

Abstract
The cellular response to excessive endoplasmic reticulum (ER) stress includes the activation of signaling pathways, which lead to apoptotic cell death. Here we show that treatment of cultured cardiac myocytes with tunicamycin, an agent that induces ER stress, causes the rapid translocation of δPKC to the ER. We further demonstrate that inhibition of δPKC using the δPKC-specific antagonist peptide, δV1-1, reduces tunicamycin-induced apoptotic cell death, and inhibits expression of specific ER stress response markers such as CHOP, GRP78 and phosphorylation of JNK. The physiological importance of δPKC in this event is further supported by our findings that the ER stress response is also induced in hearts subjected to ischemia and reperfusion injury and that this response also involves δPKC translocation to the ER. We found that the levels of the ER chaperone, GRP78, the spliced XBP-1 and the phosphorylation of JNK are all increased following ischemia and reperfusion and that δPKC inhibition by δV1-1 blocks these events. Therefore, ischemia-reperfusion injury induces ER stress in the myocardium in a mechanism that requires δPKC activity. Taken together, our data show for the first time that δPKC activation plays a critical role in the ER stress-mediated response and the resultant cell death.
Introduction
The endoplasmic reticulum (ER) is the site of synthesis and folding of secreted and membranal proteins. Various pathophysiological stimuli such as hypoxia, glucose deprivation, inhibition of glycosylation, calcium depletion from the lumen, and viral infection induce ER stress and lead to the accumulation of unfolded proteins; this process is collectively called the unfolded protein response (UPR) [1, 2]. When ER stress is prolonged and excessive, damaged cells undergo ER stress-induced apoptosis [1, 2]. This is a unique pathway, involving activation or increased expression of proteins such as the C/EBP homologous protein, CHOP [3] and caspase 12 [4].
The role of ER stress in cardiac pathology has not been extensively investigated. However, there is an increasing evidence suggesting that UPR may be activated in cardiac myocytes and expression of ER chaperone proteins increases in a rat model of pressure overload [5, 6]. Transgenic mice expressing a mutant ER localization receptor, the KDEL receptor, demonstrate that accumulation of misfolded proteins in the ER leads to the development of dilated cardiomyopathy [7]. In addition, recent evidence suggests that cardiac ischemia and hypoxia induce UPR [5, 8, 9]. Elevated UPR response is found in isolated cardiac myocytes subjected to hypoxia, as well as in a mouse model of myocardial infarction. The transcription factor 6, ATF6, is one of the ER membrane resident proteins induced by ER stress. Transgenic mice expressing ATF6 subjected to ischemia-reperfusion injury show increased cardiac levels of ER stress-induced proteins such as GRP78 and a lower level of cell death and apoptosis [9]. Therefore, ER stress appears to be an important component of ischemia and reperfusion-induced apoptosis of the myocardium.
The δPKC isozyme is a major modulator of mitochondrial-dependent apoptosis in several cell types including cardiomyocytes [10, 11]. δPKC is also a substrate of caspase 3, which activates this enzyme by proteolysis [12]. We previously demonstrated that myocardial ischemia-reperfusion results in δPKC translocation to cardiac mitochondria, inducing a pro-apoptotic response including release of cytochrome-c, caspase 3 and 9 activation and inhibition of PARP degradation, all hallmarks of mitochondria-dependent apoptosis [13]. In the present study, we demonstrate for the first time that δPKC also translocates to the ER following ischemia and reperfusion in whole hearts as well as following tunicamycin treatment of cultured cardiac myocytes. We show that translocation of δPKC to the ER contributes to the activation of the ER stress-associated response in these two different models of cardiac myocyte injury.
Materials and Methods
Materials
Tunicamycin was purchased from Sigma-Aldrich (USA). Antibodies targeted towards δPKC, CHOP (GADD153), calnexin and ANT were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). GRP78 was from Stressgen Bioreagents (Victoria BC, Canada). XBP-1 was from Biolegend (San Diego, CA, USA). GAPDH antibody, clone 6C5, was from Advanced Immunochemical (Long Beach, CA, USA). Procaspase-3, JNK and phospho-JNK were from cell signaling Biotechnology (CA, USA). The rat neonatal cardiomyocyte isolation kit was from Cellutron (Highland Park, NJ, USA). LDH assay kit was from Roche Molecular Biotechnology (Indianapolis, USA). The δPKC-specific antagonist peptide, δV1-1 (δPKC inhibitor, amino acids 8–17 [SFNSYELGSL]) was synthesized by American Peptides and conjugated to TAT-carrier peptide (amino acids 47-57) via a cysteine-cysteine S-S bond at their N termini, as previously described [14].
Isolation of rat neonatal cardiac myocytes
Animal care in this investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Cardiac myocytes were isolated by using the cardiomyocyte isolation kit from Cellutron. Cardiac myocytes represent 90–95% of total adherent cells. Cells were maintained in MEM Eagle's with Earle's BSS media (containing 50 U/ml penicillin, 80 μmol/L vitamin B12, 0.1 mM bromodeoxyuridine (BrdU) and 80 μmol/L vitamin C) with 10% serum. For all experiments, cells were transferred to serum-free media (MEM Eagle's with Earle's BSS containing 10 μg/ml insulin, 10μg/ml transferrin, 80 μmol/L vitamin C, 50 U/ml penicillin and 80 μmol/L vitamin B12) on day 4. Twenty-four hours later, fresh serum-free medium was added and the myocytes were treated with tunicamycin as indicated in the figures. The δPKC-selective antagonist peptide, δV1-1, was conjugated to the TAT47–57 carrier peptide for transmembrane delivery [14]. The unconjugated TAT47–57 carrier peptide was used as a control. δV1-1 or TAT peptide (2 μmol/L) was delivered 10 min prior to Tm treatment. For time points exceeding 3 hours, δV1-1 peptide was added again at 3 hours and 7 hours after Tm treatment.
Ex vivo model of ischemia-reperfusion
Male Wistar rats (250 to 300 g) were heparinized (2000 U/kg IP) and then anesthetized with sodium pentobarbital (100 mg/kg IP). Rapidly excised hearts were perfused through an aortic cannulation with oxygenated (95% O2, 5% CO2) Krebs-Henseleit (K-H) buffer containing (in mmol/L) NaCl 120, KCl 5.8, NaHCO3 25, NaH2PO4 1.2, MgSO4 1.2, CaCl2 1.0, and dextrose 10, pH 7.4, at 37°C in a Langendorff coronary perfusion system, as previously described [15]. The coronary flow rate was held constant at 10 mL/min throughout the experiment and hearts were equilibrated for ten minutes before the onset of global ischemia. Ischemia was induced by cessation of the flow of oxygenated buffer for 35 minutes followed by restoration of buffer flow to mimic cardiac reperfusion. To determine the time dependence of PKC translocation to endoplasmic reticulum, reperfusion was commenced for 0, 1, 5, and 15 minutes. Additionally, to determine PKC specific effects on ER function, the TAT-conjugated specific peptide inhibitor, δV1-1 (1 μmol/L), or TAT control, were perfused for the first ten minutes of reperfusion. Normoxic control hearts were perfused for the same time but were not subjected to ischemia/reperfusion. To determine cardiac necrosis, the relative amount of creatine phospho kinase (CPK) release was measured for the first 15 minutes of reperfusion. Additionally, tissue damage was assessed by triphenyl tetrazolium chloride (TTC) staining of heart sections to quantitate the amount of infarcted (dead) tissue, as was previously described [16]. All animal protocols were approved by the Institutional Animal Care and Use Committee of Stanford University.
Myocyte apoptosis and necrosis
LDH leakage assay
The viability of myocytes after 40 hrs treatment with Tm was determined by detection of lactate dehydrogenase (LDH) leakage into the culture media using a cytotoxicity detection kit (Roche Molecular Biochemical), according to the manufacturer's protocol. LDH activity was measured at the optimal density at 492 nm, and was defined as the ratio of LDH activity in the culture medium to the total activity (LDH leakage (%) = (extracellular activity)/(extracellular activity + remaining cellular activity).
Terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) Staining
Cardiac myocytes were treated with δV1-1 for 10 min before Tm treatment. After 24 hrs, apoptotic cells were identified using the TUNEL technique per the manufacturer's instructions (in situ cell death detection kit, Roche applied science). Labeled myocytes were analyzed with a fluorescent microscope.
Endoplasmic reticulum isolation
Cardiac myocytes were washed with cold phosphate-buffered saline (PBS) and incubated on ice in lysis buffer (250 mM sucrose, 20 mmol/L HEPES-NaOH, pH 7.5, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1:300 protease inhibitor cocktail, 1:300 phosphatase inhibitor cocktail) for 30 minutes. Myocytes were then scraped, disrupted by repeated aspiration through a 21 gauge needle and collected in microcentrifuge tubes. Heart tissue was minced and ground by pestle in lysis buffer. The homogenates were spun at 800g for 10 min at 4°C and the resulting supernatants were spun at 10,000g for 20 min at 4°C. The new supernatants were spun at 100,000g for 1 hour at 4°C and the pellet corresponding to the endoplasmic reticulum was resuspended in lysis buffer containing 1% Triton X-100.
Total lysate preparation
Samples were processed as discussed for endoplasmic reticulum isolation in the following lysis buffer (10 mmol/L HEPES-NaOH, pH 7.5, 150 mmol/L NaCl, 1% Triton X-100, 1:300 protease inhibitor cocktail, 1:300 phosphatase inhibitor cocktail). After 20 min of incubation on ice, homogenates were spun at 15,000 rpm for 20 min at 4°C. The supernatants correspond to the total cell lysates.
Western-blot analysis
Protein concentrations were determined by Bradford assay. Ten μg of proteins from each fraction were resuspended in Laemmli buffer, loaded on 10% SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were probed with the indicated antibody followed by visualization by ECL.
Immunocytochemistry
Rat neonatal cardiac myocytes seeded on 8 well glass chambers were washed with cold PBS, fixed in 4% formaldehyde, permeabilized with 0.1% Triton X-100, blocked with 1% normal goat serum and incubated overnight at 4°C with a monoclonal antibody against PDI (1:1000) from an ER labeling kit (invitrogen, USA) and a rabbit antibody against δPKC (1:100). Sections were washed with PBS and incubated for 60 min with TRITC-labeled goat anti-rabbit antibody and Alexa Fluor 488-labeled goat anti-mouse antibody followed by incubation with Hoechst stain (1:10000) for 15 min. Cover slips were mounted and slides were imaged by microscopy (BioRad Radiance 2100)
Electron microscopy
Endoplamic reticulum fractions were isolated from ex vivo Langendorf-perfused hearts subjected to 35 minutes of ischemia followed by 15 minutes of reperfusion, according to the method described above. The specimens were fixed in 2% formaldehyde and 0.5% glutaraldehyde for 40 minutes at 4 degree in 0.1 mol/L phosphates buffer (PBS), pH 7.2, and then rinsed 2 × in PBS buffer for 10 min each. The fixed samples were dehydrated in an ascending ethanol series up to 95% ethanol and infiltrated in a 1:1 mixture of LR White and ethanol for 1-2 hour under rotation, this was continued with a 1:2 mixture and finally, with pure LR White. Samples were embedded into gelatin capsules and cured at 55 degrees in an oven overnight. The sections were cut with an LKB V ultratome and collected on formvar-coated nickel grids. The grids were incubated for 10 min at room temperature with PBS buffer for rehydration and then treated with 1% normal goat serum (NGS) for 1 h to block non-specific reactions. The sections were incubated at room temperature with primary antibody against δPKC for 1 h, washed with PBS, and incubated for 1 h with the secondary antibody (goat-anti-rabbit IgG for δPKC coupled to 10-nm gold particles. After washing with PBS, the grids were stained with 2% uranyl acetate for 5 min and examined with a CM12 Phillips microscope.
Results
δPKC inhibition reduces ER stress-induced cell death in isolated cardiac myocytes
The involvement of δPKC in the caspase 3/9-dependent apoptotic pathway is well documented [17, 18]. However, whether δPKC participates in ER stress-induced cell death and apoptosis is not known. We therefore set out to determine whether cell death following ER stress is inhibited by the specific δPKC inhibitor peptide, δV1-1 [14]. Tunicamycin (Tm), a pharmacological agent inhibiting N-linked protein glycosylation, is commonly used to induce ER stress and subsequent cell death [4, 19-21]. Release of the cytosolic enzyme, lactose dehydrogenase (LDH), into the cell media is a marker of oncotic cell death. As shown in figure 1A, treatment with Tm for 40 hours induced an increase in LDH release compared to control cells (35% vs. 5%). Upon co-treatment with δV1-1, the Tm-induced LDH release decreased by about 25% compared to Tm treatment alone, whereas treatment with the TAT carrier peptide did not affect the release of LDH (Fig. 1A; p<0.05; n=4). Further, the number of TUNEL positive cells significantly increased in myocytes treated with Tm for 24 hours, from 20% in untreated control cells to 70% (Fig. 1B, 1C) and inhibition of δPKC completely abolished the increase in apoptosis (Fig. 1B, 1C; p<0.01; n=3). (Tm-induced increases in the number of TUNEL positive cells with or without TAT peptide were similar; data not shown).
Fig 1. δPKC activation modulates tunicamycin-induced cell death and apoptosis in cardiac myocytes.

A. Cell death after Tm-treatment (3μg/ml, 40 hours) was measured by LDH release in the presence of the δPKC antagonist peptide, δV1-1, or the TAT control peptide (1 μM). Data are mean ± S.E of four independent experiments. Student t test; * p<0.05 vs. Tm treatment.
B. Untreated or tunicamycin-treated cardiac myocytes (3 μg/ml, 24 hours) in the presence of control peptide (Tm+TAT) or the δPKC inhibitor, δV1-1 (Tm + δV1-1) were co-stained with the TUNEL method, to identify apoptotic cells, and with the nuclear dye, Hoechst, to identify the cells in the field. Arrows point out some of the TUNEL-positive cells.
C. TUNEL-positive cells were counted in a total of more than 300 myocytes over 3 random fields and expressed as percentages of the total number of nuclei. Data are expressed as mean ± S.E. of three independent experiments. Student t test; ** p<0.01 vs. Tm treatment.
D. δV1-1 inhibits caspase-3 activation induced by Tm treatment. Procaspase-3 was detected 24 hours after Tm treatment. The data are expressed as mean ± S.E of three independent experiments.
In addition, TAT or δV1-1 alone had no effect on LDH release or apoptosis (data not shown). To confirm that ER stress induced apoptosis, we also measured caspase-3 activation, another marker of apoptosis. Caspase-3 was activated 24 hours after Tm treatment, as evidenced by a decrease in the level of pro-caspase-3. A 30% reduction in activation of caspase-3 was obtained by δV1-1 treatment, as compared with Tm treatment, alone (Fig 1 D). Together, these data suggest that inhibition of δPKC by δV1-1 inhibits ER stress-induced apoptosis.
These data demonstrate that, upon Tm-induced ER stress, cultured cardiac myocytes undergo oncosis and apoptosis, both of which are significantly reduced by δPKC inhibition.
δPKC activation is involved in ER stress-induced pathways in isolated cardiac myocytes
The initial cellular response following ER stress, also termed the unfolded protein response (UPR), protects the cell through adaptive mechanisms that re-establish normal ER function [1, 2]. Given that δPKC inhibition decreased Tm-induced cell death, we sought to determine whether the ER stress pathways are modulated by δPKC. One of the hallmarks of UPR is increased expression of CHOP, a transcription factor [19, 22]. CHOP leads to ER stress-related apoptosis by decreasing Bcl2 expression [22]. We first found that induction of CHOP peaked 6 h after Tm treatment (data not shown). The δPKC inhibitor, δV1-1, but not TAT control, reduced Tm-induced increases in CHOP levels by 60% (Fig, 2A). The levels of ER chaperone proteins such as GRP78 are also increased following ER stress [23-25]. Because changes in protein levels require time, we determined GRP78 levels following 24 hours of treatment with Tm. Tm treatment increased the expression of GRP78 by almost 10 folds and inhibition of δPKC reduced this ER stress-mediated increase by ∼50% (Fig. 2B). Activated c-Jun N-terminal kinase (JNK) is one of the mediators of ER stress-induced apoptosis [26, 27]. Tm treatment triggered a two folds increase in activated (phosphorylated) JNK, which was abolished by co-treatment with δV1-1 (Fig. 2C). Taken together, our results provide evidence for the participation of δPKC in the signaling pathways triggered by ER stress in primary cultures of cardiac myocytes.
Fig 2. δPKC activation regulates signals triggered by tunicamycin-induced ER stress in cardiac myocytes.

After 6 hours (A) and 24 hours (B, C) of Tm treatment (3μg/ml) in the presence of TAT or δV1-1, total lysates of myocytes were analyzed by Western blot for the presence of the pro-apoptotic ER stress-induced protein, CHOP (A), the ER chaperone, GRP78 (B) and phosphorylation of the apoptotic kinase, JNK (C). Shown are representative blots of three independent experiments. Data are expressed as mean ± S.E. Student t test; * p < 0.05 vs. Tm treatment. GAPDH and JNK were used as internal normalization standards.
δPKC translocates to the endoplasmic reticulum following tunicamycin treatment
Various apoptotic stimuli induce δPKC translocation to mitochondria [13, 15, 28]. Because we found that δPKC regulates ER stress-mediated signaling events, we next determined whether δPKC directly translocates to the ER under these conditions. To this end, cultured cardiac myocytes were treated with Tm and ER-enriched fractions were subjected to Western blot with an anti-δPKC antibody. The levels of δPKC in the ER doubled following a 5-minute treatment with Tm (Fig. 3A). δPKC levels were normalized using calnexin, an ER resident chaperone protein. Since δPKC is known to translocate to mitochondria [13, 15], we confirmed that mitochondria are not present in this ER fraction using Western blotting against the mitochondrial protein, adenine nucleotide translocase (ANT) [17] (Fig. 3A; ANT is detected in the mitochondrial fraction, though Fig. 3D). We also confirmed the lack of cytosolic proteins in the ER fraction; the cytosolic protein, enolase, was not detected in this ER fraction (Fig. 3A), but it was detected in the cytosolic fraction (Fig. 3D). (The ability of the antibodies to detect protein markers of each fraction was confirmed using ER, mitochondria and cytosolic fractions and detecting calnexin, ANT and enolase only in the appropriate fraction (Fig. 3 D).
Fig 3. δPKC translocates to the ER of Tm-treated cardiac myocytes.

A. ER fractions from Tm-treated myocytes were subjected to Western blot analysis with an anti-δPKC antibody. The ER-specific calnexin protein was used as an internal loading control for normalization. Purity of ER fractions was confirmed using the mitochondrial marker, ANT, and the cytoplasmic marker, enolase. Data represent mean ± S.E. of three independent experiments. Student t test; * p<0.05 vs. no treatment (Con).
B. Representative confocal pictures of δPKC (red) and PDI (green, an ER marker) demonstrating increased co-localization (yellow) in Tm-treated cardiac myocytes. Data are representative of three independent experiments.
C. ER fractions from cells treated with Tm in the presence of δV1-1 or TAT were subjected to Western blot with an anti-Phospho-Ser643-δPKC (p-δPKC, a marker of active δPKC) antibody. An anti-δPKC antibody or anti-calnexin were used as internal controls for normalization.
D. The ER, mitochondria and cytosol were probed with the fraction specific markers, calnexin, ANT and enolase, respectively.
In addition, following Tm treatment, confocal microscopy analysis showed an increase in colocalization (yellow) of δPKC with protein disulfide isomerase (PDI), an ER-localized marker, as compared with control cells (Fig. 3B right panels). We also found that ER-localized δPKC was phosphorylated on Ser643, which correlates with its increased activity [29] (Fig. 3C). Pretreatment of the cells with δV1-1 abolished Tm-induced translocation of active δPKC to the ER membrane (Fig. 3C). Finally, similar levels of total δPKC in control and Tm-treated myocytes suggest that changes in the synthesis and degradation of δPKC cannot account for its increased levels at the ER (data not shown). Together, these data show that active δPKC translocates to ER upon Tm-induced ER stress.
δPKC translocation to the ER mediates, at least in part, ER stress response of the myocardium in a model of ischemia-reperfusion injury
Inhibition of δPKC protects the heart from ischemia-reperfusion injury partly through reducing mitochondria-mediated cell death [13, 16]. Increasing evidence suggests that ischemia/reperfusion in the heart also activates the ER stress response resulting in cellular injury [5, 8, 9, 30-32]. Based on our in vitro results, we next determined whether inhibition of δPKC protects the myocardium from ER stress in an ex vivo model of ischemia and reperfusion injury. Ischemia-reperfusion activates several ER stress pathways including increased GRP78, p-JNK and XBP-1 splicing (Fig. 4A, 4B). δV1-1 treatment at the onset of reperfusion partially inhibited those increases; the rise in GRP78, p-JNK and XBP1 splicing levels was inhibited by 30, 70 and 75%, respectively (Fig. 4A, 4B). Under these conditions, we did not observed induction of CHOP or activation of caspase12 (data not shown), other markers involved in ER stress-mediated apoptosis. This is likely due to the short duration of ischemia. As before [33], δV1-1 significantly reduced the extent of ischemic damage in hearts as demonstrated by a decrease in infarct size as measured by triphenyltetrazolium chloride (TTC) staining and creatine phosphokinase (CPK) release (Fig 4C; p<0.05; n=3). Therefore, this suggests that δPKC mediates ER stress response leading to cardiac ischemia and reperfusion injury.
Fig 4. δPKC modulates the ER stress response of the myocardium in a model of cardiac ischemia and reperfusion injury.
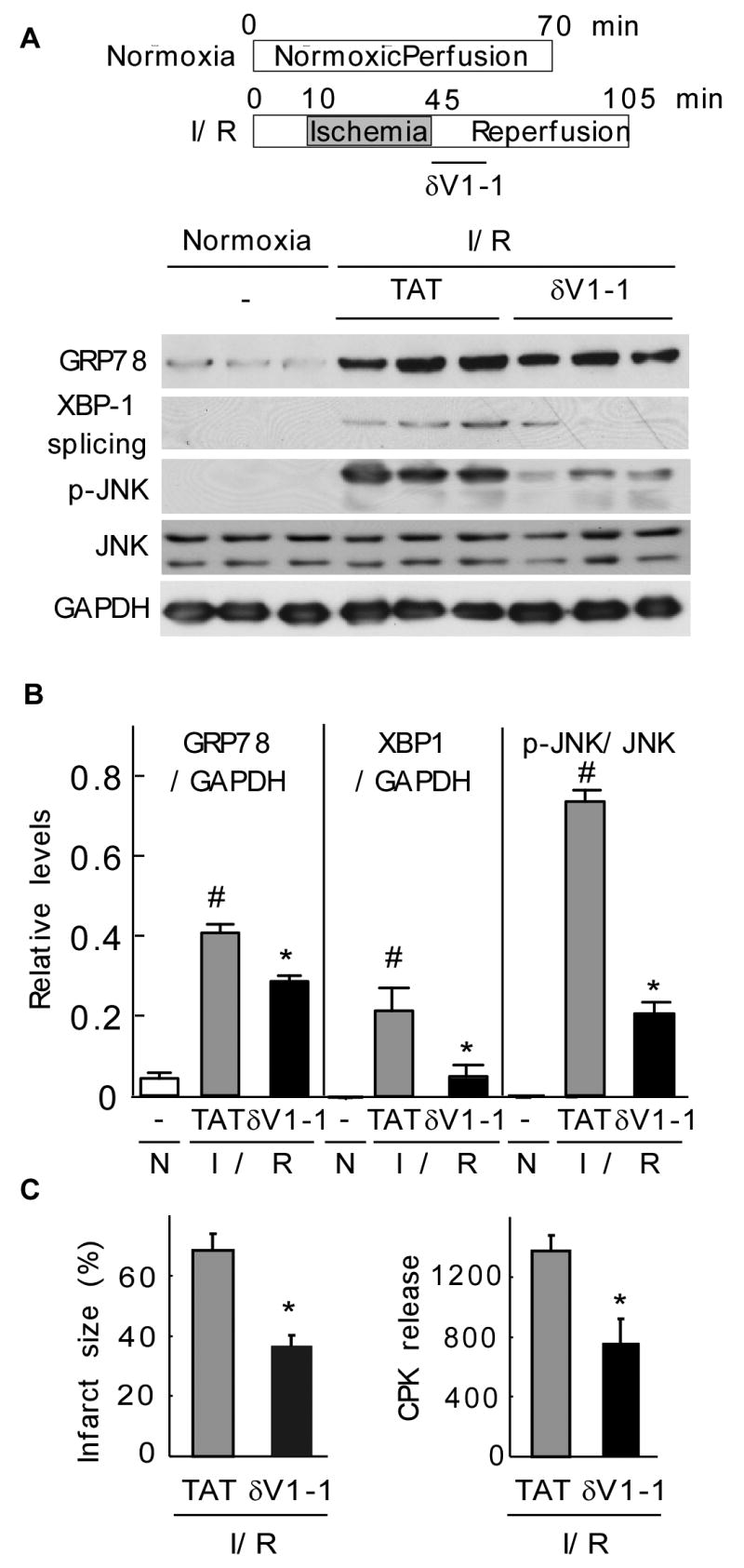
A. Normoxic control hearts and hearts that underwent ischemia and reperfusion were homogenized and total extracts were isolated. The levels of GRP78, spliced XBP1 and phospho-JNK were determined by Western blot.
B. Quantitative data of the hearts described in (A). Values represent mean ± S.E. of three animals in each group (N: normoxia; I/R: ischemia/reperfusion). Student t test; * p<0.05 vs. TAT treatment, # p< 0.05 vs. control.
C. Hearts were subjected to ischemia-reperfusion and treated at the onset of reperfusion with TAT control peptide or δV1-1 and the infarct size (left panel) and cell survival, as demonstrated by the decrease in CPK release (right panel), were determined. Data are expressed as mean ± S.E. of three animals in each group. Student t test; * p<0.05 vs. TAT treatment.
Next, we determined whether ischemia-reperfusion induces δPKC translocation to ER relative to normoxic and ischemic hearts. Western blot analysis of ER fractions from reperfused hearts displayed a significant increase in δPKC immunoreactivity (Fig. 5A). Following ischemia, δPKC translocation to ER was observed after 5 minutes of reperfusion and was increased by ∼4 fold after 15 minutes of reperfusion (Fig. 5A; p<0.05; n=3). Since the total levels of calnexin, an ER-resident chaperone protein, did not change during ischemia-reperfusion (data not shown), δPKC translocation and levels in the ER were normalized to this protein. We previously reported that ischemia-reperfusion injury induces δPKC translocation to the mitochondria [15]. However, in the current study, there is no significant contamination of mitochondria in the ER-enriched fraction as attested by the lack of ANT, a mitochondrial protein, during ischemia/reperfusion (Fig. 5A). The increased association of δPKC with the ER upon ischemia-reperfusion of the heart was further demonstrated by electron microscopy. Immunogold staining of δPKC in ER-enriched fractions of injured hearts following ischemia and 15 min of reperfusion increased two-folds as compared to normoxic hearts (Fig. 5B).
Fig 5. δPKC translocates to the ER in an ex vivo model of ischemia-reperfusion injury.
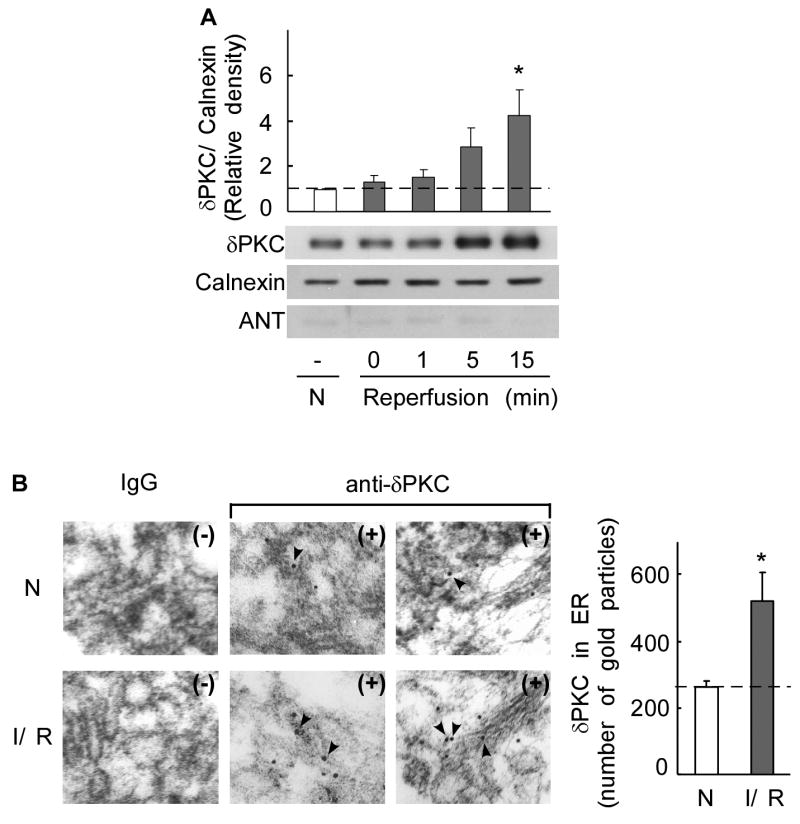
A. ER fractions of normoxic and ischemia-reperfusion injured hearts were subjected to Western blot analysis with an anti-δPKC antibody. Whereas ischemia (0 min reperfusion) did not induce translocation of δPKC to the ER as compared with normoxic control hearts (N), reperfusion significantly induced δPKC translocation to the ER by 5 minutes. Shown are representative Western blots (bottom) and a histogram depicting the amount of δPKC associated with the ER in heart samples (top). δPKC levels were normalized to the ER marker, calnexin. Purity of the ER fractions was confirmed by the lack of the mitochondrial marker, ANT. Student t test; * p < 0.05 vs. control normoxia.
B. ER localization of δPKC as evidenced by immuno-electron microscopy. Representative electron micrographs of δPKC staining in the ER fractions from normoxic control hearts (N) and hearts subjected to 35 min of ischemia followed by 15 min of reperfusion (I/R). (magnification: × 35000). Samples were probed in the presence (+) or absence (-) of δPKC antibody. Arrows indicate δPKC-positive staining with gold particles. Quantitative data of gold particles associated with ER lumen are provided in the right histogram. Five random fields of each section from two animals were counted. The data represent mean ± SD of two animals in each group. Student t test; * p < 0.05 vs. control normoxia.
Discussion
In the current study, we determined the role of δPKC in cellular responses triggered by the disruption of ER homeostasis. We demonstrated that tunicamycin-induced ER stress in isolated neonatal cardiac myocytes led to the translocation of active δPKC to the ER and that δPKC, at least in part, mediates tunicamycin-induced oncosis and apoptosis. Furthermore, we found that in an ex vivo model of cardiac ischemia/reperfusion, δPKC translocates to the ER, where it participates in the activation of ER stress-induced signaling cascades.
δPKC has been shown to have pro-apoptotic effects [34-36]. Using dominant negative mutants of δPKC, broad spectrum inhibitors or a specific δPKC antagonist peptide, δPKC was further demonstrated to be a mediator of apoptosis [13, 18, 37, 38]. Few studies have reported an anti-apoptotic role for δPKC [39, 40]. δPKC role in mediating cellular apoptosis may result from stimulus-dependent translocation of δPKC to different subcellular sites. Indeed, one of the key factors in determining PKC function is its subcellular location following activation [41]. Our present work demonstrates that δPKC translocates to the ER in response to ER stress in cardiac myocytes and provides the first evidence that δPKC regulates the ER stress-induced responses; when δPKC was selectively inhibited by the antagonist peptide, δV1-1, ER stress-induced signaling pathways and cell death were significantly inhibited both in tunicamycin-treated neonatal cardiac myocytes in culture and in cardiac ischemia-reperfusion.
Ischemia-reperfusion injury occurs during cardiac arrest and transient arterial occlusion and is associated with cell death. We have shown previously that δPKC inhibition at the onset of reperfusion protected hearts against ischemia-reperfusion injury [33]. This was later determined to be due to inhibition of δPKC translocation to cardiac mitochondria resulting in diminished cytochrome c release and subsequent prevention of apoptosis [13]. However, few studies have investigated the role of ER stress in the induction of apoptosis during ischemia/reperfusion. Hypoxia induces the expression of XBP1 splicing and GRP78 in cultures of rat neonatal cardiac myocytes [5, 8]. In a mouse model of myocardial infarction, an increased expression of GRP78 was observed in cells near the infarct four days after occlusion of the left anterior descending coronary artery [5]. However, in our study, we found that the levels of GRP878 increased after 35 min ischemia followed by 60 min of reperfusion and after 24 hours of Tm treatment in cultured myocytes. This early rise in GRP78 expression after ischemia and reperfusion may be due to the experimental model used. Indeed, ER stress-induced injury occurs with different kinetics under pathological conditions [6, 8].
Here we show that ER stress is activated by a δPKC-dependent pathway during ischemia-reperfusion injury and that inhibition of δPKC at the onset of reperfusion significantly reduced ER stress responses. Studies in transgenic mice expressing the activation transcription factor 6, ATF6, also suggest that ER stress may participate in ischemia-reperfusion injury [9]. During ischemia-reperfusion injury, ER stress may be excessively activated, since in addition to the increased expression of the chaperone protein, GRP78, there is a significant increase in the pro-apoptotic kinase, phosphorylated-JNK. We found that in the same experimental model, δPKC also translocates to the mitochondria where it triggers mitochondrial-dependent cytochrome c-mediated apoptosis [13]. Our present study suggests that translocation of δPKC to the ER in hearts subjected to ischemia/reperfusion may provide an additional novel regulatory role of δPKC in ER stress-induced apoptosis. Our data showing that tunicamycin-induced ER stress is reduced when δPKC is inhibited in cardiac myocytes suggests that during ischemia-reperfusion, δPKC translocation to the ER may mediate ER stress. The finding that ER stress pathways are all reduced following δPKC inhibition at the onset of reperfusion strongly supports this hypothesis. The involvement of oxidative stress in ER dysfunction has been well documented [42, 43]. One possibility may be that δV1-1 reduces ER stress-induced oxidative stress, which subsequently initiates ER stress response pathways.
Our previously published work [13] and the present study show that ischemia-reperfusion injury triggers δPKC translocation to two distinct organelles and thus argues in favor of a dual role of δPKC in both mitochondrial-and ER stress-induced apoptosis in the ischemic hearts. Whether those two apoptotic pathways are redundant parallel functions of δPKC or whether the mitochondrial-induced apoptosis contributes to the ER stress-induced cell death or vice-versa, remains to be determined. In addition, as described before [44, 45], δPKC was also found in the nucleus. It is possible that the nuclear δPKC is involved in regulating the expression of the transcription factor, CHOP, and the molecular chaperone, GRP78. In summary, our data show that δPKC activation plays a critical role in two different models of ER stress-mediated responses and provides new insight into the role of ER stress into the pathophysiology of the heart.
Acknowledgments
SOURCES OF FUNDING: This work was supported by NIH HL76675 to DM-R. Dr. A. Vallentin was supported in part by postdoctoral awards from La Fondation pour la Recherche Médicale and from the American Heart Association.
Footnotes
DISCLOSURES: Dr. Mochly-Rosen is the founder of KAI Pharmaceuticals, Inc, a company that plans to bring PKC regulators to the clinic. However, none of the work described here is in collaboration with or supported by the company.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13:363–73. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- 2.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–64. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 4.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 5.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–82. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 6.Zhang PL, Lun M, Teng J, Huang J, Blasick TM, Yin L, et al. Preinduced molecular chaperones in the endoplasmic reticulum protect cardiomyocytes from lethal injury. Ann Clin Lab Sci. 2004;34:449–57. [PubMed] [Google Scholar]
- 7.Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y, et al. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol. 2004;24:8007–17. doi: 10.1128/MCB.24.18.8007-8017.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554–75. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–93. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Zhang HY, McPherson BC, Baman T, Roth S, Shao Z, et al. Role of opioid delta1 receptors, mitochondrial K(ATP) channels, and protein kinase C during cardiocyte apoptosis. J Mol Cell Cardiol. 2001;33:2007–14. doi: 10.1006/jmcc.2001.1464. [DOI] [PubMed] [Google Scholar]
- 11.Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase c delta. Apoptosis. 2003;8:19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- 12.Cross T, Griffiths G, Deacon E, Sallis R, Gough M, Watters D, et al. PKC-delta is an apoptotic lamin kinase. Oncogene. 2000;19:2331–7. doi: 10.1038/sj.onc.1203555. [DOI] [PubMed] [Google Scholar]
- 13.Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D. Protein kinase Cdelta activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem. 2004;279:47985–91. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, et al. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci U S A. 2001;98:11114–9. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Churchill EN, Szweda LI. Translocation of deltaPKC to mitochondria during cardiac reperfusion enhances superoxide anion production and induces loss in mitochondrial function. Arch Biochem Biophys. 2005;439:194–9. doi: 10.1016/j.abb.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Inagaki K, Hahn HS, Dorn GW, 2nd, Mochly-Rosen D. Additive protection of the ischemic heart ex vivo by combined treatment with delta-protein kinase C inhibitor and epsilon-protein kinase C activator. Circulation. 2003;108:869–75. doi: 10.1161/01.CIR.0000081943.93653.73. [DOI] [PubMed] [Google Scholar]
- 17.Kaul S, Kanthasamy A, Kitazawa M, Anantharam V, Kanthasamy AG. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: relevance to oxidative stress in dopaminergic degeneration. Eur J Neurosci. 2003;18:1387–401. doi: 10.1046/j.1460-9568.2003.02864.x. [DOI] [PubMed] [Google Scholar]
- 18.Niwa K, Inanami O, Yamamori T, Ohta T, Hamasu T, Karino T, et al. Roles of protein kinase C delta in the accumulation of P53 and the induction of apoptosis in H2O2-treated bovine endothelial cells. Free Radic Res. 2002;36:1147–53. doi: 10.1080/1071576021000016409. [DOI] [PubMed] [Google Scholar]
- 19.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Gene Dev. 1998;12:982–95. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi X, Hosoi T, Okuma Y, Kaneko M, Nomura Y. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol. 2004;66:899–908. doi: 10.1124/mol.104.001339. [DOI] [PubMed] [Google Scholar]
- 21.Qi X, Okuma Y, Hosoi T, Kaneko M, Nomura Y. Induction of murine HRD1 in experimental cerebral ischemia. Brain Res. 2004;130:30–8. doi: 10.1016/j.molbrainres.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 22.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–59. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–32. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 24.Lujan HD, Mowatt MR, Conrad JT, Nash TE. Increased expression of the molecular chaperone BiP/GRP78 during the differentiation of a primitive eukaryote. Biology of the cell / under the auspices of the European Cell Biology Organization. 1996;86:11–8. doi: 10.1111/j.1768-322x.1996.tb00950.x. [DOI] [PubMed] [Google Scholar]
- 25.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–4. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- 26.Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K, et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005;12:19–24. doi: 10.1038/sj.cdd.4401528. [DOI] [PubMed] [Google Scholar]
- 27.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–6. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 28.Fryer RM, Wang Y, Hsu AK, Gross GJ. Essential activation of PKC-delta in opioid-initiated cardioprotection. Am J Phys. 2001;280:1346–53. doi: 10.1152/ajpheart.2001.280.3.H1346. [DOI] [PubMed] [Google Scholar]
- 29.Shimohata T, Zhao H, Sung JH, Sun G, Mochly-Rosen D, Steinberg GK. Suppression of deltaPKC activation after focal cerebral ischemia contributes to the protective effect of hypothermia. J Cereb Blood Flow Metab. 2007:1–13. doi: 10.1038/sj.jcbfm.9600450. [DOI] [PubMed] [Google Scholar]
- 30.Azfer A, Niu J, Rogers LM, Adamski FM, Kolattukudy PE. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am J Phys. 2006;291:1411–20. doi: 10.1152/ajpheart.01378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szegezdi E, Duffy A, O'Mahoney ME, Logue SE, Mylotte LA, O'Brien T, et al. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006;349:1406–11. doi: 10.1016/j.bbrc.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–12. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 33.Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, et al. Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. 2003;108:2304–7. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- 34.Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, et al. Proteolytic activation of protein kinase C delta by an ICE/CED 3-like protease induces characteristics of apoptosis. J Exp Med. 1996;184:2399–404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, et al. Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells. EMBO J. 1995;14:6148–56. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knox KA, Johnson GD, Gordon J. A study of protein kinase C isozyme distribution in relation to Bcl-2 expression during apoptosis of epithelial cells in vivo. Exp Cell Res. 1993;207:68–73. doi: 10.1006/excr.1993.1164. [DOI] [PubMed] [Google Scholar]
- 37.Liao YF, Hung YC, Chang WH, Tsay GJ, Hour TC, Hung HC, et al. The PKC delta inhibitor, rottlerin, induces apoptosis of haematopoietic cell lines through mitochondrial membrane depolarization and caspases' cascade. Life Sci. 2005;77:707–19. doi: 10.1016/j.lfs.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 38.Shizukuda Y, Helisch A, Yokota R, Ware JA. Downregulation of protein kinase cdelta activity enhances endothelial cell adaptation to hypoxia. Circulation. 1999;100:1909–16. doi: 10.1161/01.cir.100.18.1909. [DOI] [PubMed] [Google Scholar]
- 39.Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res. 2003;63:780–6. [PubMed] [Google Scholar]
- 40.McCracken MA, Miraglia LJ, McKay RA, Strobl JS. Protein kinase C delta is a prosurvival factor in human breast tumor cell lines. Mol Cancer Ther. 2003;2:273–81. [PubMed] [Google Scholar]
- 41.Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 1995;268:247–51. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- 42.Hanada S, Harada M, Kumemura H, Bishr Omary M, Koga H, Kawaguchi T, et al. Oxidative stress induces the endoplasmic reticulum stress and facilitates inclusion formation in cultured cells. J Hepatol. 2007;47:93–102. doi: 10.1016/j.jhep.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 43.Horke S, Witte I, Wilgenbus P, Kruger M, Strand D, Forstermann U. Paraoxonase-2 reduces oxidative stress in vascular cells and decreases endoplasmic reticulum stress-induced caspase activation. Circulation. 2007;115:2055–64. doi: 10.1161/CIRCULATIONAHA.106.681700. [DOI] [PubMed] [Google Scholar]
- 44.Basu A. Involvement of protein kinase C-delta in DNA damage-induced apoptosis. J Cell Mol Med. 2003;7:341–50. doi: 10.1111/j.1582-4934.2003.tb00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gordon AS, Yao L, Jiang Z, Fishburn CS, Fuchs S, Diamond I. Ethanol acts synergistically with a D2 dopamine agonist to cause translocation of protein kinase C. Mol Pharmacol. 2001;59:153–60. doi: 10.1124/mol.59.1.153. [DOI] [PubMed] [Google Scholar]