesterol at C. parvum infection sites (Fig. 1A2 and B2). To ensure that the labelling of SEM components is host cell-associated and not parasite in origin, C. parvum sporozoites were mounted on slides and labelled with cholera toxin-FITC and Fillipin as described. Parasites alone only showed a very limited fluorescence, approximately 97% less fluorescence compared with the parasite–host interface shown in Fig. 1A2 and B2, suggesting that the strong labelling of cholera toxin-FITC and Fillipin at the host–parasite interface is predominately from the host cells. In contrast, no significant accumulation of transferrin receptor, whose membrane distribution is not SEM-associated (Jing et al., 1990), was observed at infection sites (Fig. 1C). In addition, caveolae-dependent endocytosis is associated with SEMs and often hijacked by intracellular microbes to enter host cells including SV40 (Shin et al., 2000; Pelkmans et al., 2001; Duncan et al., 2002). To test whether SEM-associated caveolae-dependent endocytotic pathway is involved in C. parvum infection process, we tested the accumulation of caveolin-1 at C. parvum infection sites. No obvious recruitment of caveolin-1 was found at infection sites (Fig. 1D). In contrast, a significant accumulation of caveolin-1 was detected at the infection sites of SV40 (Fig. 1E). Quantitative analysis of GM1, cholesterol, transferrin receptor and caveolin-1 accumulation was shown in Fig. 1F. These data suggested that C. parvum infection selectively recruits components of SEMs to infection sites.
Fig. 1. Cryptosporidium parvum selectively recruits components of host-cell SEMs to infection sites.
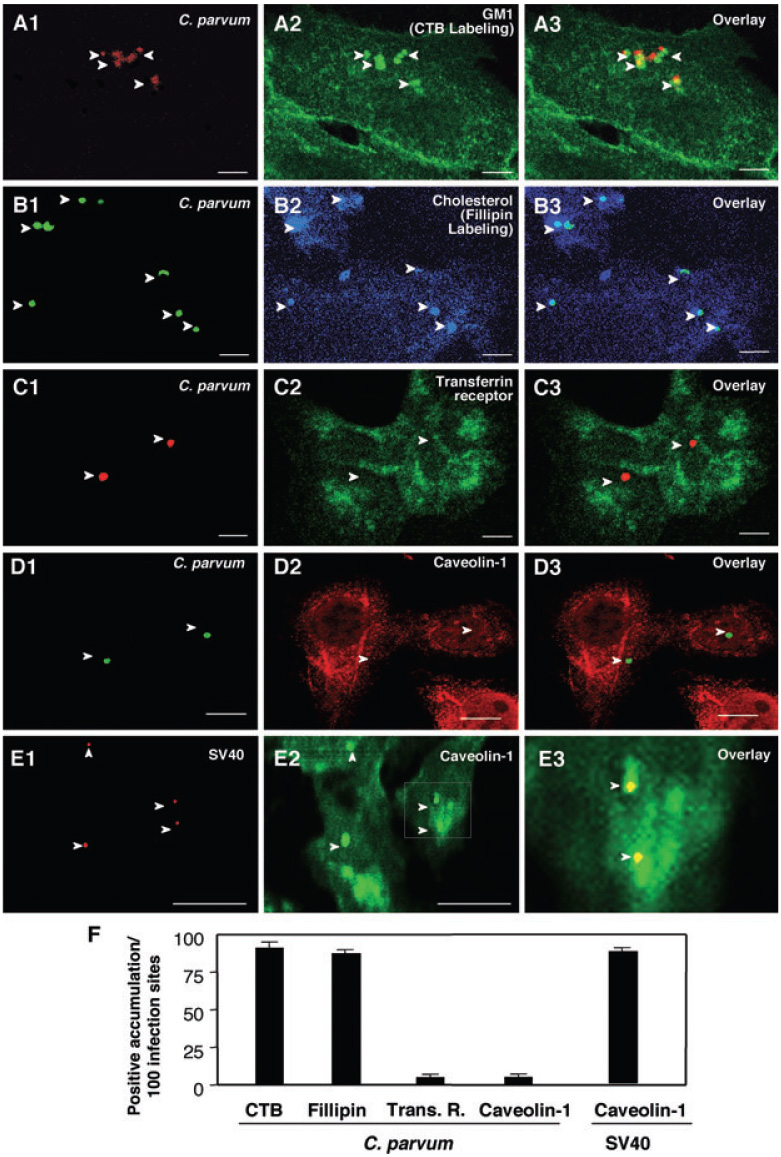
A–E. H69 cells were infected with C. parvum and labelled with various markers to identify SEMs. C. parvum was labelled using an specific antibody [arrowheads in (A1)–(D1)]. Accumulation of GM1 (labelled by cholera toxin B) and cholesterol (labelled by Fillipin), components of SEMs, was observed at infection sites [arrowheads in (A2) and (B2) or in the overlay images of (A3) and (B3)]. Transferrin receptor, a non-SEM-associated membrane receptor, showed no significant accumulation at infection sites ([arrowheads in (C2) and (C3)]. Moreover, caveolin-1, a protein that exists in some SEMs and is associated with caveolae, did not accumulate at infection sites [arrowheads in (D2) and (D3)]. As a positive control, a significant accumulation of caveolin-1 was detected at the infection sites of SV40 [arrowheads in (E1) and (E2)]. (E3) is an overlay image of high magnification of the boxed region in (E2). Bar = 5 µm.
F. Quantitative analysis as determined by number of sites with positive accumulation per 100 infection sites. CTB, cholera toxin B.
ASM is activated and translocated to the membrane surface during C. parvum infection
Aggregation of SEMs is usually associated with the activation of ASM, which translocates to the membrane surface when activated and subsequently, converts membrane-associated sphingomyelin into ceramide resulting in larger SEM platform formation (Brown and London, 1998; Grassme et al., 2003). To test whether ASM and ceramide are also recruited to infection sites, we used antibodies to ASM and ceramide to stain cells after exposure to C. parvum. Whereas non-infected control cells or areas of infected cells not directly adjacent to the parasite–host cell interface showed no obvious localized accumulation of ASM and ceramide (Fig. 2A and B), a strong accumulation of both ASM and ceramide was found at infection sites (Fig. 2A and B). Parasites alone showed no detectable positive staining to the anti-ceramide antibody but showed a very limited fluorescence to the anti-ASM antibody with the approach. However, the fluorescence was approximately 85% less compared with the parasite–host interface shown in Fig. 2A and B. Localized accumulation of host-cell ceramide was further confirmed as assessed by BoDipy-ceramide tracking. Cells were first treated with the BoDipy-ceramide followed by extensive washing prior to C. parvum infection, an approach that enables us to trace the distribution of host-cell ceramide in cells following C. parvum infection. As predicted, a strong accumulation of BoDipy-ceramide was detected at infection sites (Fig. 2C). Quantitative analysis of ASM and ceramide accumulation was shown in Fig. 2E. To determine whether the accumulation of ceramide is due to de novo synthesis from activated ASM at infection sites or from aggregation of pre-existing ceramide to infection sites, we tested whether inhibition of ASM blocks C. parvum-induced aggregation of ceramide. Cultured cholangiocytes were transfected with an ASM-siRNA (small interfering RNA) that decreased ASM protein expression by up to 65% as confirmed by Western blot (Fig. 2D). Cells were then exposed to C. parvum. Aggregation of ceramide was assessed by dual labelling with C. parvum and ceramide antibodies. A significant reduction of ceramide accumulation at infection sites was found in cells transfected with the ASM-siRNA compared with cells transfected with a scrambled siRNA (Fig. 2E), suggesting that a significant portion of the ceramide accumulated at the infection sites may result directly from the activation of ASM. The results suggest that ASM and ceramide, molecules associated with SEM aggregation to form larger platforms on the membrane, are also recruited to infection sites.
Fig. 2. Cryptosporidium parvum recruits host-cell ASM and ceramide to infection sites.
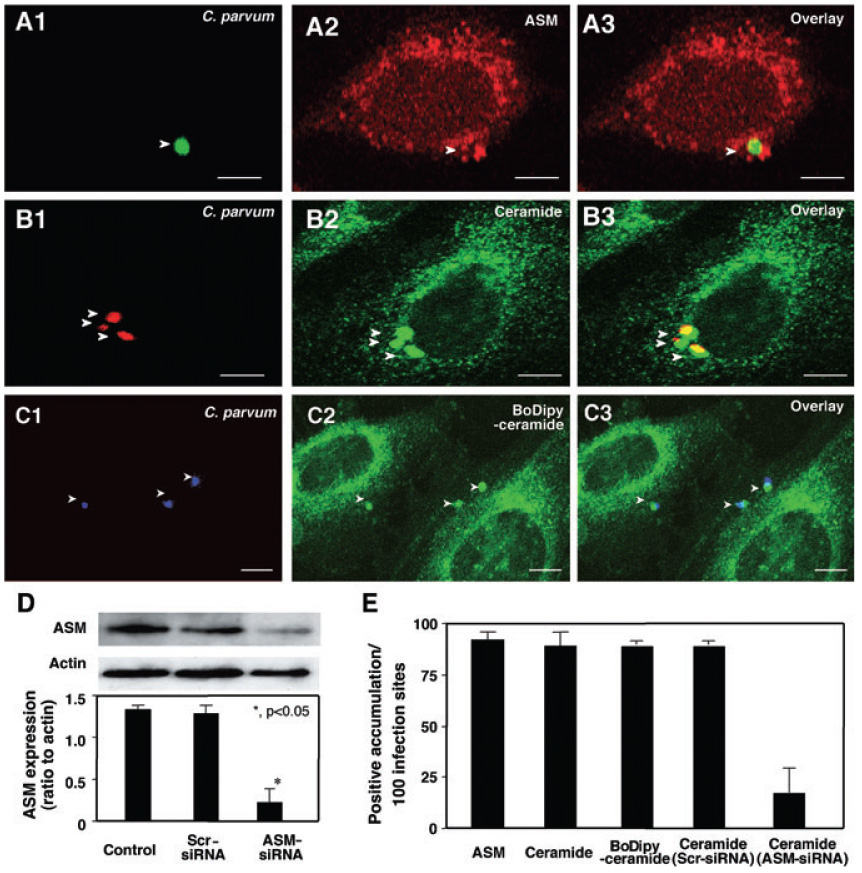
A–C. H69 cells were infected with C. parvum and labelled with antibodies to ASM or ceramide or BoDipy-ceramide followed by fluorescent microscopy. C. parvum was labelled using an specific antibody [arrowheads in (A1)–(C1)]. Accumulation of ASM [arrowhead in (A2) and (A3)] and ceramide [arrowheads in (B2) and (B3)] was observed at infection sites. Accumulation of ceramide was further confirmed by the recruitment of BoDipy-ceramide to infection sites [arrowheads in (C2) and (C3)]. Bar = 5 µm.
D. Suppression of ASM protein expression in H69 cells by ASM-siRNAs was confirmed by Western blot analysis. H69 cells were transfected with 10 nM ASM-siRNA and corresponding scrambled siRNAs for 24 h followed by Western for ASM and actin. Representative immunoblots and quantitative analysis show a significant suppression of ASM protein levels by ASM-siRNA but not scrambled siRNAs. *P < 0.05, compared with control.
E. Quantitative analysis as determined by number of sites with positive accumulation per 100 infection sites.
Membrane translocation of ASM was further confirmed by flow cytometry analysis. After infection with C. parvum for 45 min, the cells were labelled with ASM antibody without membrane permeabilization and analysed by flow cytometry to detect surface ASM levels. As a positive control, some cells were permeabilized, an approach that would allow the antibody to bind to intracellular ASM as well as surface ASM. No change of cell surface fluorescence for ASM was found in the non-infected cells compared with sham-infected cells. In contrast, cells infected with C. parvum showed a significant (P < 0.05) increase of surface ASM fluorescence compared with sham-infected cells as indicated by a peak shift to the right (Fig. 3A) and by quantitative fluorescent intensity analysis (Fig. 3B), suggesting an increase in surface ASM in infected cells compared with sham-infected cells. No significant difference in fluorescence was detected between permeabilized infected cells and permeabilized sham-infected cells (Fig. 3A and B), suggesting that no increase in total ASM protein occurs immediately after infection.
Fig. 3. Cryptosporidium parvum infection induces activation and membrane translocation of ASM in infected cells.
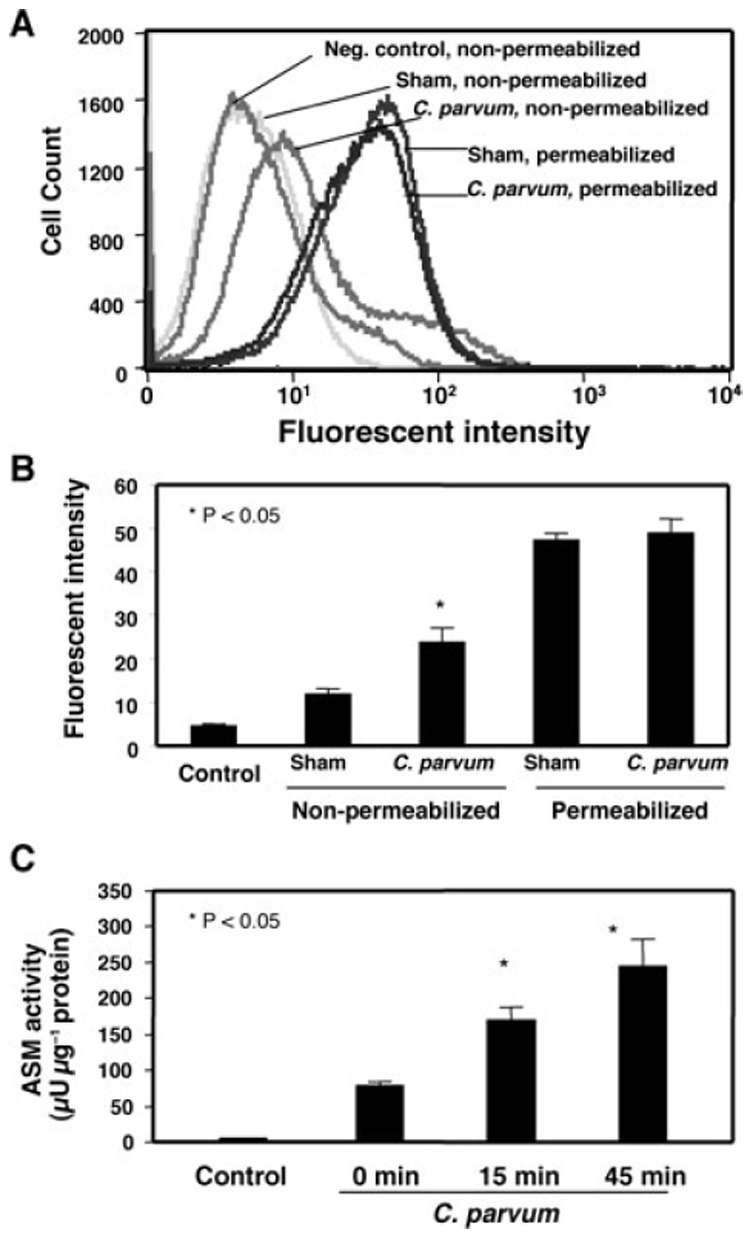
A. A representative flow cytometry plot of ASM labelling in H69 cells. Right shift of fluorescence to ASM was found in non-permeabilized cells after exposure to C. parvum. No change of fluorescence was found in membrane-permeabilized cells.
B. Quantitative analysis of fluorescent intensity to ASM by flow cytometry from three independent experiments.
C. Activation of ASM as assessed by measuring the production of phosphorylcholine, a product of sphingomyelin hydrolation by ASM. A significant increase in phosphorylcholine production reflecting ASM activity was detected in cells after exposure to C. parvum for 15 min and 45 min.
To test whether ASM is actually activated in cholangiocytes during C. parvum infection, cells were infected with C. parvum at 15 min intervals to determine the level of active ASM. Activation of ASM was determined by measuring the amount of sphingomyelin converted to ceramide using a sphingomyelinase assay kit that measures phosphorylcholine, a product of sphingomyelin hydrolysis by ASM (He et al., 2002). After incubating cells with C. parvum for 15 min, a time-dependent increase of ASM activity was detected (Fig. 3C). These results suggest that ASM is activated and accumulated to infection sites during C. parvum infection.
Disruption of SEM components and associated membrane aggregation decreases C. parvum attachment to and entry of cholangiocytes
To test whether aggregation of SEMs at the infection sites are involved in C. parvum host-cell attachment and cellular entry, we used various pharmacological reagents that disrupt SEM components and associated membrane aggregation (Kruth et al., 1986; Okada et al., 1988; Mays et al., 1995; Nichols, 2003) and tested their effects on C. parvum host-cell attachment and cell entry. When cells were fixed and then exposed to C. parvum, a model to test the attachment of host-cell surface by C. parvum (Chen and LaRusso, 2000), we found that cells treated with 10 mM methyl-β-cyclodextrin (a chemical agent that has been shown to disrupt microdomains by sequestering cholesterol), fumonisin B1 (inhibits sphingomyelin synthesis) and threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) (inhibits glycosphingolipid synthesis) (Okada et al., 1988; Mays et al., 1995) showed a significant (P < 0.05) decrease in the attachment of C. parvum (Fig. 4A). Disruption of host-cell SEM components and associated membrane aggregation by those reagents was confirmed by confocal microscopy (data not shown). We further tested whether inhibition of aggregation of SEMs by blocking the production of ASM would reduce C. parvum attachment. Cholangiocytes were treated with an siRNA to ASM and then infected with C. parvum. A significant (P < 0.05) decrease of C. parvum attachment was detected in cells treated with siRNA to ASM (Fig. 4A). Transfection of cells with a scrambled siRNA did not show any effect on C. parvum attachment. Moreover, when unfixed living cells were exposed to C. parvum sporozoites, a model in which the organism can both attach to and enter into host cells (Chen and LaRusso, 2000), significantly fewer parasites were detected in cells transfected with ASM-siRNA or in the presence of various pharmacological reagents (Fig. 4B). No change of C. parvum cellular invasion was found in cells transfected with a scrambled siRNA to ASM. Taken together, our results suggest that disruption of SEM components and associated membrane aggregation decreases C. parvum attachment to and entry of cholangiocytes.
Fig. 4. Disruption of aggregation of SEMs hinders C. parvum attachment to and entry of cholangiocytes. H69 cells treated with pharmacological agents known to disrupt SEMs or siRNA to ASM were exposed to C. parvum for 2 h followed by immunofluorescent microscopy.
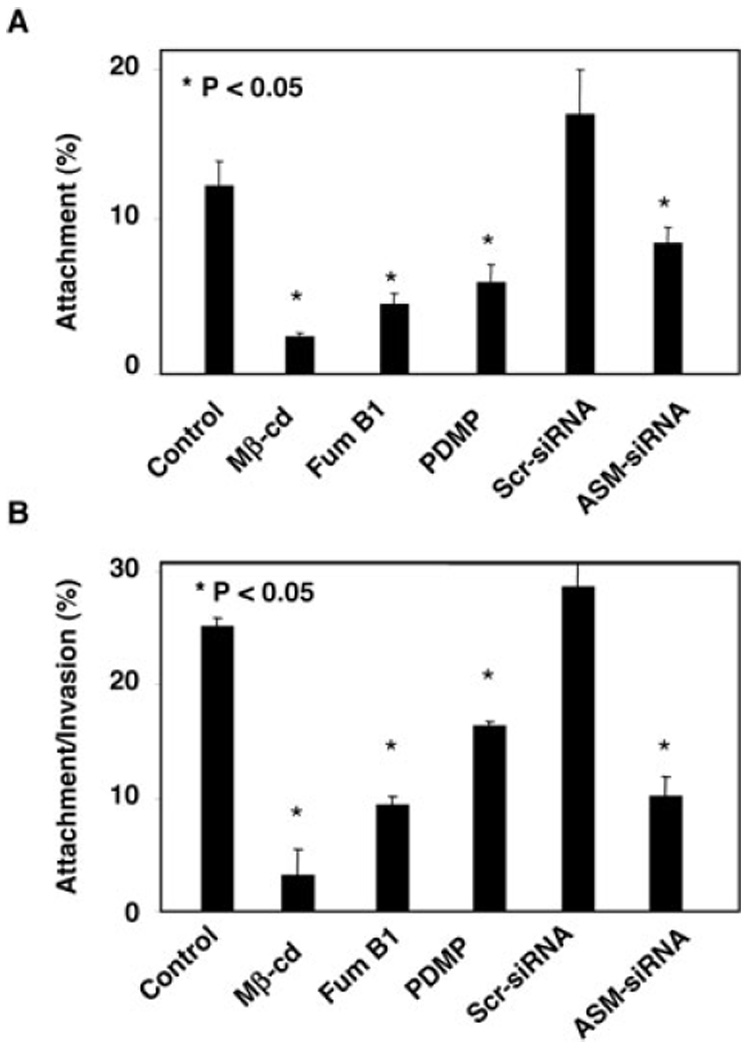
A. Attachment assay in prefixed cells. Cells treated with methyl-β-cyclodextrin (Mβ-cd), Fumonocin B1 (Fum B1), PDMP or ASM-siRNA each showed a significant decrease of C. parvum attachment to host cells.
B. Attachment/invasion assay in non-fixed cells. Cells treated with Mβ-cd, Fum B1, PDMP or ASM-siRNA each showed a significant decrease of C. parvum attachment/invasion rate. Scr-siRNA, scrambled siRNA.
Accumulation of Gal/GalNAc glycoproteins for C. parvum attachment is associated with SEM aggregation at infection sites
To test whether host-cell Gal/GalNAc epitopes of membrane glycoproteins are aggregated to infection sites during C. parvum infection, we used a FITC-tagged PNA, a lectin that specifically binds to Gal/GalNAc epitopes, to label membrane-associated Gal/GalNAc epitopes during C. parvum infection as we previously reported (Chen and LaRusso, 2000). We found a strong accumulation of FITC-PNA fluorescence at infection sites (Fig. 5A). Furthermore, disruption of SEM components by methyl-β-cyclodextrin treatment and knockdown of ASM activation by ASM-siRNA transfection blocked C. parvum-associated accumulation of Gal/GalNAc epitopes at infection sites (Fig. 5B and C). Cells transfected with ASM-siRNA were identified by the tagged Cy3 fluorescence in the cytoplasm, consistent with our previous studies (Chen et al., 2005). Parasites alone showed no obvious fluorescence to PNA (data not shown). Quantitative analysis of Gal/GalNAc epitope accumulation at infection sites was shown in Fig. 5E. In addition, transfection of cells with a scrambled siRNA failed to block C. parvum-induced accumulation of Gal/GalNAc epitopes at infection sites (Fig. 5E). These data indicate that SEMs are involved in C. parvum attachment to host cells, possibly via accumulation or clustering of associated host-cell membrane receptors to infection sites.
Fig. 5. Cryptosporidium parvum induces accumulation of Gal/GalNAc at infection sites in a SEM-dependent manner.
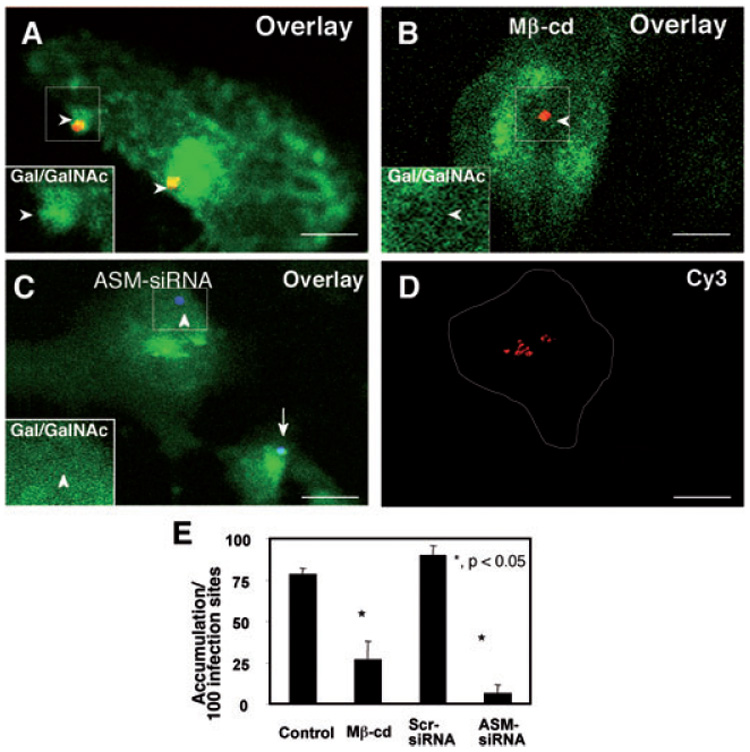
A–D. Cultured cholangiocytes were pre-treated with either methyl-β-cyclodextrin (Mβ-cd) or ASM-siRNA and then exposed to C. parvum sporozoites followed by dual labelling fluorescence with a polyclonal antibody to C. parvum and FITC-tagged PNA lectin to label Gal/GalNAc epitopes as we previously reported (Chen and LaRusso, 2000). The left lower insets in (A)–(C) show labelling of Gal/GalNAc in the boxed regions. (A) Untreated cells showed strong labelling of FITC-tagged PNA lectin at infection sites [arrowheads in (A) and inset]. Cells treated with 10 mM Mβ-cd for 1 h prior to C. parvum infection did not show strong labelling of FITC-tagged PNA lectin at infection sites [arrowheads in (B) and inset]. Cells transfected with Cy3-tagged ASM-siRNA showed a significant decrease in Gal/GalNAc accumulation at infection sites [arrowheads in (C) and inset], whereas non-transfected cells showed strong Gal/GalNAc accumulation [arrow in (C)]. Cells transfected with ASM-siRNA were identified by positive fluorescence of Cy3 [as outlined in (D)]. Bar = 5 µm.
E. Quantitative analysis as determined by number of sites with positive accumulation per 100 infection sites. *P < 0.05, compared with control. Scr-siRNA, scrambled siRNA.
SEMs are required for C. parvum-induced accumulation and activation of PI-3K at infection sites
Entry of host cells by C. parvum is dependent on class IA PI-3K-mediated host-cell actin remodelling at infection sites. Thus, we tested whether SEMs are required for C. parvum-induced accumulation and activation of class IA PI-3K. We found that both disruption of SEM components by methyl-β-cyclodextrin and knockdown of ASM via siRNA transfection blocked C. parvum-induced PI-3K accumulation at infection sites (Fig. 6A–D). Transfection of cells with a scrambled siRNA failed to block C. parvum-induced PI-3K accumulation at infection sites (Fig. 6E). Consistent with our previous studies (Chen et al., 2004a), parasites alone showed no positive reaction to the PI-3K antibody with our staining approach. To test whether SEMs are required for activation of PI-3K during C. parvum infection, we measured the phosphorylation of immunoprecipitated PI-3K via Western blot after disruption of SEMs. The Western blot analysis shows that cells infected with C. parvum for 1 h displayed an increase in phosphotyrosine of PI-3K compared with sham-infected controls, confirming our previous studies (Chen et al., 2004a). Cells treated with methyl-β-cyclodextrin before C. parvum infection showed no increase in phosphotyrosine of PI-3K compared with control cells. Cells transfected with siRNA to ASM also showed no increase in phosphotyrosine of PI-3K after 1 h infection with C. parvum. However, cells transfected with scrambled siRNAs showed an increase in PI-3K phosphotyrosine after 1 h infection with C. parvum, similar to untransfected cells exposed to the parasite (Fig. 6F). These results suggest that SEM aggregation is required for C. parvum-induced accumulation and activation of PI-3K in infected cells.
Fig. 6. Inhibition of SEM aggregation hinders C. parvum-induced accumulation and activation of PI-3K in infected cells.
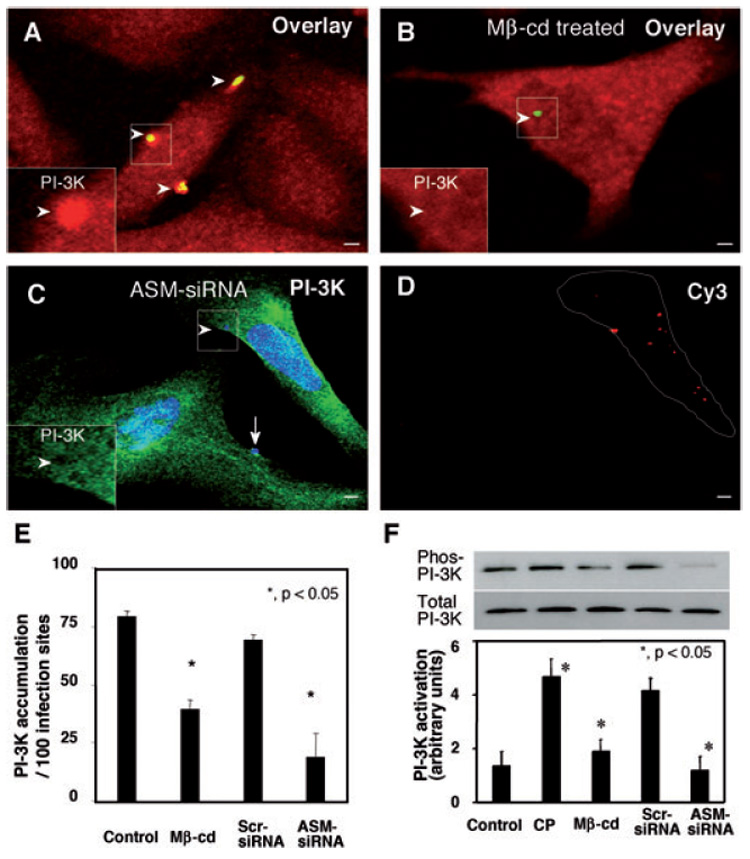
A–D. H69 cells were treated with 10 mM methyl-β-cyclodextrin (Mβ-cd) or ASM-siRNA and then infected with C. parvum sporozoites. Accumulation and activation of PI-3K were determined by dual immunofluorescent labelling and Western blot analysis of tyrosine-phosphorylated p85 subunit of class IA PI-3K as we previously reported (Chen et al., 2004a). The left lower insets in (A)–(C) show labelling of PI-3K in the boxed regions. Whereas non-treated cells showed strong accumulation of PI-3K at infection sites [arrowheads in (A) and inset], treatment of cells with Mβ-cd reduced accumulation of PI-3K [arrowhead in (B) and inset]. Inhibition of PI-3K accumulation was also detected in cells transfected with Cy3-tagged ASM-siRNA [arrowheads in (C) and inset]. In contrast, non-transfected cells showed strong PI-3K accumulation [arrow in (C)]. Cells transfected with ASM-siRNA were identified by positive fluorescence of Cy3 [as outlined in (D)]. Bar = 5 µm.
E. Quantitative analysis of PI-3K accumulation by dual immunofluorescent labelling.
F. Activation of PI-3K as assessed by Western blot analysis of tyrosine-phosphorylated p85 subunit of PI-3K. The PI-3K p85 subunit was immunoprecipitated from whole-cell lysates, separated by SDS-PAGE and then probed for phosphotyrosine. The same whole-cell lysates were also probed for p85 to demonstrate unchanged total p85 protein level. *P < 0.05, compared with control. CP, C. parvum; Scr-siRNA, scrambled siRNA.
SEMs are required for Cdc42-medicated actin remodelling at infection sites
Activation of PI-3K is required for C. parvum cellular entry via activation of Cdc42-mediated actin remodelling at infection sites. To further test the role of SEMs in C. parvum cellular entry, we tested whether disruption of SEMs and associated membrane aggregation blocks PI-3K/Cdc42-mediated actin remodelling at infection sites. H69 cells were treated with methyl-β-cyclodextrin or siRNA to ASM and then infected with sporozoites. Consistent with our previous studies (Chen et al., 2004b), parasites alone showed no positive staining to the Cdc42 antibody or fluorescein-phallooidin with our staining approach. The accumulation of Cdc42 and actin at the host–parasite interface was assessed by double immunofluorescent staining. Strong accumulation of Cdc42 (Fig. 7A) and actin (Fig. 7C) was found at the host–parasite interface in the non-treated cells as previously reported (Forney et al., 1999; Chen and LaRusso, 2000; Elliott and Clark, 2000; Chen et al., 2004b). In cells pre-treated with methyl-β-cyclodextrin, a reduced accumulation of Cdc42 (Fig. 7B) and actin (Fig. 7D) was found at infection sites. Host cells transfected with ASM-siRNA also showed a significant reduction in actin accumulation at infection sites compared with nearby non-transfected cells (Fig. 7E and F). In contrast, cells transfected with a scrambled siRNA show a comparable actin accumulation as non-transfected cells (Fig. 7G and H). Quantitative analysis of Cdc42 and actin accumulation at infection sites was shown in Fig. 7I. These data suggest that SEMs are required for C. parvum-induced and PI-3K/Cdc42-medicated actin remodelling at infection sites.
Fig. 7. Inhibition of SEM aggregation hinders C. parvum-induced accumulation of Cdc42 and actin at infection sites.
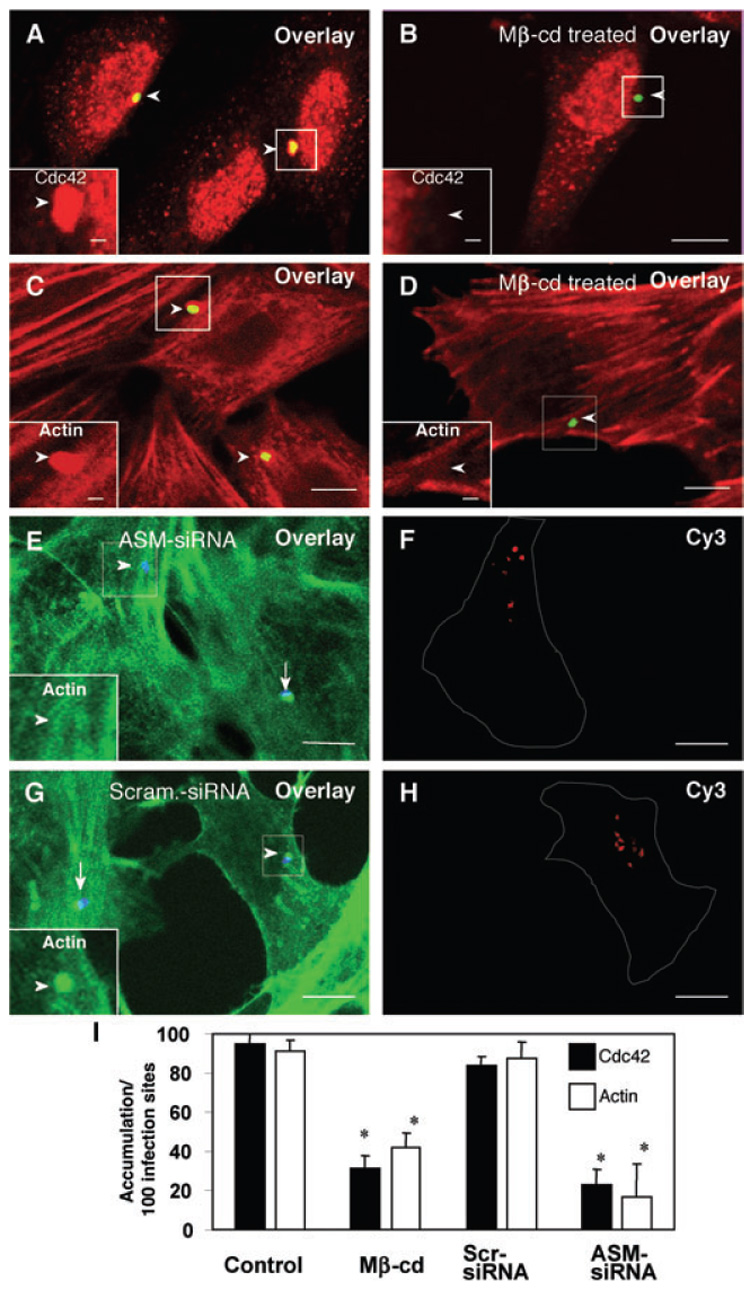
A–H. H69 cells were treated with 10 mM methyl-β-cyclodextrin (Mβ-cd) or siRNA to ASM and then infected with sporozoites. Accumulation of Cdc42 and actin were determined by dual immunofluorescent labelling. The left lower insets in (A)–(E) and (G) show labelling of Cdc42 (A and B) or actin (C–E and G) in the boxed regions. Whereas non-treated cells showed strong accumulation of Cdc42 [arrowheads in (A) and inset] and actin [arrowheads in (C) and inset], treatment of cells with Mβ-cd reduced accumulation of Cdc42 [arrowhead in (B) and inset] and actin [arrowhead in (D) and inset] at infection sites. Inhibition of actin accumulation was also detected in cells transfected with Cy3-tagged ASM-siRNA [arrowheads in (E)] but not in adjacent non-transfected cells [arrow in (E)] or in cells transfected with a scrambled siRNA [arrowheads in (G)]. Cells transfected with Cy3-tagged siRNAs were identified by positive fluorescence of Cy3 [as outlined in (F) and (H)]. Bar = 5 µm.
I. Quantitative analysis of Cdc42 and actin accumulation by dual fluorescent labelling. *P < 0.05, compared with control. Scr-siRNA, scrambled siRNA.
Discussion
The results of our studies provide the first evidence suggesting an important role of SEMs in C. parvum attachment to and entry of host epithelial cells. Using an in vitro model of human biliary cryptosporidiosis, we found that: (i) C. parvum infection activates host-cell ASM to recruit selective SEM components to infection sites, (ii) disruption of known SEM components and associated SEM membrane aggregation decreases C. parvum attachment to and entry of cholangiocytes, (iii) recruitment of SEM components is associated with accumulation of Gal/GalNAc-associated membrane-binding molecules for parasite attachment at infection sites and (iv) recruitment of SEM components is involved in C. parvum-induced activation of the PI-3K/Cdc42 pathway and subsequent actin remodelling at the infection sites. Thus, SEMs are required for C. parvum attachment to and entry of host cells.
Sphingolipid-enriched membrane microdomains are key to the regulation of protein interactions on the cell membrane as components of SEMs can be aggregated to form larger platforms upon stimulation. This process is associated with selective recruitment of functionally related proteins and has been implicated in various membrane functions, including activation of intracellular signalling pathways and caveolae-dependent endocytosis (Brown and London, 1998; Bollinger et al., 2005). Intracellular pathogens, including bacteria, virus and parasites, have developed sophisticated strategies to enter host cells and one strategy appears to be to hijack the SEM machinery on the host-cell membrane surface (Mordue et al., 1999; Shin et al., 2000; Duncan et al., 2002; Haldar et al., 2002; Manes et al., 2003; Lafont and van der Goot, 2005). Here, we found that components of SEMs are recruited to infection sites during C. parvum infection of cultured human cholangiocytes. ASM, an enzyme that converts sphingomyelin into ceramide resulting in aggregation of SEMs into larger platforms (Grassme et al., 2003; Gulbins et al., 2004), is also recruited to infection sites. In addition, membrane translocation and activation of ASM were detected in cells exposed to C. parvum. Indeed, ceramide accumulated at the infection sites as well. Thus, accumulation of SEM components and aggregation of SEMs to form larger platforms occur at C. parvum infection sites. Interestingly, disruption of SEM components and knockdown of ASM to inhibit aggregation of SEMs decrease both C. parvum attachment to and entry of cultured cholangiocytes, suggesting that both C. parvum attachment and cellular entry require host-cell SEMs.
Cryptosporidium parvum attachment to the host-cell surface is mediated via poorly characterized parasite ligands which bind to the host-cell membrane receptors. Obviously, multiple host-cell membrane molecules are involved in this process and so far, Gal/GalNAc glycoprotiens, gp14/40 and TRAP C1 have been implicated as binding molecules for C. parvum attachment (Sultan et al., 1997; Spano et al., 1998; Chen and LaRusso, 2000). Interestingly, disruption of host-cell SEM components and knockdown of ASM partially blocked C. parvum attachment to cultured cholangiocytes (i.e. prefixed cells for C. parvum attachment), suggesting that host-cell membrane molecules for C. parvum attachment may be SEM-associated. Moreover, using FITC-PNA lectin to label the Gal/GalNAc epitopes on the cell surface, we found accumulation of fluorescence at infection sites when non-fixed cells were exposed to C. parvum, suggesting recruitment of Gal/GalNAc-associated membrane molecules for C. parvum attachment to infection sites. Obviously, this process would further favour the parasite host-cell attachment as more receptors at the infection site would assure stronger binding of the parasite to the host-cell membrane surface.
Cellular entry by C. parvum is dependent upon host-cell membrane protrusion mediated by the activation of class IA PI-3K/Cdc42 resulting in actin remodelling at infection sites. Induced membrane protrusions encapsulate the parasite and form a parasitophorous vacuole where C. parvum resides and further develops. How C. parvum activates host-cell class IA PI-3K kinase to induce actin remodelling at infection sites is unclear. Our current study provides the first evidence suggesting that SEMs are required for C. parvum-induced activation of class IA PI-3K and subsequent actin remodelling at infection sites. We found that disruption of SEM components and knockdown of ASM decrease C. parvum-induced accumulation of class IA PI-3K, Cdc42 and actin at infection sites. Moreover, phosphorylation of class IA PI-3K is also inhibited by SEM disruption and knockdown of ASM. Thus, activation of the PI-3K/Cdc42 signalling pathways and subsequent actin remodelling at the infection sites induced by C. parvum requires aggregation of SEMs, a process facilitating parasite entry of host cells. SEM-mediated actin remodelling via activation of Cdc42/Arp2/3 signalling pathway has also been reported during infection by other pathogens including hepatitis C virus and Listeria monocytogenes (Seveau et al., 2004). In addition, involvement of SEMs in the activation of PI-3K has been reported in insulin-like growth factor-I-dependant PI-3K/Akt recruitment and phosphorylation within SEMs (Remacle-Bonnet et al., 2005).
Many pathogenic microbes enter host cells via endocytosis. We previously demonstrated that neither clathrin nor Dynamin II-mediated endocytosis is involved in C. parvum cellular internalization (Chen et al., 2003). The caveolae-dependent endocytotic pathway, a process mediated by a portion of SEMs on the cell surface, is involved in the host-cell entry by some intracellular pathogens including bacteria and viruses, such as FimH-expressing Escherichia coli and SV40 (Shin et al., 2000; Pelkmans et al., 2001; Duncan et al., 2002). However, we did not detect accumulation of caveolin-1, a major component in caveolae, at C. parvum infection sites, suggesting that the caveolae-dependent endocytotic pathway may not be involved in C. parvum infection. Whether neutral sphingomyelinase, an enzyme that converts sphingomyelin into ceramide from the inner leaflet of the plasma membrane (Rouquette-Jazdanian et al., 2005), is also activated and involved in C. parvum infection requires further investigation.
In conclusion, using an in vitro model of biliary cryptosporidiosis, we demonstrated that C. parvum hijacks host-cell membrane SEM machinery for parasite attachment to and entry of host epithelial cells. Our results support the hypothesis that, upon contact with the host-cell membrane surface, C. parvum activates host-cell ASM to generate ceramide resulting in aggregation of small SEMs into larger platforms at infection sites. This process clusters membrane receptors and triggers actin remodelling via activation of the PI-3K/Cdc42 signalling pathway at infection sites and thus, facilitates parasite attachment to and entry of host cells. Future efforts should define the molecular mechanisms by which C. parvum activates host-cell ASM and clarify the role of SEMs in other host epithelial cell responses against C. parvum infection.
Experimental procedures
H69 cells and C. parvum
H69 cells, a gift of Dr Jefferson (Tufts University, Boston, MA), are a human cholangiocyte cell line derived from normal liver harvested for transplant. They were immortalized by simian virus 40 transformation and have been extensively characterized. Cells maintained for three passages without co-culture cells to ensure that the culture was free of 3T3 fibroblasts and between passage 23 and passage 30 were used for experiments. C. parvum oocysts of the Iowa strain were purchased from a commercial source (Bunch grass farms, Deary, ID). Oocysts are treated with 1% sodium hypochlorite at 4°C for 20 min and then washed twice with PBS and once with DMEM-F12 medium (Life Technologies, Carlsbad, CA). The oocysts are then incubated with excitation media consisting of 0.75% taurodeoxycholate and 0.25% trypsin at 37°C for 30 min. Excysted sporozoites were then collected to infect cells.
Infection models and infection assay
Cryptosporidium parvum infection was performed as described previously (Chen and LaRusso, 2000). Briefly, H69 cells were grown to 70–80% confluence and exposed to a culture medium of DMEM-F12 containing 100 units ml−1 penicillin, 100 µg ml−1 streptomycin (Sigma-Aldrich, St Louis, MO) and freshly excysted sporozoites. Inactivated sporozoites (treated at 65°C for 30 min) were used for sham infection controls (O’Donoghue, 1995). The infection was then carried out at 37°C for 1 or 2 h and then fixed with 2% paraformaldehyde for 30 min followed by immunofluorescence microscopy.
Two models were used as previously reported (Chen and LaRusso, 2000): the attachment model and attachment/invasion model. For the attachment model, the cells are fixed before infecting with C. parvum; for the attachment/invasion model, the cells are fixed following the infection. Infection assay was carried out using an indirect immunofluorescent approach (Chen and LaRusso, 2000; Zhu et al., 2000). The number of cells and parasites were counted randomly and the results were expressed as attachment or attachment/invasion rate [(the number of parasites per total number of cells) × 100]. A total of > 1000 cells were counted for each slide, and all assays were performed in triplicate.
Labelling of SEM components and associated membrane aggregation
Cholera toxin B was used to label GM1 and Fillipin to label cholesterol as reported by others (Kruth et al., 1986; Nichols, 2003). Accumulation of ceramide was measured either by transfection of cells with Bodipy-FL-Ceramide (Molecular Probes, Eugen, OR) or by immunostaining with a specific antibody (25 µg ml−1, Sigma-Aldrich) (Okazaki et al., 1989; Pagano et al., 1991). Accumulation of ASM was measured by immunostaining with a specific antibody (30 µg ml−1, Santa Cruz Biotechnology, Santa Cruz, CA). To test whether SEM-associated caveolae-mediated endocytosis is involved in C. parvum infection, caveolin-1 was stained using a monoclonal antibody (1 µg ml−1, Santa Cruz) as we previously reported (Mazzone et al., 2006). A monoclonal antibody to transferrin receptor (2 µg ml−1, Santa Cruz), a protein known to be not associated with SEM, was used as the control (Jing et al., 1990). For labelling, H69 cells were grown in culture to 60–75% confluence on four-well or eight-well chamber slides (Falcon). After exposure to C. parvum sporozoites for 2 h, cells were fixed and incubated with antibodies or Fillipin (Sigma-Aldrich) followed by confocal microscopy. For labelling with cholera toxin B and Bodipy-FL-Ceramide, cells were first incubated with cholera toxin B FITC conjugate (10 µg ml−1, Sigma-Aldrich) for 30 min at 37°C or with Bodipy-FL-Ceramide (200 µM) for 30 min on ice. Treated cells were then exposed to C. parvum sporozoites for 2 h followed by fixation and confocal microscopy. For the SV40 infection, H69 cells were grown on four-well glass slides and incubated with the SV40 virus at 37°C for 3 h. The cells were then washed with DMEM and fixed followed by immunofluorescent confocal microscopy as described above.
Confocal microscopy
Cells were fixed [0.1 mol l−1 piperazine-1,4-bis[2-ethanesulphonic acid] (Sigma-Aldrich), pH 6.95, 1 mM ethylene glycol-bis [2-oiminoether]N,N,N′,N′-tetraacetic acid] (Sigma-Aldrich), 3 mM magnesium sulphate (Sigma-Aldrich) and 2% paraformaldehyde] at 37°C for 20 min and then permeabilized with 0.2% (v/v) Triton X-100 in PBS. Cells labelled with cholera toxin B FITC conjugate, Fillipin or Bodipy-FL-Ceramide were incubated with the polyclonal antibody to C. parvum. For dual immunofluorescent labelling using two antibodies, fixed cells were incubated with primary monoclonal antibodies to associated molecules mixed with the polyclonal antibody against C. parvum sporozoite (Zhu et al., 2000). Some cells were incubated with polyclonal antibodies to associated molecules mixed with a monoclonal antibody against C. parvum (2H2, ImmunuCell, Portland, ME). Labelled cells were then mounted with mounting medium (H-1000, Vector Laboratories) followed by confocal microscopy. To test accumulation of PI-3K and Cdc42 at the infection sites, monoclonal antibodies to PI-3K (5 ng ml−1, Upstate) or Cdc42 (5 ng ml−1, Santa Cruz) were used. For localization of actin with C. parvum, fluoresceinphalloidin (Sigma-Aldrich) was incubated for 30 min at room temperature after incubation with the polyclonal antibody to C. parvum. The numbers of parasites with and without accumulation of associated molecules of interest at the infection sites were counted separately for quantitative analysis as previously described (Chen et al., 2004c). The results were expressed as the accumulation percentage (the number of parasites with an accumulation of the molecules at the infection site/total number of parasites × 100). Between 500 and 1000 C. parvum cells were counted randomly for each assay.
Functional disruption of SEM components and associated membrane aggregation
Disruption of SEM components was performed by either pharmaceutical agents or siRNAs against ASM. To disrupt rafts by cholesterol depletion, the cells were treated with 10 mM methyl-β-cyclodextrin in DMEM F-12 at 37°C for 1 h. Cells were treated at 37°C for 24 h with Fumonisin B1 (25 µg ml−1; Sigma-Aldrich) to inhibit sphingomyelin synthesis or with PDMP (25 µg ml−1; Sigma-Aldrich) to inhibit glycosphingolipid synthesis (Okada et al., 1988; Mays et al., 1995). No cytotoxic effects on H69 cells or on C. parvum sporozoites after treatment of these reagents were detected as assessed by the viability assay as we previously reported (Chen and LaRusso, 2000; Chen et al., 2004c).
To generate siRNAs to ASM, a web-based software program from Ambion® (Austin, TX) was used to select several appropriate RNA target sequences from ASM mRNA and to generate their respective sense and antisense oligonucleotide sequences. The oligonucleotides were constructed at the Mayo Molecular Core facility (Rochester, MN) and ASM-siRNAs were synthesized using Silencer™ siRNA construction kit (Ambion). A non-specific siRNA containing the same nucleotides but in irregular sequence (i.e. scrambled siRNA) was used as the control. To test the inhibitive capacity of the constructed ASM-siRNAs, H69 cells were transfected with the siRNAs using a siPort lipid (Ambion) according to manufacture’s protocol. Expression of ASM in transfected cells was then measured by Western blot. An ASM-siRNA with a sequence of AATTTGAGGTCTTCTATGATG showed significant inhibition of ASM protein expression (up to 65%) was selected for the study. This ASM-siRNA was further labelled with Cy3 using the Silencer siRNA labelling Kit (Ambion) and used for confocal microscopy. Cells were transfected with the siRNA-ASM 24 h prior to exposure to C. parvum.
Activation assay for ASM
Activation of ASM was carried out using the Amplex ®Red Sphingomyelinase Assay Kit (Molecular Probes). Cells were exposed to C. parvum for 15 and 45 min in T75 flasks and then lysed with Cell Disassociation Solution® (Sigma-Aldrich) for 15 min. ASM was immunoprecipitated using a polycolonal antibody to ASM (Santa Cruz) and ASM activity was assessed according the protocol from the manufacturer. A standard curve was made using ASM enzyme supplied with the kit to convert absorbance readings into enzymatic units per microlitre. The protein concentration of the samples was measured using a Bradford assay and used to confirm equal loading of the samples.
Membrane translocation of ASM as assessed by flow cytometry
T25 flasks with H69 cells grown to 95% confluence were infected with 4 × 107 C. parvum sporozoites and incubated at 37°C for 1 h. The cells were then treated with cell disassociation solution (Sigma-Aldrich) for 15 min at 37°C and followed by membrane permeabilization with 0.2% Triton X-100 for 2 min. Some cells were processed without membrane permeabilization only to reveal membrane surface labelling of ASM. Cells were then incubated with a rabbit polyclonal antibody to ASM (30 µg ml−1, Santa Cruz) for 1.5 h at 37°C. After washing with PBS, the cells were incubated with a FITC-labelled anti-rabbit secondary antibody (Molecular Probes) at room temperature for 30 min followed by analysis with a FACscalibur flow cytometer. Cells incubated with the secondary antibody only were used as a negative control.
Immunoprecipitation and phosphotyrosine of PI-3K
H69 cells were treated with either ASM-siRNA or methyl-β-cyclodextrin as previously described, and then exposed to C. parvum sporozoites for 1 h. PI-3K immunoprecipitates were obtained using an antibody against the p85 subunit of class IA PI-3K (10 µg ml−1, Upstate) at 4°C overnight to immunoprecipitate class IA PI-3K as we previously described (Chen et al., 2004a). The immunoprecipitated protein then was released with Western sample buffer (20% glycerol, 4% SDS, 10% β-mercaptoethanol, 0.05% bromphenol blue and 1.25 M Tris, pH 6.8), incubated at 95°C for 5 min, separated by SDS-PAGE, and immunoblotted with an antibody to phosphotyrosine (Clone 4G10, Upstate). Immunoreactive bands were developed using a chemiluminescent substrate (ECL Plus, Amersham Biosciences).
Statistical analysis
All values are given as means plus standard errors. Means of groups were compared with the Student’s t-test (unpaired) or anova test when appropriate. P-values less than 0.05 were considered statistically significant.
Acknowledgements
This work was supported by National Institutes of Health Grants GM22942 (R.E.P.), AI071321 (X.-M.C.) and DK57993 and DK24031 (N.F.L.) and by the Mayo Foundation. We thank P.L. Splinter and A. Mazzone for their helpful and stimulating discussions; and Ms D. Hintz for secretarial assistance.
References
- Bollinger CR, Teichgraber V, Gublins E. Ceramide-enriched membrane domains. Biochim Biophys Acta. 2005;1746:284–294. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- Chen XM, LaRusso NF. Mechanisms of attachment and internalization of Cryptosporidium parvum to biliary and intestinal epithelial cells. Gastroenterology. 2000;118:368–379. doi: 10.1016/s0016-5085(00)70219-8. [DOI] [PubMed] [Google Scholar]
- Chen XM, Keithly JS, Paya CV, LaRusso NF. Cryptosporidiosis. N Engl J Med. 2002;346:1723–1731. doi: 10.1056/NEJMra013170. [DOI] [PubMed] [Google Scholar]
- Chen XM, Huang BQ, Splinter PL, Cao H, Zhu G, McNiven M, LaRusso N. Cryptosporidium parvum invasion of biliary epithelia requires host cell tyrosine phosphorylation of cortactin via c-Src. Gastroenterology. 2003;125:216–228. doi: 10.1016/s0016-5085(03)00662-0. [DOI] [PubMed] [Google Scholar]
- Chen XM, Splinter PL, Tietz PS, Huang BQ, Billadeau DD, LaRusso NF. Phosphatidylinositol 3-kinase and frabin mediate Cryptosporidium parvum invasion via activation of Cdc42. J Biol Chem. 2004a;279:31671–31678. doi: 10.1074/jbc.M401592200. [DOI] [PubMed] [Google Scholar]
- Chen XM, Huang BQ, Splinter PL, Orth JD, Billadeau D, MicNiven MA, LaRusso NF. Cdc42 and the actin-related protein/neural Wiskott–Aldrich syndrome protein network mediate cellular invasion by Cryptosporidium parvum. Infect Immun. 2004b;72:3011–3021. doi: 10.1128/IAI.72.5.3011-3021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X-M, O’Hara SP, Huang BQ, Nelson JB, Lin J, Zhu G, et al. Apical organelle discharge by Cryptosporidium parvum is temperature, cytoskeleton, and intracellular calcium dependant and required for host cell invasion. Infect Immun. 2004c;72:6806–6816. doi: 10.1128/IAI.72.12.6806-6816.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X-M, O’Hara SP, Huang BQ, Splinter PL, Nelson JB, LaRusso NF. Localized glucose and water influx facilitates Cryptosporidium parvum cellular invasion by means of modulation of host-cell membrane protrusion. Proc Natl Acad Sci USA. 2005;102:6338–6343. doi: 10.1073/pnas.0408563102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DP. New insights in human cryptosporidiosis. Clin Microbiol Rev. 1999;12:554–563. doi: 10.1128/cmr.12.4.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan MJ, Shin J-S, Abraham SN. Microbial entry through caveolae: variations on a theme. Cell Microbiol. 2002;4:783–791. doi: 10.1046/j.1462-5822.2002.00230.x. [DOI] [PubMed] [Google Scholar]
- Elliott D, Clark DP. Cryptosporidium parvum induces host cell actin accumulation at the host–parasite interface. Infect Immun. 2000;68:2315–2322. doi: 10.1128/iai.68.4.2315-2322.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forney JR, DeWald DB, Yang S, Speer CA, Healey MC. A role for host phosphoinositide 3-kinase and cytoskeletal remodeling during Cryptosporidium parvum infection. Infect Immun. 1999;67:844–852. doi: 10.1128/iai.67.2.844-852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassme H, Jendrossek V, Riehle A, von Kurthy G, Berger J, Schwarz H, et al. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat Med. 2003;9:322–330. doi: 10.1038/nm823. [DOI] [PubMed] [Google Scholar]
- Gulbins E, Dreschers S, Wilker B, Grassme H. Ceramide, membrane rafts and infections. J Mol Med. 2004;82:357–363. doi: 10.1007/s00109-004-0539-y. [DOI] [PubMed] [Google Scholar]
- Haldar K, Mohandas N, Samuel BU, Harrison T, Hiller NL, Akompong T, Cheresh P. Protein and lipid trafficking induced in erythrocytes infected by malaria parasites. Cell Microbiol. 2002;4:383–395. doi: 10.1046/j.1462-5822.2002.00204.x. [DOI] [PubMed] [Google Scholar]
- He X, Chen F, McGovern MM, Schuchman EH. A fluorescence-based, high throughput sphingomyelin assay for the analysis of Niemann–Pick disease and other disorders of sphingomyelin metabolism. Anal Biochem. 2002;306:115–123. doi: 10.1006/abio.2002.5686. [DOI] [PubMed] [Google Scholar]
- Jing SQ, Spencer T, Miller K, Hopkins C, Trowbridge IS. Role of the human transferrin receptor cytoplasmic domain in endocytosis: localization of a specific signal sequence for internalization. J Cell Biol. 1990;110:283–294. doi: 10.1083/jcb.110.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruth HS, Comly ME, Butler JD, Vanier MT, Fink JK, Wenger DA, et al. Type C Nimann-Pick disease. Abnormal metabolism of low density lipoprotein in homozygous and heterozygous fibroblasts. J Biol Chem. 1986;261:16769–16774. [PubMed] [Google Scholar]
- Kwiatkowska K, Frey J, Sobota A. Phosphorylation of FcγRIIS is required for the receptor-induced actin rearrangement and capping: the role of membrane rafts. J Cell Sci. 2003;116:537–550. doi: 10.1242/jcs.00254. [DOI] [PubMed] [Google Scholar]
- Lafont F, van der Goot FG. Bacterial invasion via lipid rafts. Cell Microbiol. 2005;7:613–620. doi: 10.1111/j.1462-5822.2005.00515.x. [DOI] [PubMed] [Google Scholar]
- Lafont F, Tran Van Nhieu G, Hanada K, Sansonetti P, van der Goot FG. Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44–IpaB interaction. EMBO J. 2002;21:4449–4457. doi: 10.1093/emboj/cdf457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes S, del Real G, Martinez-A C. Pathogens: raft hijackers. Nat Rev Immunol. 2003;3:557–568. doi: 10.1038/nri1129. [DOI] [PubMed] [Google Scholar]
- Mays RW, Siemers KA, Fritz BA, Lowe AW, van Meer G, Nelson WJ. Hierarchy of mechanisms involved in generating Na/K-ATPase polarity in MDCK epithelial cells. J Cell Biol. 1995;130:1105–1115. doi: 10.1083/jcb.130.5.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzone A, Tietz P, Jefferson J, Pagano R, LaRusso NF. Isolation and characterization of lipid microdomains from apical and basolateral plasma membranes of rat hepatocytes. Hepatology. 2006;43:287–296. doi: 10.1002/hep.21039. [DOI] [PubMed] [Google Scholar]
- Mordue DG, Desai N, Dustin M, Sibley LD. Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J Exp Med. 1999;190:1783–1792. doi: 10.1084/jem.190.12.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols BJ. GM1-containing lipid rafts are depleted in clathrin-coated pits. Curr Biol. 2003;13:686–690. doi: 10.1016/s0960-9822(03)00209-4. [DOI] [PubMed] [Google Scholar]
- O’Donoghue PJ. Cryptosporidium and cryptosporidiosis in man and animals. Int J Parasitol. 1995;25:139–195. doi: 10.1016/0020-7519(94)e0059-v. [DOI] [PubMed] [Google Scholar]
- Okada Y, Radin N, Hakomori S. Phenotypic changes in 3T3 cells associated with the change of sphingolipid synthesis by a ceramide analog, 2-decanoylamino-3-morpholino-1-phenylpropanol (compound RV538) FEBS Lett. 1988;235:25–29. doi: 10.1016/0014-5793(88)81227-4. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Bell RM, Hannun YA. Sphingomyelin turnover induced by vitamin D3 in HL-60 cells. Role in cell differentiation. J Biol Chem. 1989;264:19076–19080. [PubMed] [Google Scholar]
- Pagano RE, Martin OC, Kang HC, Haugland RP. A novel fluorescent ceramide analogue for studying membrane traffic in animal cells: accumulation at the Golgi apparatus results in altered spectral properties of the sphingolipid precurser. J Cell Biol. 1991;113:1267–1279. doi: 10.1083/jcb.113.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol. 2001;3:473–483. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- Remacle-Bonnet M, Garrouste F, Baillat G, Andre F, Marvaldi J, Pommier G. Membrane rafts segregate pro- from anti-apoptotic insulin-like growth factor I receptor signaling in colon carcinoma cells stimulated by members of the tumor necrosis factor superfamily. Am J Pathol. 2005;167:761–773. doi: 10.1016/S0002-9440(10)62049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotolo JA, Zhang J, Donepudi M, Lee H, Fuks Z, Kolesnick R. Caspase-dependent and -independent activation of acid sphingomyelinase signaling. J Biol Chem. 2005;280:26425–26434. doi: 10.1074/jbc.M414569200. [DOI] [PubMed] [Google Scholar]
- Rouquette-Jazdanian AK, Foussat A, Lamy L, Pelassy C, Lagadec P, Breittmayer JP, Aussel C. Cholera Toxin-B subunit prevents activation and proliferation of human CD4+ T cells by activation of a neutral sphingomyelinase in lipid rafts. J Immunol. 2005;175:5637–5648. doi: 10.4049/jimmunol.175.9.5637. [DOI] [PubMed] [Google Scholar]
- Seveau S, Bierne H, Giroux S, Prevost MC, Cossart P. Role of lipid rafts in E-cadherin- and HGF-R/Met-mediated entry of Listeria monocytogenes into host cells. J Cell Biol. 2004;166:743–753. doi: 10.1083/jcb.200406078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JS, Gao Z, Abraham SN. Involvement of cellular caveolae in bacterial entry into mast cells. Science. 2000;289:785–788. doi: 10.1126/science.289.5480.785. [DOI] [PubMed] [Google Scholar]
- Spano F, Putignani L, Naitza S, Puri C, Wright S, Crisanti A. Molecular cloning and expression analysis of a Cryptosporidium parvum gene encoding a new member of the thrombospondin family. Mol Biochem Parasitol. 1998;92:147–162. doi: 10.1016/s0166-6851(97)00243-0. [DOI] [PubMed] [Google Scholar]
- Sultan AA, Thathy V, Frevert U, Robson KJ, Crisanti A, Nussenzweig V, et al. TRAP is necessary for gliding motility and infectivity of plasmodium sporozoites. Cell. 1997;90:511–522. doi: 10.1016/s0092-8674(00)80511-5. [DOI] [PubMed] [Google Scholar]
- Thompson RC, Olson ME, Zhu G, Enomoto S, Abrahamsen MS, Hijjawi NS. Cryptosporidium and cryptosporidiosis. Adv Parasitol. 2005;59:77–158. doi: 10.1016/S0065-308X(05)59002-X. [DOI] [PubMed] [Google Scholar]
- Tzipori S, Griffiths JK. Natural history and biology of Cryptosporidium parvum. Adv Parasitol. 1998;40:5–36. doi: 10.1016/s0065-308x(08)60116-5. [DOI] [PubMed] [Google Scholar]
- Wuhib T, Silva TM, Newman RD, Garcia LS, Pereira ML, Chaves CS, et al. Cryptosporidial and microsporidial infections in human immunodeficiency virus-infection patients in northeastern Brazil. J Infect Dis. 1994;170:494–497. doi: 10.1093/infdis/170.2.494. [DOI] [PubMed] [Google Scholar]
- Zhu G, Marchewka MJ, Ennis JG, Keithly JS. Direct Isolation of DNA from patient stools for polymerase chain reaction detection of Cryptosporidium parvum. J Infect Dis. 1998;177:1443–1446. doi: 10.1086/517834. [DOI] [PubMed] [Google Scholar]
- Zhu G, Marchewka MJ, Woods KM, Upton SJ, Keithly JS. Molecular analysis of a Type I fatty acid synthase in Cryptosporidium parvum. Mol Biochem Parasitol. 2000;105:253–260. doi: 10.1016/s0166-6851(99)00183-8. [DOI] [PubMed] [Google Scholar]