

Abstract
CXCL9 and CXCL10 mediate the recruitment of T lymphocytes and NK cells known to be important in viral surveillance. The relevance of CXCL10 in comparison to CXCL9 in response to genital HSV-2 infection was determined using mice deficient in CXCL9 (CXCL9−/−) and CXCL10 (CXCL10−/−) along with wild type (WT) C57BL/6 mice. An increased sensitivity to infection was found in CXCL10−/− mice in comparison to CXCL9−/− or WT mice as determined by detection of HSV-2 in the central nervous system (CNS) at day 3 post infection. However, by day 7 post infection both CXCL9−/− & CXCL10−/−mice possessed significantly higher viral titers in the CNS in comparison to WT mice consistent with mortality (18–35%) of these mice within the first 7 days after infection. Even though CXCL9−/− and CXCL10−/− mice expressed elevated levels of CCL2, CCL3, CCL5, and CXCL1 in the spinal cord in comparison to WT mice, there was a reduction in NK cell and virus-specific CD8+ T cell mobilization to this tissue suggesting CXCL9 and CXCL10 are critical for recruitment of these effector cells to the spinal cord following genital HSV-2 infection. Moreover, leukocytes from the spinal cord but not draining lymph nodes or spleens of infected CXCL9−/− or CXCL10−/− mice displayed reduced CTL activity in comparison to effector cells from WT mice. Thus, the absence of CXCL9 or CXCL10 expression significantly alters the ability of the host to control genital HSV-2 infection through the mobilization of effector cells to sites of infection.
Introduction
Herpes simplex virus type 2 (HSV-2)3 is a significant human pathogen and the most common cause of genital ulcerations in humans (1–5). With more than 1.6 million Americans infected annually, it is one of the most prevalent sexually transmitted diseases in the US and worldwide (3,4). In addition, the viral pathogen can be fatal in newborns and immunocompromised persons (4). Recent studies suggest acquisition of HIV increases 2–3 fold in HSV-2 infected individuals underscoring the contribution of this virus in facilitating increased susceptibility to other microbial pathogens (5–7).
The mobilization of effector cells (NK and T cells) to active sites of infection is driven by a number of soluble factors including chemokines, a family of small secreted proteins that are produced in response to viral infection (3–5). However, the role of chemokines in recruitment of effector cells relative to genital HSV-2 infection is largely unknown. It has been found that CXC type chemokines that lack the ELR sequence including CXCL9 and CXCL10 are potent chemoattractants for activated T cells, NK cells, monocytes, dendritic cells, and B cells (8–11). Both CXCL9 and CXCL10 selectively bind the same G-protein coupled receptor CXCR3 which is highly expressed on activated T cells. Moreover, CXCR3 signaling has been demonstrated to be essential for generating an effective antiviral response against many viral infections by promoting a Th1 response (12–19).
T lymphocytes have been reported to be critical in suppressing viral replication in the mouse model of genital HSV-2 infection (20–26). These effector cells can lyse susceptible virus infected cells by exocytosis of granzyme containing cytoplasmic granules and perforins or by Fas-mediated apoptosis (27–30). Likewise, NK cells monitor HSV-2 infection in mice operating through similar cytolytic processes in lysing infected cells (31, 32). The cellular processes that control virus infection are complemented by soluble factors including IFN- which is critical for resolution of lesions and clearance of the virus following genital infection (33, 34).
Many chemokines and their receptors are expressed in a tissue specific manner (35,36). Previous studies have shown CXCL10 is expressed early in the central nervous system (CNS) in response to virus infection (11, 12). We hypothesized the early expression of CXCL10 in infected tissues is critical to the outcome of HSV-2 genital infection by facilitating the recruitment of effector cells (NK and T cells) to the active sites of viral replication. In fact, an initial experiment found CXCL10 but not CXCL9 was significantly elevated above basal levels within 24 hr post infection (pi) in the vaginal tissue. In contrast, CXCL9 but not CXCL10 levels rose significantly within the first 72 hr within the draining lymph nodes following genital HSV-2 infection. Taken together, it would appear the early tissue-specific chemokine response may provide a basis for redundancy is chemokines that operate thru the lone receptor for CXCL9 and CXCL10, CXCR3. As a means to compare/contrast the nature of CXCL9 and CXCL10 expression in the immune response to a viral pathogen, mice deficient in CXCL9 (CXCL9−/−) and CXCL10 (CXCL10−/−) along with wild type (WT) mice were evaluated for resistance to HSV-2 genital infection focusing on the inflammatory immune response, phenotypic analysis of leukocytes, and functional analysis of effector cells. The results suggest both CXCL9 and CXCL10 expression is necessary for optimal recruitment of NK cells and CTLs to the spinal cord as well as clearance of HSV-2 from the CNS following genital infection.
Materials and Methods
Virus and cells
A clinical isolate of HSV-2 obtained from Charity Hospital (New Orleans, LA) was propagated in Vero cells (African green monkey kidney fibroblasts, ATCC CCL-81, American Type Culture Collection, Manassas, VA). Virus stock (4.8 x 107 plaque forming units (pfu/ml) was stored at −80° C and diluted in RPMI-1640 medium immediately before infection. Vero cells were propagated in RPMI-1640 medium containing 10% FBS, gentamicin, and anti-mycotic-antibiotic solution (complete medium) (Invitrogen, Carlsbad, CA) at 37° C, 5% CO2, and 95% humidity.
Mice and infection
WT C57BL/6 female mice (6–8 weeks old, The Jackson Laboratory, Bar Harbor, ME), CXCL9−/− (9), and CXCL10−/− (10) female mice backcrossed to the C57BL/6 genetic background for 8–9 generations for this study. DepoProvera (Pharmacia and Upjohn Co., Kalamazoo, MI) inoculated mice (2.0 mg/mice) were infected with 2000 pfu HSV-2 (20 μl) intravaginally. Mice were euthanized at time points pi to determine virus titer, phenotypic analysis of leukocytes and cytokine/chemokine content within infected tissues. All procedures were approved by The University of Oklahoma Health Sciences Center and Dean A. McGee Eye Institute animal care and use committee.
Virus plaque assay
Tissues (vagina, spinal cord, and brain stem) were removed from infected mice at times pi (day 3 and day 7), placed into complete medium (500 hl) and homogenized using a tissue homogenizer (Fisher Scientific, Pittsburgh, PA). Supernatants were clarified (10,000 x g, 1 min) and assessed for viral titer by plaque assay as described previously (37).
Suspension Array and ELISA
Detection of MCP-1 (CCL2), MIP-1a (CCL3), RANTES (CCL5), KC (CXCL1), IL-12, and IFN-γwere performed using a suspension array system (Bioplex, Bio-Rad, Hercules, CA). Samples were analyzed in duplicate along with a standard provided. The weight of each tissue was used to normalize amount of cytokine/chemokine per milligram (mg) of tissue weight. Measurement of TNF-α, CXCL9 and CXCL10 were performed by ELISA (R&D Systems, Minneapolis, MN). Known amounts of each analyte provided were used to generate standard curves to extrapolate the amount of each unknown sample.
Flow cytometry
At the designated time, mice were exsanguinated and vagina, spinal cord, brain stem, spleen and lymph nodes were removed. Tissues were processed to generate single cell suspensions as described (32) and were transferred into 5 ml polystyrene round-bottomed tubes (Becton Dickinson, Franklin Lakes, NJ). The cells were incubated with 2 μl of anti-mouse CD16/32 (Fc III/II receptor, 2.4G2, BD Pharmingen, San Diego, CA) for 20 min on ice. For measuring CD4 T, CD8 T, and NK cell content, cells were labeled with 1–2 μg of FITC-conjugated anti-CD3 (17A2), and PE-conjugated anti-CD4 (RM4–5) or anti-CD8α (53–6.7) or anti-NK1.1 (PK136), and PE-Cy5-anti-CD45 (30-F11) and allowed to incubate on ice in the dark. After a 30 min incubation, the cells were washed (300 x g, 5 min at 4° C) and resuspended in 1% paraformaldehyde for 60 min. Next, the cells were resuspended in 3 ml of 1x PBS containing 1% BSA. A known number of beads (20,800) (Invitrogen, Eugene, OR) was immediately added to the sample which were then briefly vortexed and then analyzed on a Coulter Epics XL flow cytometer (Beckman Coulter, Miami, FL). Cells were gated on CD45high expressing cells, and a second gate was established to capture the number of beads that passed through during the sampling time. The absolute number of leukocytes (CD45high) in the tissue was determined by multiplying the ratio of the number of beads collected per sample over the total number of beads added X the number of CD45high events X sample dilution factor. Isotypic control antibodies were included in the analysis to establish background fluorescence levels. Likewise, samples from uninfected mice were also analyzed to determine the degree of contamination from incomplete perfusion. For measuring inguinal/iliac lymph nodes (I/ILN) and spleen T and NK cell content, single cell suspensions (1x106 cells) were transferred into 5 ml polystyrene round-bottomed tubes and labeled with antibodies and analyzed by flow cytometry. The absolute number of cells was determined by multiplying the percentage of T or NK cell populations found in each lymphoid organ by the total number of cells recovered.
Tetramer staining
For tetramer staining, cells were labeled with 1–2 μg of the HSV peptide gB498–505 (SSIEFARL) specific MHC tetramer (MHC Tetramer Lab, Houston, Tx) for 60 min. The cells were washed (300 x g, 5 min at 4° C) and labeled with 1–2 hg of FITC-conjugated anti-CD8 and PE-Cy5-conjugated anti-CD45. Following a 30 min incubation, cells were washed (300 x g, 5 min at 4° C) and resuspended in 1% paraformaldehyde. After a 60 min incubation, the cells were resuspended in 1x PBS. Cells were subsequently analyzed by flow cytometry as described above.
CTL assay
MC57G (CRL-2295TM, ATCC, Manassas, VA) were infected with HSV-2 with a multiplicity of infection of 3 for 8 hrs at 37°C, 5% CO2, and 95% humidity. Following infection, ten thousand MC57G cells were resuspended in pre-warmed (37°C) PBS containing 0.25 μM carboxyfluorescein diacetate succinimidyl ester (CFSE, Invitrogen) and incubated for 15 min at 37°C. Cells were washed with 1x PBS and resuspended in fresh pre-warmed complete medium. The desired number of isolated leukocytes from the processed spinal cord, spleen, and I/ILN of mice was added to the CFSE-labeled HSV-2 infected MC57G target cells in 96 well microtiter plate wells in a total volume of 200 μl of complete medium at an effector-to-target cell ratio of 10:1. After a 4 hr incubation, 0.5 hl of propidium iodide (0.5 hg) was added to cells followed by a 15 min incubation at 37°C, 5% CO2 and 95% humidity. Cells were then washed and resuspended in 1x PBS and immediately analyzed by XL flow cytometry. The gate was set for CFSE-expressing cells. The percent (%) cytotoxicity was calculated by dividing the number of propidium iodide labeled CFSE-expressing cells by the total number of CFSE-expressing cells multiplied by 100. The Background level was determined by using target cells without effector cells and target cells incubated with spleen cells from uninfected mice. The assay was repeated using leukocytes from spinal cord of infected CXCL9−/− mice incubated with recombinant CXCL9 (2.0 ng, PeproTech Inc., Rocky Hill, NJ) for the duration of the assay. Leukocytes from the spinal cord of infected WT mice were also included in the assay.
Statistics
All statistical analyses were carried out using the GBSTAT program (Dynamic Microsystems, Silver Springs, MD). One-way ANOVA and Tukey’s post hoc t-test were used to determine significant (p<0.05) differences between WT, CXCL9−/− and CXCL10−/−mice. Man-Whitney U test was used for analysis of survival to determine significant (p<0.05) differences between WT, CXCL9−/− and CXCL10−/− mice.
Results
CXCL9−/− and CXCL10−/− mice are susceptible to genital HSV-2 infection
If both CXCL9 and CXCL10 operate through the only receptor for these chemokines, are there noticeable differences in the sensitivity to infection? To address this question, WT, CXCL9−/−, and CXCL10−/− mice were evaluated for virus titer in infected tissue following exposure to HSV-2 (2,000 pfu/vagina). CXCL10−/−, but not CXCL9−/− or WT mice were found to harbor infectious virus in spinal cord at day 3 pi (Fig. 1a). In addition, CXCL9−/−and CXCL10−/− mice showed significantly higher viral loads in the spinal cord and brain stem at day 7 pi compared to WT controls (Fig. 1b). These results are consistent with the incidence of mortality in the CXCL9−/− and CXCL10−/− mice during this same time period (Fig. 1c). Specifically, 18% (8/44) of CXCL9−/− and 35% (21/59) of CXCL10−/− mice succumbed to infection in comparison to 0/52 WT mice by day 7 pi (Fig. 1c). There was no virus recovered from brain stem of WT, CXCL9−/− or CXCL10−/− mice at day 3 pi and no significant difference in viral loads found in the vaginal tissue at day 3 or day 7 pi comparing all groups of mice (Fig. 1a). Taken together, CXCL10−/− mice appear to be more sensitive to genital HSV-2 infection based on an increase in mortality and appearance of HSV-2 in the spinal cord earlier than in CXCL9−/− mice.
Figure 1.
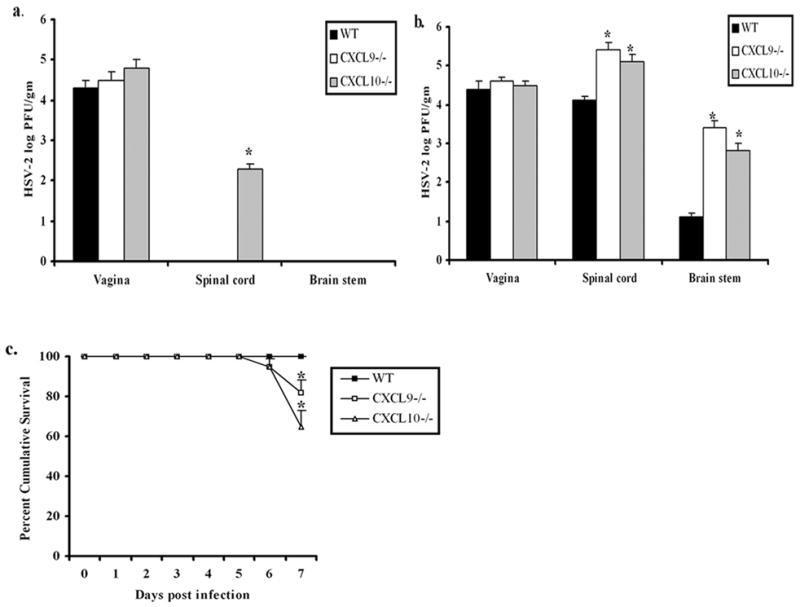
CXCL9−/− and CXCL10−/− mice are highly susceptible to HSV-2 genital infection. Wild type (WT), CXCL9−/− and CXCL10−/− mice (n=6 mice/group) were infected with HSV-2 (2000 pfu/vagina). At indicated times day 3 pi (a), and day 7 pi (b), mice were exsanguinated and vaginal tissue, brain stem and spinal cord were processed and assayed for viral titer by standard plaque assay and viral titer is expressed as mean log PFU ± SEM. (c), WT, CXCL9−/− and CXCL10−/− mice (n=44–59 mice/group) were infected with HSV-2 (2000 pfu/vagina) and were monitored and recorded for survival. The bars represent the mean ± SEM summarizing the results of three independent experiments. *, p<.05 comparing WT to CXCL9−/− and CXCL10−/− mice.
CXCL9 & CXCL10 levels rapidly increase in tissue specific manner following infection with HSV-2 in WT mice
Since CXCL10−/− mice were found to be more sensitive to virus infection, we next asked if there were differences in the expression of CXCL9 and CXCL10 at critical sites following infection that might coincide with an increase in sensitivity. While both CXCL9 & CXCL10 were constitutively expressed in vaginal tissue, CXCL10 levels increased significantly within 24 hr pi whereas CXCL9 levels did not change in WT mice (Fig. 2a & 2b). By day 3 pi, CXCL9−/− mice possessed similar levels of CXCL10 in the vaginal tissue compared to WT mice (Fig. 2b) whereas CXCL9 levels in CXCL10−/− mice were reduced compared to WT mice (Fig. 2a). Both CXCL10 and CXCL9 levels were diminished in CXCL9−/− and CXCL10−/− mice respectively in the I/ILN compared to WT mice at day 3 and day 7 pi (Fig. 2c & 2d). By comparison, CXCL9 and CXCL10 levels were not significantly elevated in the nervous system of infected mice until the day 7 pi (Fig. 2e-h). CXCL9 levels were significantly increased in the brain stem and spinal cord of WT mice compared to CXCL10−/− mice at day 7 pi (Fig. 2 e &g). However, CXCL10 levels in the spinal cord and brain stem did not vary between WT and CXCL9−/− mice (Fig. 2 f &h). We interpret the results to suggest CXCL10−/− mice cannot respond to an equivalent level with the corresponding CXCR3 chemokine, CXCL9, in the CNS, in comparison to CXCL9−/− mice which are capable of producing similar levels of the corresponding CXCR3 chemokine, CXCL10, in the CNS relative to WT mice. These results demonstrate a deficiency pronounced in the CXCL10−/− mice in comparison to CXCL9−/− mice.
Figure 2.
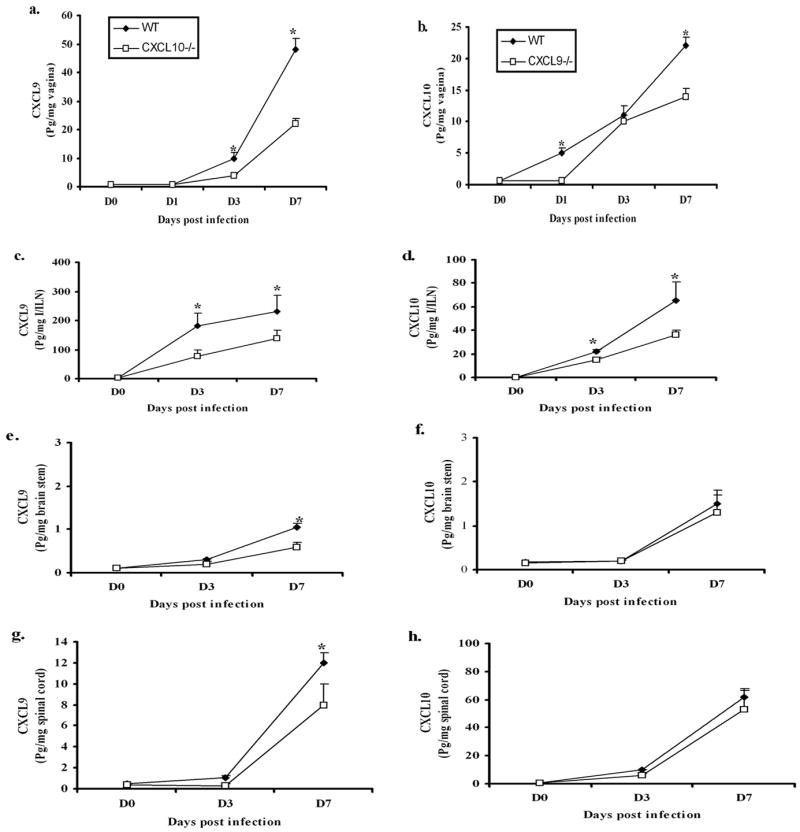
CXCL9 & CXCL10 levels are elevated in tissue specific manner. WT, CXCL9−/− and CXCL10−/− (n=6/group) were infected with HSV-2 (2000 pfu/vagina). At indicated times, the mice were exsanguinated and vaginal tissue (a & b), Inguinal/Iliac lymph node (I/ILN) (c & d), brain stem (e & f), and spinal cord (g & h) were removed, processed and assayed for CXCL9 (left panel) and CXCL10 (right panel) content using ELISA. The weight of the tissue was used to normalize the amount of chemokine per milligram of tissue (expressed as pg per mg of tissue). Day 0 time point represents uninfected controls. Each point represents the mean ± SEM summarizing the results of two independent experiments. *, p<.05 comparing WT to CXCL9−/− and CXCL10−/− groups.
CXCL9−/− and CXCL10−/− mice express elevated chemokine/cytokine levels in infected tissues
Many CC type chemokines (i.e. CCL2, CCL3, CCL5), inflammatory cytokines (e.g. IFN- ) and pro-inflammatory cytokine (e.g. TNF-α) are expressed locally in response to viral infection (15,18,32). To determine if additional cytokine/chemokine levels were modified in the CXCL9−/− and CXCL10−/− mice following HSV-2 infection, candidate cytokines/chemokines were surveyed at times pi. Even though there was no difference in viral titers found in the vaginal tissue comparing the WT, CXCL9−/− & CXCL10−/− mice at day 7 pi, there was a significant increase in chemokine expression including CCL2 (MCP-1), CCL3 (MIP-1a), CCL5 (RANTES), and CXCL1 (KC) as well as TNF-α in the vaginal tissue of CXCL10−/− mice in comparison to CXCL9−/− and WT mice (Fig. 3). The IFN- and IL-12p70 were also included in the analysis; however, no significant differences were found (data not shown). By comparison, there was an increase in CCL2, CCL3, CCL5, and CXCL1 as well as TNF-α levels in the spinal cord of chemokine knockout mice compared to WT mice at day 5 (except TNF-α) and day 7 pi (Fig. 4). TNF-α was the only chemokine or cytokine significantly elevated in the brain stem in both chemokine knockout mice compared to WT controls following viral infection (Fig. 4). Collectively, the results suggest an increase in cytokine/chemokine expression in the CNS but not vagina is driven by the elevation in infectious virus recovered in this tissue in the CXCL9−/− and CXCL10−/− mice.
Figure 3.
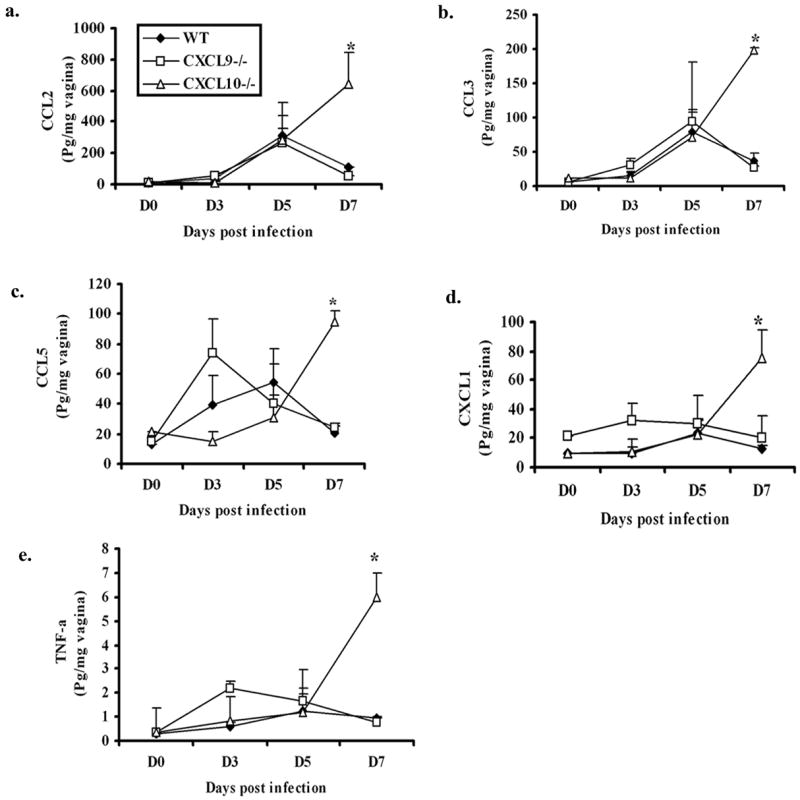
Chemokine/cytokine levels are elevated in the vaginal tissue of CXCL10−/−mice. WT, CXCL9−/−, CXCL10−/− mice (n=6/group) were infected with HSV-2 (2000 pfu/vagina). At indicated times, the mice were exsanguinated and vaginal tissues were removed, processed and assessed for CCL2, CCL3, CCL5, CXCL1, TNF-α, and IFN-and IL-12p70 (not shown) content using a suspension array system and ELISA. Samples were analyzed in duplicate along with standard provided to generate standard curves for each analyte. The weight of the tissue was used to normalize amount of cytokine/chemokine per milligram of tissue weight. Day 0 time point represents uninfected controls. Each point represents the mean ± SEM summarizing the results of two independent experiments. *, p<.05 comparing the WT to CXCL9−/− and CXCL10−/−groups.
Figure 4.
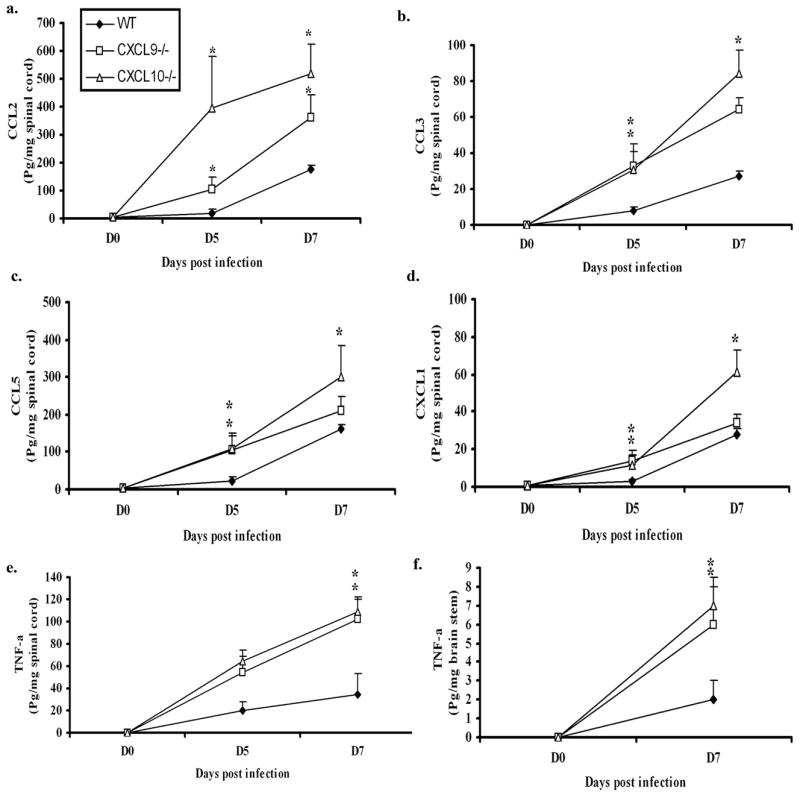
Chemokine/cytokine levels are elevated in the nervous system of chemokine knockout mice. WT, CXCL9−/− , CXCL10−/− mice (n=6/group) were infected with HSV-2 (2000 pfu/vagina). At indicated times, the mice were exsanguinated and the spinal cords and brain stems were removed, processed and assessed for CCL2, CCL3, CCL5, CXCL1 and TNF-α content using a suspension array system and ELISA. As there were no differences found in other analytes measured, only TNF-α level is shown for brain stem. Samples were analyzed in duplicate along with standard provided to generate standard curves for each analyte. The weight of the tissue was used to normalize amount of cytokine/chemokine per milligram of tissue weight. Day 0 time point represents uninfected controls. Each point represents the mean ± SEM summarizing the results of two independent experiments. *, p<.05 comparing the WT to CXCL9−/− and CXCL10−/−groups.
Effector cell mobilization into the central nervous system is impaired in CXCL9−/− and CXCL10−/− mice
Since chemokine levels were significantly altered in CXCL9−/− and CXCL10−/− mice in comparison to WT animals, would these changes influence leukocyte mobilization? In contrast to the predicted outcome, there was no significant difference in the absolute number of CD45high, NKT, CD4+ T, or total CD3+ T cell populations infiltrating the CNS or vagina comparing the WT to chemokine knockout mice in response to acute genital HSV-2 infection (data not shown). However, there was a significant but transient reduction in the number of NK cells infiltrating the CNS and vagina of knockout mice in comparison to WT animals at day 3 (vagina only) and/or day 5 pi (Fig. 5). It should also be noted CXCL9−/− mice had significantly more NK cells within the vaginal tissue at day 3 pi compared to CXCL10−/− mice (Fig. 5). Likewise, total CD8+ T cell numbers were also increased in the spinal cord of WT mice compared to chemokine knockout mice at day 7 pi (Fig. 5). A similar trend in CD8+ T cell number was found in the brain stem and vaginal tissue although the levels did not reach significance (data not shown). Collectively, the results demonstrate a deficiency in CXCL9 or CXCL10 significantly impacts on the recruitment of CD8+ T cells and NK cells to the spinal cord whereas the increase in other chemokines observed in this tissue including CCL2, CCL3, and CCL5 do not play a significant role in response to HSV-2 infection (based on their level of expression in the chemokine deficient mice).
Figure 5.
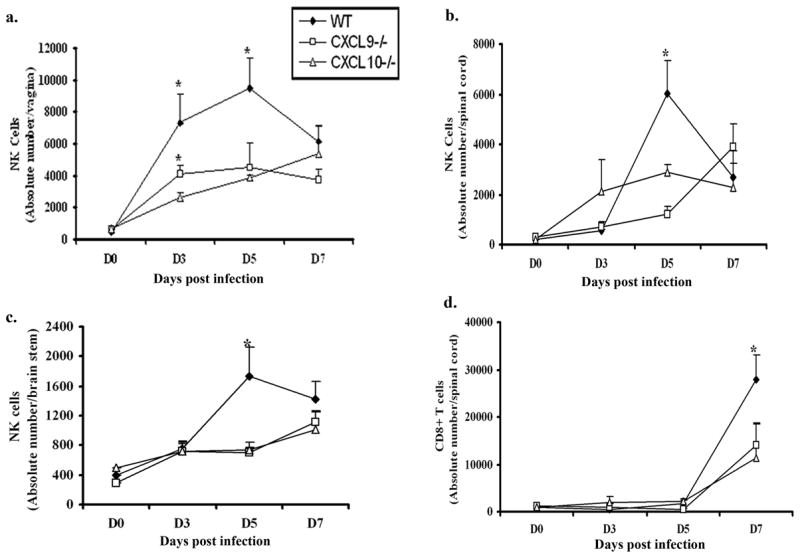
NK and CD8+ T cells infiltration into infected tissue of chemokine knockout mice is reduced or delayed. WT, CXCL9−/− and CXCL10−/− mice (n= 6/group) were infected with HSV-2 (2000 pfu/vagina) and subsequently exsanguinated at indicated times post infection (pi) and (a) vaginal tissue, (b) spinal cord, and (c) brain stem samples were processed and analyzed for NK cell (NK1.1+CD3−CD45high) content by flow cytometry. (d) Similarly, spinal cords were processed and analyzed for CD8+ T (CD3+CD8+CD45high) cells content using flow cytometry. Day 0 time point represents uninfected controls. Each point represents the mean ± SEM summarizing the results of three independent experiments. *, p<0.05 comparing WT to CXCL9−/− and CXCL10−/−.
Virus specific T cell mobilization is reduced in the CNS of CXCL9−/− and CXCL10−/− mice
The ability to identify the antigen specific CD8+ T cells in a given population of lymphocytes is made possible by the use of tetramer (38). Since a reduction in total CD8+ T cell numbers was found in the spinal cord of CXCL9−/− and CXCL10−/− mice following genital HSV-2 infection, would these changes also be reflected in the number and function of HSV-specific CD8+ effector T cells as well? In the case of HSV-2, we analyzed the HSV-gB specific tetramer positive CD8+ T cells residing in the spinal cord, brain stem, vaginal tissue, I/ILN, and spleen following HSV-2 infection. There were no detectable HSV-gB+ CD8+ T cells tissue prior to infection. However, there were more tetramer positive CD8+ T cells residing in the spinal cord and brain stem of WT mice at day 7 pi in comparison to CXCL9−/− and CXCL10−/− animals (Fig. 6a). In contrast, there was no significant loss of HSV gB-specific CD8+ T cells found in the vaginal tissue of WT mice in comparison to chemokine knockout mice (data not shown). To establish the functional role of resident CD8+ T cells, leukocytes isolated from spinal cord of WT, CXCL9−/− and CXCL10−/− mice were evaluated for CTL activity. Cells extracted from spinal cord preparations of CXCL9−/− and CXCL10−/− mice were found to exhibit significantly less cytolytic activity against HSV-2 specific target cells in comparison to WT (Fig. 6b). By comparison, spleen and I/ILN cells from WT, CXCL9−/− and CXCL10−/−HSV-2-infected mice showed similar cytolytic levels against HSV-2 infected targets (Fig. 6c & d). To determine whether the reduced cytolytic activity in the chemokine deficient mice could be corrected with the addition of exogenous chemokine, spinal cord leukocytes obtained from HSV-2-infected CXCL9−/− mice were evaluated for cytolytic activity in the presence or absence of recombinant CXCL9 (2 ng/culture). The results show no difference between effector cells from mock-treated cultures (11% cytolysis of target cells) in comparison to effector cells from CXCL9-treated cultures (12% cytolysis of target cells) relative to effector cells from WT cultures (26% cytolysis of target cells). Taken together, the loss of HSV-specific CD8+ T cells in the CNS of CXCL9−/− and CXCL10−/− mice correlates with a loss in CTL function by the enriched leukocytes from the spinal cord.
Figure 6.
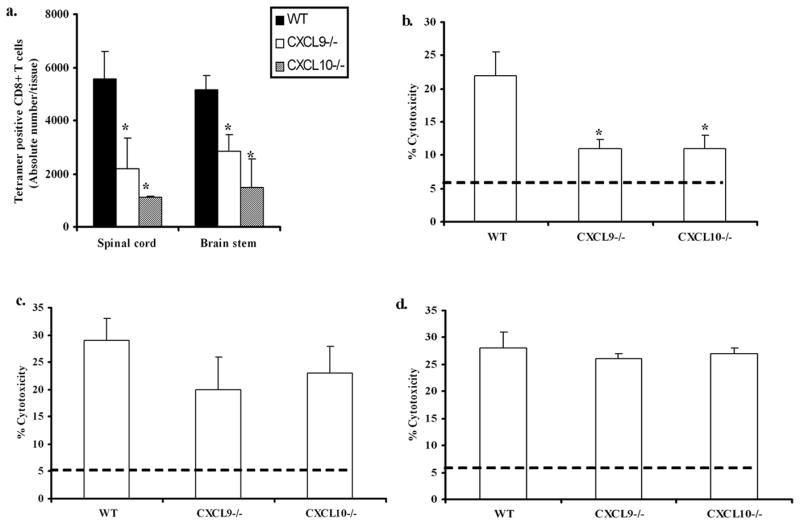
Reduced HSV-2 specific CD8+ T cell recruitment is associated with decreased cytolytic activity. WT, CXCL9−/− and CXCL10−/− mice were (n= 6/group) were infected with HSV-2 (2000 pfu/vagina). At indicated times, mice were exsanguinated and spinal cord, Spleen and I/ILN were processed and analyzed (a), for tetramer positive T cells at day 7 and (b), for CTL activity using percoll gradient enriched SC leukocytes, (c), for CTL activity using I/ILN lymph node cells and (d), for CTL activity using spleen cells. For tetramer staining, MHC I tetramer specific for HSV peptide gB498–505 (SSIEFARL) was used to assay the total tetramer positive cells in the spinal cord. For CTL assay, HSV-2 infected target cells (MC57G) were labeled with CFSE dye and incubated with percoll gradient enriched SC leukocytes or spleen cells or lymph node cells at a E:T ratio of 10:1 for 4 hr at 37°C. Propidium iodide was added after the 4 hr incubation and percent lysis was determined using flow cytometry. The line in b-d indicates the background PI incorporation into CSFE-labeled targets incubated only or in the presence of spleen cells from uninfected mice. The bars represent the mean ± SEM summarizing the results of two independent experiments. *, p<.05 comparing WT to CXCL9−/− and CXCL10−/−groups.
Discussion
The present study underscores the role of two specific chemokines CXCL9 and CXCL10 in mounting a host response against genital HSV-2 infection. In the absence of CXCL9 or CXCL10, mice showed heightened sensitivity to genital HSV-2 infection compared to WT controls even though both chemokines operate through the same receptor, CXCR3. Relative to CXCL9 and CXCL10 expression in the vaginal tissue of WT mice, CXCL10 was found to be expressed rapidly within 24 hrs following HSV-2 infection while CXCL9 expression was not noticeably elevated until day 3 pi. Furthermore, CXCL9 levels were significantly reduced in CXCL10−/− mice in comparison to WT mice. We propose the absence of CXCL10 and delay in CXCL9 within the vaginal tissue results in the reduction in NK cell mobilization to the vagina leading to early viral infection of the spinal cord of CXCL10−/− mice. In comparison to these studies, CCR5 deficient mice show no deficiency in NK cell recruitment to the vagina following HSV-2 infection and no difference in virus recovered in the spinal cord early pi in comparison to WT animals (32). Consequently, we suggest CXCL10 but not CXCL9 or CCR5 ligands is the principal chemoattractant molecule liberated early post genital HSV-2 infection that serves to signal NK cell mobilization to the vaginal tissue.
The increase in HSV-2 found in the spinal cord of CXCL9−/− and CXCL10−/− mice correlates with elevated chemokine levels including CXCL1, CCL2, CCL3, CCL5 chemokines. Similar observations have also been reported in other CNS virus infections, including vesicular stomatitis virus, lymphocytic choriomeningitis virus, mouse hepatitis virus, as well as experimental autoimmune encephalomyelitis (39–42). Our previous findings suggest that CCL2 or CCL5 levels within the CNS do not correspond to virus-mediated mortality as a result of ocular HSV-1 infection (43–44) while one study reports CNS levels of CCL2 are highly correlative with HSV encephalitis (45). Likewise, CCL3 has been linked to a number of CNS inflammatory infections including dengue virus, progressive multifocal leukoencephalopathy associated with AIDS, and Listeria meningoencephalitis (46–48).
We consistently found TNF-α elevated in the vagina, spinal cord, and brain stem of the CXCL9−/− and CXCL10−/− mice. Similar results were also reported in CCR5 deficient mice which were also found to have a higher mortality rate compared to the WT controls following genital HSV-2 infection (32). As TNF-α can be neurotoxic (49), it is tempting to speculate the increase in CNS TNF-α levels in the chemokine deficient mice may be the primary contributory factor resulting in a higher mortality rate in comparison to WT animals. Although mortality rates were not evaluated, early studies suggest the neutralization of TNF-α has no effect on virus resolution following genital HSV-2 infection (50–52).
A significant reduction in NK cell mobilization at day 3 and day 5 pi and reduction in CD8+ T cell trafficking to the spinal cord at day 7 pi were observed in the CXCL9−/− and CXCL10−/− mice. We previously reported depletion of NK cells following genital HSV-2 infection resulted in an elevation in virus recovered in the vaginal tissue and brain stem but not spinal cords of WT mice (32). Furthermore, previous studies have shown that the reduction of NK cell activity through antibody-mediated depletion of IL-12, IL-15 and IL-18 results in higher virus titers and increased mortality following HSV-2 infection (4, 27, 28, 53). Collectively, we interpret these findings to suggest NK cells maintain virus surveillance within the genital tract and brain stem following vaginal HSV-2 infection while CD8+ T cells monitor infection within the spinal cord.
Previous studies have suggested CXCL10 plays a central role in recruiting CD8+ T cells to the lymphocytic choriomeningitis virus-infected CNS controlling virus infection through a perforin-mediated lysis of infected cells (25,35,54). Other studies have linked Fas-FasL-mediated apoptosis in control of Theiler’s virus whereas in vaccinia virus, vesicular stomatitis virus or Semliki virus infections, neither Fas-FasL or perforin pathways were required for CNS clearance (55–56). While we have not formally proven the cytolytic process involved in CD8+ CTL effector activity for cells infiltrating the CNS following genital HSV-2 infection, FasL does not appear to be expressed on CD8+ T cells residing in the spinal cord of HSV-2 infected mice (Thapa & Carr, unpublished observation). The deficiency in CTL activity in the CNS of CXCL9−/− and CXCL10−/−mice is most likely due to an insufficient number of effector cells (based on HSV gB-specific tetramer stain) mobilized to the infected tissue as opposed to aberrant cytolytic effector molecules expressed by effector cells since similar CTL levels were observed in the spleen and lymph node cell populations comparing all genotypes. In addition, recombinant murine CXCL9 did not restore CTL activity by effector cells obtained from the spinal cord of CXCL9−/− mice suggesting the reduced cytolytic activity is not due to chemokine deficiency.
Collectively, the present study indicates an increase in sensitivity to genital HSV-2 infection in mice deficient in CXCL9 and CXCL10 is associated with a reduction in the recruitment of an optimal number of NK cells and HSV-specific CD8+ T cells to sites of infection. We interpret the non-redundant role of CXCL9 and CXCL10 to be a result of tissue-specific and temporal expression represented by levels measured in the draining lymph nodes and infected vaginal tissue of mice. It has yet to be determined whether the source of the chemokines is principally non-hematopoietic or hematopoietic-derived cells. It is also unknown whether CXCR3 expression is modified in the absence of CXCL9 or CXCL10 which could also explain changes in the mobilization of cells. However, the present observation does suggest a hierarchy of CXCR3 ligand expression that influences both HSV gB-specific CTL mobility and NK cell recruitment to vaginal tissue and the CNS. A rapid expression of CXCL10 in the infected genitalia may augment NK cell mobilization to the vagina ultimately resulting in a reduction or delay in virus trafficking to the sacral ganglia and spinal cord reducing the likelihood of the establishment of latency.
Acknowledgments
The authors acknowledge Dr. Josh Farber & Dr. Andrew Luster for the CXCL9−/− and CXCL10−/− mice and are thankful to Todd Wuest and Gabriel Nyugen for their excellent technical help. The authors are grateful for constructive criticism by Dr. Paul Kincade (Oklahoma Medical Research Foundation, Oklahoma City, OK) in reading the manuscript.
Footnotes
This work was supported by USPHS grant AI067309.
Abbreviations used in this paper: HSV-2, herpes simplex virus type 2; CNS, central nervous system; WT, wild type; pfu, plaque forming units; pi, post infection; I/ILN, inguinal/iliac lymph nodes; CSFE, carboxyfluorescein diacetate succinimidyl ester.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Inagaki K, Daikoku T, Goshima F, Nashiyama Y. Impaired induction of protective immunity by highly virulent herpes simplex type 2 in a murine model of genital herpes. Arch Virol. 2000;145:1989–2002. doi: 10.1007/s007050070035. [DOI] [PubMed] [Google Scholar]
- 2.Harandi AM, Svennerholm B, Holmgren J, Eriksson K. Differential roles of B cells and IFN- -secreting CD4+ T cells in innate and adaptive immune control of genital herpes simplex virus type 2 infection in mice. J Gen Virol. 2001;82:845–853. doi: 10.1099/0022-1317-82-4-845. [DOI] [PubMed] [Google Scholar]
- 3.Whitley RJ, Miller RL. Immunologic approach to Herpes Simplex Virus. Viral Immunol. 2001;14(2):111–118. doi: 10.1089/088282401750234484. [DOI] [PubMed] [Google Scholar]
- 4.Duerst RJ, Morrison LA. Innate immunity to herpes simplex virus type 2. Viral Immunol. 2003;16(4):475–490. doi: 10.1089/088282403771926300. [DOI] [PubMed] [Google Scholar]
- 5.MasCasullo V, Fam E, Keller MJ, Herold BC. Role of mucosal immunity preventing genital herpes infection. Viral Immunol. 2005;18(4):595–606. doi: 10.1089/vim.2005.18.595. [DOI] [PubMed] [Google Scholar]
- 6.Celum CL. The interaction between herpes simplex virus and human immunodeficiency virus. Herpes. 2004;11(Supplement 1):36A–45A. [PubMed] [Google Scholar]
- 7.Strick LB, Wald A, Celum C. Management of Herpes Simplex Virus Type 2 Infection HIV Type 1–Infected Persons. Clinical Infectious Diseases. 2006;43:347–356. doi: 10.1086/505496. [DOI] [PubMed] [Google Scholar]
- 8.Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 2000;95(10):3032–3043. [PubMed] [Google Scholar]
- 9.Park MK, Amichay D, Love P, Elizabeth W, Fang L, Grienberg A. The CXC chemokine murine monokine induced by IFN- (CXC chemokine ligand 9) is made by APCs, targets lymphocytes including activated B cells, and supports antibody responses to a bacterial pathogen in vivo. J Immunol. 2002;169:1433–1443. doi: 10.4049/jimmunol.169.3.1433. [DOI] [PubMed] [Google Scholar]
- 10.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN- - inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168:3195–3204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- 11.Liu MT, Chen BP, Oertel P, Buchmeier MJ, Armstrong D, Hamilton TA, Lane TE. The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J Immunol. 2000;165:2327–2330. doi: 10.4049/jimmunol.165.5.2327. [DOI] [PubMed] [Google Scholar]
- 12.Kolb SA, Sporer B, Lahrtz F, Koedel U, Pfister HW, Fontana A. Identification of a T cell chemotactic factor in the cerebrospinal fluid of HIV-1 infected individuals as interferon- inducible protein 10. J Neuroimmunol. 1999;93:172–181. doi: 10.1016/s0165-5728(98)00223-9. [DOI] [PubMed] [Google Scholar]
- 13.Khan I, MacLean JA, Lee F, Casciotti L, DeHaan E, Schwartzman J, Luster A. The IP-10 chemokine is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity. 2000;12:483–494. doi: 10.1016/s1074-7613(00)80200-9. [DOI] [PubMed] [Google Scholar]
- 14.Liu MT, Keirstead HS, Lane TE. Neutralization of the chemokine CXCL10 reduces inflammatory cell invasion and demyelination and improves neurological function in a viral model of multiple sclerosis. J Immunol. 2001;167:4091–4097. doi: 10.4049/jimmunol.167.7.4091. [DOI] [PubMed] [Google Scholar]
- 15.Melchjorsen J, Sorensen LN, Paludan SR. Expression and function of chemokines during viral infections: from molecular mechanisms to in vivo function. J Leukocyte Biol. 2003;74:331–343. doi: 10.1189/jlb.1102577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apolinario A, Majano PL, Alvarez-Perez E, Saez A, Lozano C, Varqas J, Garcia-Monzon C. Increased expression of T cell chemokines and their receptors in chronic hepatitis C: relationship with the histological activity of liver disease. Am J Gastroenterol. 2002;97:2861–2870. doi: 10.1111/j.1572-0241.2002.07054.x. [DOI] [PubMed] [Google Scholar]
- 17.Mahalingam S, Farber JM, Karupiah G. The interferon-inducible chemokines MuMig and Crg-2 exhibit antiviral activity in vivo. J Virol. 1998;73:147. doi: 10.1128/jvi.73.2.1479-1491.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salazar-Mather TP, Hamilton TA, Biron CA. A chemokine to cytokine to chemokine cascade critical in antiviral defense. J Clin Invest. 2000;105:985. doi: 10.1172/JCI9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu MT, Armstrong D, Hamilton TA, Lane TE. Expression of Mig (monokine induced by interferon-y) is important in T lymphocyte recruitment and host defense following viral infection of central nervous system. J Immunol. 2001;166:1790–1795. doi: 10.4049/jimmunol.166.3.1790. [DOI] [PubMed] [Google Scholar]
- 20.McDermott MR, Goldsmith CH, Rosenthal KL, Brais LJ. T lymphocytes in genital lymph nodes protect mice from intravaginal infection with herpes simplex virus type 2. J Infectious Diseases. 1989;159:460–466. doi: 10.1093/infdis/159.3.460. [DOI] [PubMed] [Google Scholar]
- 21.Parr MB, Parr EL. Mucosal immunity to herpes simplex virus type 2 in the mouse vagina is impaired by in vivo depletion of T lymphocytes. J Virol. 1998;72:2677–2685. doi: 10.1128/jvi.72.4.2677-2685.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koelle DM, Posavad CM, Barnum GR, Johnson ML, Frank JM, Corey L. Clearance of HSV-2 from recurrent genital lesions correlates with infiltration of HSV-specific cytotoxic T lymphocytes. J Clinical Investigation. 1998;101:1500–1508. doi: 10.1172/JCI1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrison LA, Zhu L, Thebeau LG. Vaccine-induced serum immunoglobulin contributes to protection from herpes simplex virus type 2 genital infection in the presence of immune T cells. J Virol. 2001;75:1195–1204. doi: 10.1128/JVI.75.3.1195-1204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cunningham AL, Mikloska Z. The holy grail: immune control of human herpes simplex virus infection and disease. Immune control of HSV infection and Disease Herpes. 2001;8(supplement 1):6A–9A. [PubMed] [Google Scholar]
- 25.Dobbs ME, Strasser JE, Chu C, Chalk C, Milligan GN. Clearance of Herpes Simplex Virus Type 2 by CD8+ T cells requires gamma interferon and either perforin- or fas-mediated cytolyitc mechanisms. J Virol. 2005;79(23):14546–14554. doi: 10.1128/JVI.79.23.14546-14554.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pardo J, Balkow S, Anel A, Simon MM. The differential contribution of granzyme A and granzyme B in cytotoxic T lymphocyte-mediated apoptosis is determined by the quality of target cells. Eur J Immunol. 2002;32(7):1980–1985. doi: 10.1002/1521-4141(200207)32:7<1980::AID-IMMU1980>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 27.Biron CA, Nyugen KB, Pien GC, Cousins LP, Salazar-Mather TP. Natural Killer Cells in Antiviral Defense: functions and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 28.Ashkar AA, Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol. 2003;10:168–171. doi: 10.1128/JVI.77.18.10168-10171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milligan GN, Bernstein DI, Bourne N. T Lymphocytes are required for protection of the vaginal mucosae and sensory ganglia of immune mice against reinfection with Herpes Simplex Virus Type 2. J Immunol. 1998;160:6093–6100. [PubMed] [Google Scholar]
- 30.Sin JI, Kim JJ, Pachuk C, Satishchandran C, Weiner DB. DNA vaccines encoding interleukin-8 and RANTES enhance antigen-specific Th1-type CD4+T-cell-mediated protective immunity against herpes simplex virus type 2 in vivo. J Virol. 2000;74:11173–11180. doi: 10.1128/jvi.74.23.11173-11180.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Santo JP. Natural Killer Cell Developmental Pathways: A Question of Balance. Annu Rev Immunol. 2006;24:257–286. doi: 10.1146/annurev.immunol.24.021605.090700. [DOI] [PubMed] [Google Scholar]
- 32.Thapa M, Kuziel WA, Carr DJJ. Susceptibility of CCR5 mice is linked to NK cell mobilization. J Virol. 2007;81:3704–3713. doi: 10.1128/JVI.02626-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh R, Kumar A, Creery WD, Ruben M, Giulivi A, Diaz-Mitoma F. Dysregulated expression of IFN-and IL-10 and impaired IFN- -mediated responses at different disease stages in patients with genital herpes simplex virus-2 infection. Clin Exp Immunol. 2003;133:97–107. doi: 10.1046/j.1365-2249.2003.02183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parr MB, Parr EL. The role of gamma interferon in immune resistance to vaginal infection by herpes simplex virus type 2 in mice. Virology. 1999;258:282–294. doi: 10.1006/viro.1999.9739. [DOI] [PubMed] [Google Scholar]
- 35.Christensen JE, Lemos CD, Moos T, Christensen JP, Thomsen AR. CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J Immunol. 2006;176:4235–4243. doi: 10.4049/jimmunol.176.7.4235. [DOI] [PubMed] [Google Scholar]
- 36.Ghersa P, Gelati M, Colinge J, Feger G, Power C, Papoin R, Salmaggi A. MIG-differential gene expression in mouse brain endothelial cells. Neuroreport. 2002;13:9–14. doi: 10.1097/00001756-200201210-00007. [DOI] [PubMed] [Google Scholar]
- 37.Härle P, V, Cull F, Agbaga MP, Silverman RF, Williams BR, James C, Carr DJ. Differential effect of murine alpha/beta interferon transgenes on antagonization of herpes simplex virus type 1 replication. J Virol. 2002;76:6558–6567. doi: 10.1128/JVI.76.13.6558-6567.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skinner PJ, Daniels M, Schmidt CS, Jameson SC, Haase AT. Cutting edge: In situ tetramer staining of antigen-specific T cells in tissues. J Immunol. 2000;165(2):613–617. doi: 10.4049/jimmunol.165.2.613. [DOI] [PubMed] [Google Scholar]
- 39.Ireland DDC, Reiss CS. Gene expression contributing to recruitment of circulating cells in response to vesicular stomatitis virus infection of CNS. Viral Immunol. 2006;19(3):535–545. doi: 10.1089/vim.2006.19.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asensio VC, I, Campbell L. Chemokine gene expression in brains of mice with lymhocytic choriomeninigitis. J Virol. 1997;71:7832–7840. doi: 10.1128/jvi.71.10.7832-7840.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glass WG, Chen BP, Liu MT, Lane TE. Mouse hepatitis virus infection of the central nervous system: chemokine mediated regulation of host defense and disease. Viral Immunol. 2002;15:262–272. doi: 10.1089/08828240260066215. [DOI] [PubMed] [Google Scholar]
- 42.Dos Santos AC, Barsante MM, Esteves Arantes RM, Bernard CC, Teixeira MM, Carvalho-Tavares J. CCL2 and CCL5 mediate leukocyte adhesion in experimental autoimmune encephalomyelitis: An intravital microscopy study. J Neuroimmunol. 2005;162:122–129. doi: 10.1016/j.jneuroim.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 43.Wickham SB, Lu B, Ash J, Carr DJJ. Chemokine receptor deficiency is associated with increased chemokine expression in the peripheral and central nervous systems and increased resistance to herpetic encephalitis. J Neuroimmunol. 2005;162:51–59. doi: 10.1016/j.jneuroim.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Carr DJJ, Ash J, Lane TE, Kuziel WA. Abnormal immune response of CCR5-deficient mice to ocular infection with herpes simplex virus type 1. J Gen Virol. 2006;87:490–499. doi: 10.1099/vir.0.81339-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RM. Herpes simplex virus 1 interaction with toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci. 2004;101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanchez-Burgos G, Hernandez-Pando R, Campbell IL, Ramos-Castenada J, Ramos C. Cytokine production in brain of mice experimentally infected with dengue virus. Neuroreport. 2004;15:37–42. doi: 10.1097/00001756-200401190-00009. [DOI] [PubMed] [Google Scholar]
- 47.Bonwetsch R, Croul S, Richardson MW, Lorenzana C, Valle LD, Sverstiuk AE, Amini S, Morgello S, Khalili Rappaport K. Role of HIV-1 tat and CC chemokine Mip-1alpha in the pathogenesis of HIV associated central nervous system disorders. J Neurovirol. 1999;5:685–694. doi: 10.3109/13550289909021297. [DOI] [PubMed] [Google Scholar]
- 48.Seebach J, Bartholdi D, Frei K, Spanaus KS, Ferrero E, Widmer U, Isenmann S, Strieter RM, Schwab M, Pfister H. Experimental Listeria meninigoencephalitis: Macrophage inflammatory protein-1 and -2 produced intrathecally and mediate chemotactic activity in cerebrospinal fluid of infected mice. J Immunol. 1995;155:4367–4375. [PubMed] [Google Scholar]
- 49.Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 2005;034:11–24. doi: 10.1016/j.brainres.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 50.Milligan GN, Dudley-McClain KL, Young CG, Chu CF. T-cell-mediated mechanisms involved in resolution of genital herpes simplex virus type 2 (HSV-2) infection of mice. J Reprod Immunol. 2004;61:115–127. doi: 10.1016/j.jri.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 51.Nuovo GJ, Defaria DL, Chanona-Vilchi JG, Zhang Y. Molecular detection of rabies encephalitis and correlation with cytokine expression. Mod Pathol. 2005;18:62–67. doi: 10.1038/modpathol.3800274. [DOI] [PubMed] [Google Scholar]
- 52.Sternberg JM, Rodgers J, Bradley B, Maclean L, Murray M, Kennedy PG. Meningoencephalitic African trypanosomiasis: Brain IL-10 and IL-6 are associated with protection from neuron-inflammatory pathology. J Neuroimmunol. 2005;167:81–89. doi: 10.1016/j.jneuroim.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 53.Harandi AM, Svennerholm B, Holmgren J, Eriksson K. Interleukin-12 (IL-12) and IL-18 are important in innate defense against genital herpes simplex virus type 2 infection in mice but are not required for the development of acquired gamma interferon-mediated protective immunity. J Virol. 2001;75:6705–6709. doi: 10.1128/JVI.75.14.6705-6709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 55.Rossi CP, McAllister A, Tanguy M, Kagi D, Brahic M. Theiler’s virus infection of perforin deficient mice. J Virol. 1998;72:4515–4519. doi: 10.1128/jvi.72.5.4515-4519.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kagi D, Hengartner H. Different roles for cytotoxic T cells in the control of infections with cytopathic versus noncytopathic viruses. Curr Opin Immunol. 1996;8:472–477. doi: 10.1016/s0952-7915(96)80033-1. [DOI] [PubMed] [Google Scholar]