

Abstract
Ribonuclease L (RNase L) is an antiviral endoribonuclease that cleaves hepatitis C virus (HCV) RNA at single-stranded UA and UU dinucleotides throughout the open reading frame (ORF). To determine whether RNase L exerts evolutionary pressure on HCV we examined the frequencies of UA and UU dinucleotides in 162 RNA sequences from the Los Alamos National Labs HCV Database (http://hcv.lanl.gov). Considering the base composition of the HCV ORFs, both UA and UU dinucleotides were less frequent than predicted in each of 162 HCV RNAs. UA dinucleotides were significantly less frequent than predicted at each of the three codon positions while UU dinucleotides were less frequent than predicted predominantly at the wobble position of codons. UA and UU dinucleotides were among the least abundant dinucleotides in HCV RNA ORFs. Furthermore, HCV genotype 1 RNAs have a lower frequency of UA and UU dinucleotides than genotype 2 and 3 RNAs, perhaps contributing to increased resistance of HCV genotype 1 infections to interferon therapy. In vitro, RNase L cleaved both HCV genotype 1 and 2 RNAs efficiently. Thus, RNase L can cleave HCV RNAs efficiently and variably reduced frequencies of UA and UU dinucleotides in HCV RNA ORFs are consistent with the selective pressure of RNase L.
Introduction
Hepatitis C virus (HCV) has infected more than 4 million adults in the United States and 3.2 million are chronically infected at present (Armstrong et al., 2006). Although HCV provokes both innate and acquired immune responses, HCV effectively evades host defenses in most people to establish subclinical chronic infections (Thimme et al., 2006). After decades of inapparent infection, accumulating liver damage becomes clinically relevant in the forms of cirrhosis, liver failure, and hepatocellular carcinoma (Alter, 1997).
Interferon, in combination with the antiviral drug ribavirin, can cure chronic HCV infections (McHutchison et al., 1998). Unfortunately, many HCV infections are resistant to interferon therapy (reviewed in (McHutchison and Hoofnagle, 2000)). In particular, HCV genotype 1 infections resist interferon therapy more often than HCV genotype 2 and 3 infections (McHutchison et al., 1998). 48 weeks of pegylated interferon and ribavirin therapy results in a sustained viral response (SVR) in 42% of patients with HCV genotype 1 infections while 24 weeks of therapy results in a SVR in >80% of patients with genotype 2 or 3 infections (Manns et al., 2001). Compounding the problem of poor SVR for genotype 1 infections is the fact that HCV genotype 1 infections are much more common in the US than HCV genotype 2 or 3 infections. In addition to HCV genotype several clinical factors correlate with failure of interferon therapy including high viral load, obesity, and increased age (McHutchison and Hoofnagle, 2000). The magnitude of decrease in viral load initially after beginning interferon therapy correlates well with successful therapy (Davis et al., 2003; McHutchison et al., 2001; Terrault et al., 2005) yet little is known about the specific molecular factors that affect the alternate magnitudes of viral load decrease following induction of interferon/ribavirin therapy.
Interferon activates the expression of numerous antiviral proteins and HCV replicons are sensitive to interferon in vitro (Guo et al., 2001; Guo et al., 2003). Nonetheless, HCV encodes several mechanisms to evade both the production of interferon and individual interferon-regulated antiviral pathways (Gale and Foy, 2005). HCV NS3/4a protease cleaves Cardif/IPS-1/MAVS, a protein in a viral-RNA-triggered signal transduction cascade associated with the production of interferon (Evans and Seeger, 2006; Meylan et al., 2005). The 5′ triphosphate of HCV RNA may be a viral ligand recognized by RIG-I before subsequent interaction of RIG-I with Cardif/IPS-1/MAVS (Hornung et al., 2006; Pichlmair et al., 2006). Cleavage of Cardif/IPS-1/MAVS in HCV infected cells inhibits the activation of NF-kB and IRF-3, thereby inhibiting the expression of several important chemokines and cytokines, including interferon (Foy et al., 2005; Johnson and Gale, 2006; Loo et al., 2006). Furthermore, HCV can directly inhibit PKR, an interferon-regulated dsRNA-activated antiviral protein (Gale et al., 1998). HCV NS5a binds to and inhibits the antiviral activity of PKR, however, polymorphisms in NS5a do not correlate with outcomes of interferon therapy (Sarrazin et al., 2000; Yang et al., 2003). Likewise, HCV genotype 1 E2 can bind and inhibit PKR (Pavio et al., 2002; Taylor et al., 1999; Taylor et al., 2001). Nonetheless, polymorphisms in the PKR-eIF2a phosphorylation homology domain of HCV E2 do not correlate with the variable efficacy of interferon therapy in HCV genotype 1 infections (Watanabe et al., 2003). Thus, molecular explanations for the variable response of individual HCV infections to interferon therapy are incomplete and there are no viral or host polymorphisms which reliably predict the success of interferon therapy.
RNase L is a latent endoribonuclease in an interferon-regulated dsRNA-activated antiviral pathway (reviewed in (Player and Torrence, 1998)). 2-5A, the product of dsRNA-activated oligoadenylate synthetase (OAS), binds to ankyrin repeats within monomeric RNase L leading to the dimerization and activation of RNase L endoribonuclease (Dong et al., 2001; Tanaka et al., 2004). RNase L is expressed in most tissues, including liver (Zhou et al., 2005). In chimpanzees, the magnitude of HCV infection correlates with the magnitude of 2′-5′ OAS expression (Thimme et al., 2002). Using cytoplasmic extracts from HeLa cells, we discovered that HCV RNA was detected and destroyed by the interferon-regulated dsRNA-activated 2′-5′ OAS/RNase L antiviral pathway (Han and Barton, 2002). In contrast to HCV, we found that group C enteroviruses possess an RNA structure that inhibits the endoribonuclease activity of RNase L (Han et al., 2007). RNase L cleaves HCV RNA efficiently at several single-stranded UA and UU dinucleotides throughout the viral RNA ORF (Han et al., 2004); however, the RNase L cleavage sites identified in one HCV genotype 1a strain are not conserved in the majority of HCV ORFs due to the large amount of sequence variability among HCV strains. HCV RNA sequences vary by as much as 31 to 34%, with a significant amount of the variation at wobble positions of codons (Simmonds, 2004) (Smith and Simmonds, 1997). Intriguingly, we observed that several representative HCV RNAs had fewer UA and UU dinucleotides than expected and that UA and UU dinucleotides within wobble positions of codons in HCV RNAs evolved rapidly in patients during interferon therapy (Han and Barton, 2002).
Individual HCV isolates are related to one of six major genotypes (HCV 1, HCV 2, HCV 3, etc.) and more than 50 subtypes (HCV 1a, 1b, 1c; HCV 2a, 2b, etc) (Simmonds, 2000). In this study we report that all strains of HCV RNA have fewer UA and UU dinucleotides than expected, consistent with the selective pressure of RNase L, and that most genotype 1 strains have fewer UA and UU dinucleotides than genotype 2 and 3 strains. The frequency of UA and UU dinucleotides in particular HCV RNAs could affect the efficiency of cleavage by RNase L and thereby contribute to the efficacy of interferon therapy.
Methods
HCV RNA Sequences
162 full-length HCV RNA sequences were collected from the HCV Sequence Database hosted through Los Alamos National Labs (http://hcv.lanl.gov) (Kuiken et al., 2005). The ORF from each sequence was isolated with “getorf”, a utility from the European Molecular Biology Open Software Suite (http://emboss.sourceforge.net) using the EMBOSS graphical controls hosted at the Singapore Biomedical Computing Resource (http://sbcr.bii.a-star.edu.sg/emboss/) (Rice et al., 2000). CLC FreeWorkbench (http://www.clcbio.com) was also used to isolate ORFs from sequences.
Base composition and dinucleotide analyses (Methods 1 & 2)
HCV RNA ORF sequences were packaged by HCV genotype in FASTA format. Nucleotide analyses were performed using the “Simmonics” application (Simmonds and Smith, 1999). The composition scan utility was used and results were presented as base composition and dinucleotide frequency as a function of either the base composition of the ORF (Method 1) or base composition of the three codon positions [1-2, 2-3 (wobble), 3-1 (between codons)] (Method 2). For Method 1, the predicted frequency of particular dinucleotides within an ORF was calculated by multiplying the fractional frequency of each base in the ORF by the length of the ORF [ for instance, 0.213 U × 0.199 A × 9036 bases in the ORF = 383 predicted UA dinucleotides for the ORF of AF009606]. The observed frequency of dinucleotides in each ORF were counted directly. The observed to predicted dinucleotide ratios presented in Tables 1S and Figure 1 were generated using Method 1. Alternatively, predicted dinucleotide frequency was determined considering the base composition of the codon position of each ORF by the formula: (frequency of base in codon position 1) × (frequency of base in codon position 2) × (number of codons in ORF) = predicted frequency of dinucleotide in codon position 1-2. The comparable formula was used for codon positions 2-3 and 3-1 respectively. (Supplemental Data in Figure 1S were derived using Method 2).
Figure 1. Observed to predicted dinucleotide ratios in HCV RNA ORFs.
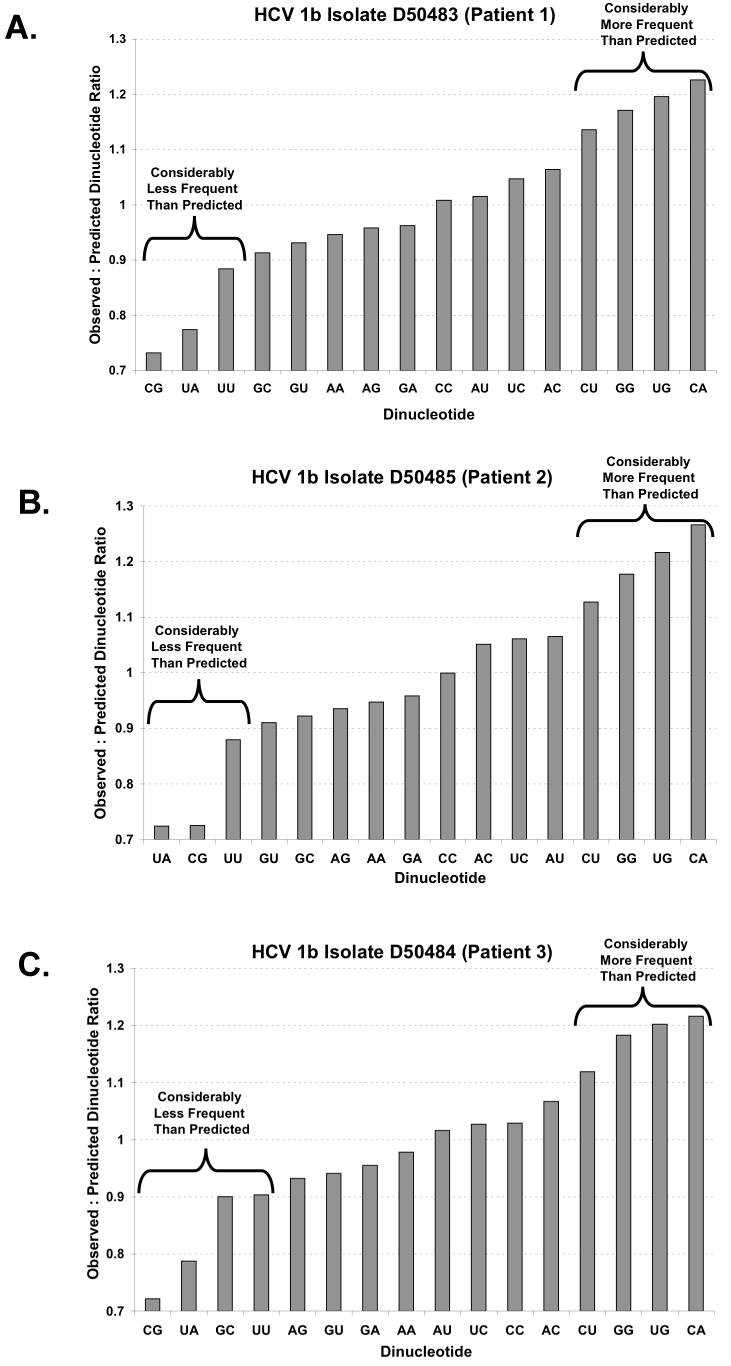
A-C There are 16 potential dinucleotides in an ORF (x-axis). The ratio of observed to predicted dinucleotide frequency (y-axis) was calculated (as described in Methods, Method 1) for three interferon-resistant HCV 1b isolates (Enomoto et al., 1995). Ratios below 1.0 correspond to fewer observed dinucleotides than predicted while ratios above 1.0 correspond to more observed dinucleotides than predicted by the base composition of the codon position.
Permuted HCV RNA ORF sequences (Method 3)
HCV RNA ORF sequences were permuted to provide control sequences for comparison with real HCV ORFs. HCV RNA ORF sequences were permuted randomly at each codon position. The permutation procedure created new HCV ORF-like sequences which preserved the size of the ORF, the absence of stop codons in the ORF, and the cummulative base composition of each codon position in the ORF. The permutations did not preserve dinucleotide biases or codon usage. The observed dinucleotide counts for each permuted ORF were then obtained and compared to the dinucleotide frequencies in the HCV ORF from which the permuted sequences were generated. We compared the average of 100 permuted ORF sequences to each real HCV ORF (as in Figures 2 and 6). The ratio of observed dinucleotide frequencies in real HCV ORFs to dinucleotide frequencies in 100 permuted sequence controls were similar to the ratios derived by mathematical calculations (Compare data in Figures 1 A-C with that in Figures 2 A-C). A key advantage of Method 3 relative to Methods 1 & 2 is that Method 3 excludes stops codons within the permuted ORFs used to predict dinucleotide frequencies, whereas Methods 1 & 2 do not exclude stop codons from the predicted frequencies. Because of this advantage, statistical significance was determined with data derived using Method 3 (Figures 3 & 6).
Figure 2. Observed to permuted dinucleotide ratios in HCV RNA ORFs.
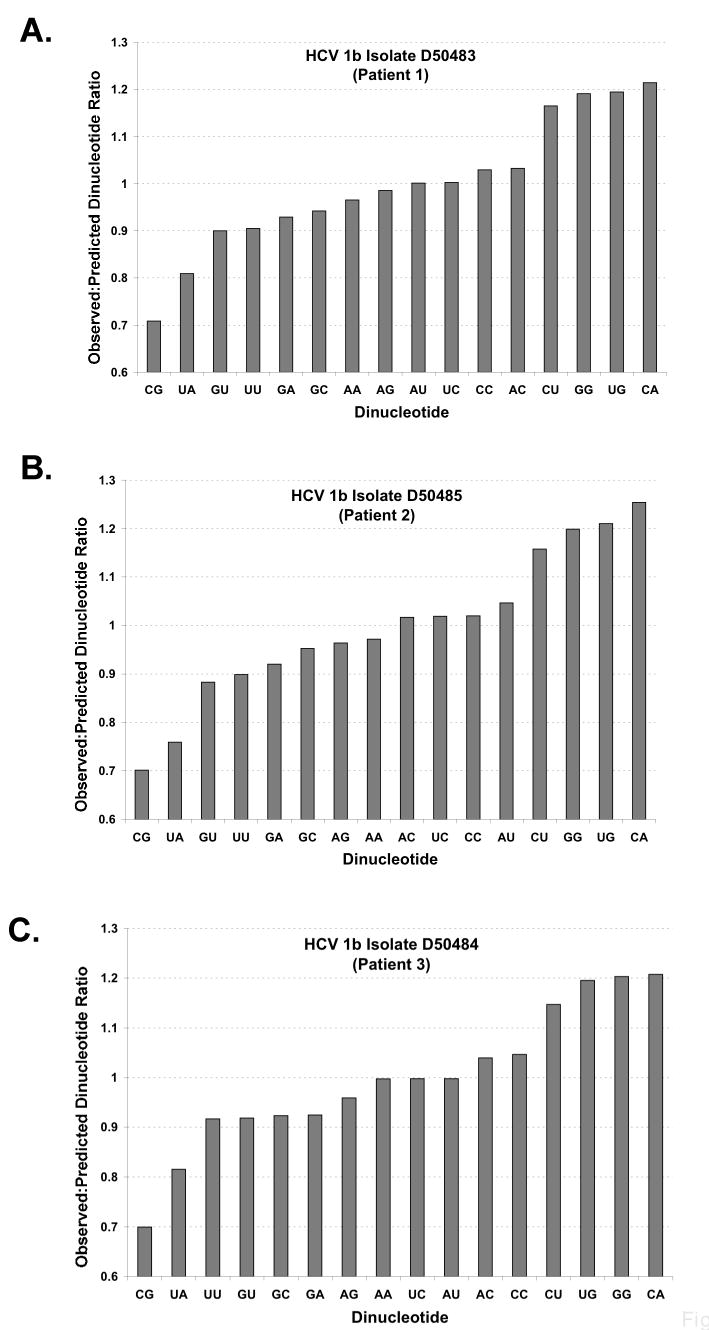
A-C There are 16 potential dinucleotides in an ORF (x-axis). The ratio of observed to permuted dinucleotide frequency (y-axis) was calculated (as described in Methods, Method 3) for three interferon-resistant HCV 1b isolates (Enomoto et al., 1995). Ratios below 1.0 correspond to fewer observed dinucleotides than in the average of 100 permuted sequences while ratios above 1.0 correspond to more observed dinucleotides than in the average of 100 permuted ORF sequences.
Figure 6. Predicted and observed UA and UU dinucleotide frequencies based on codon position.
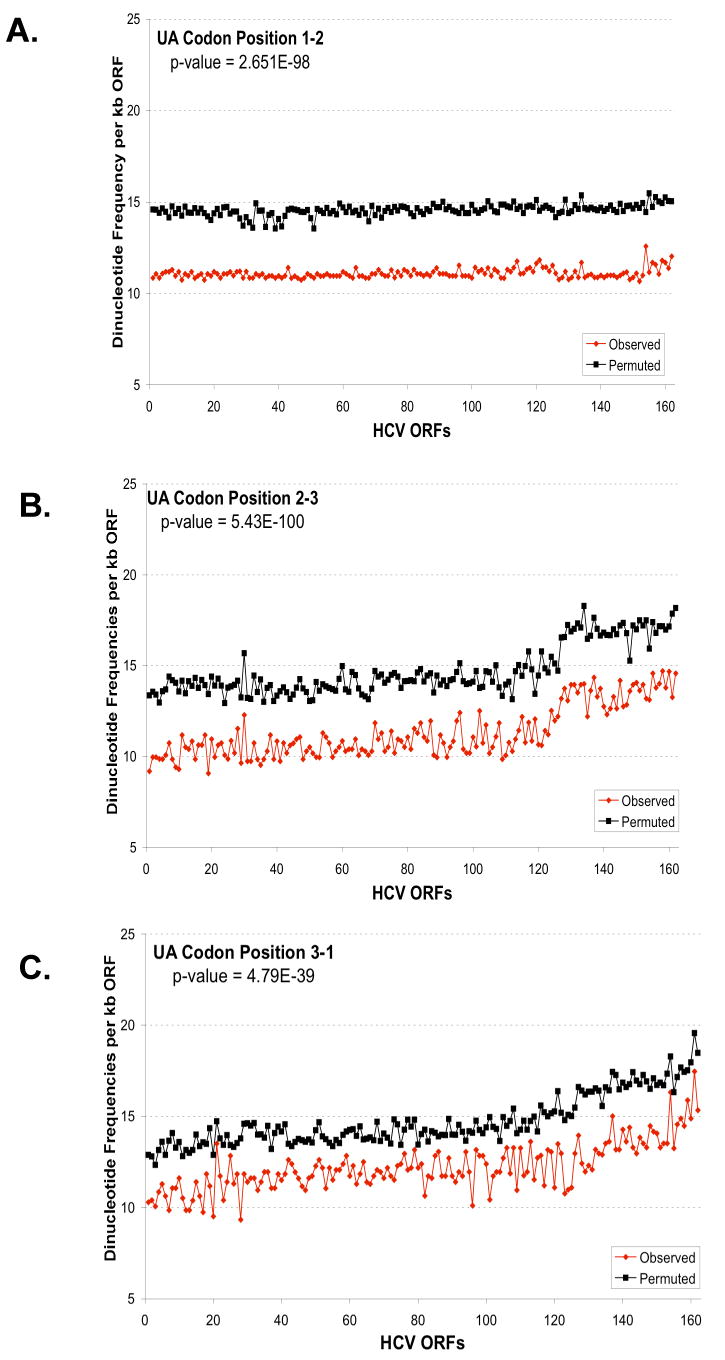
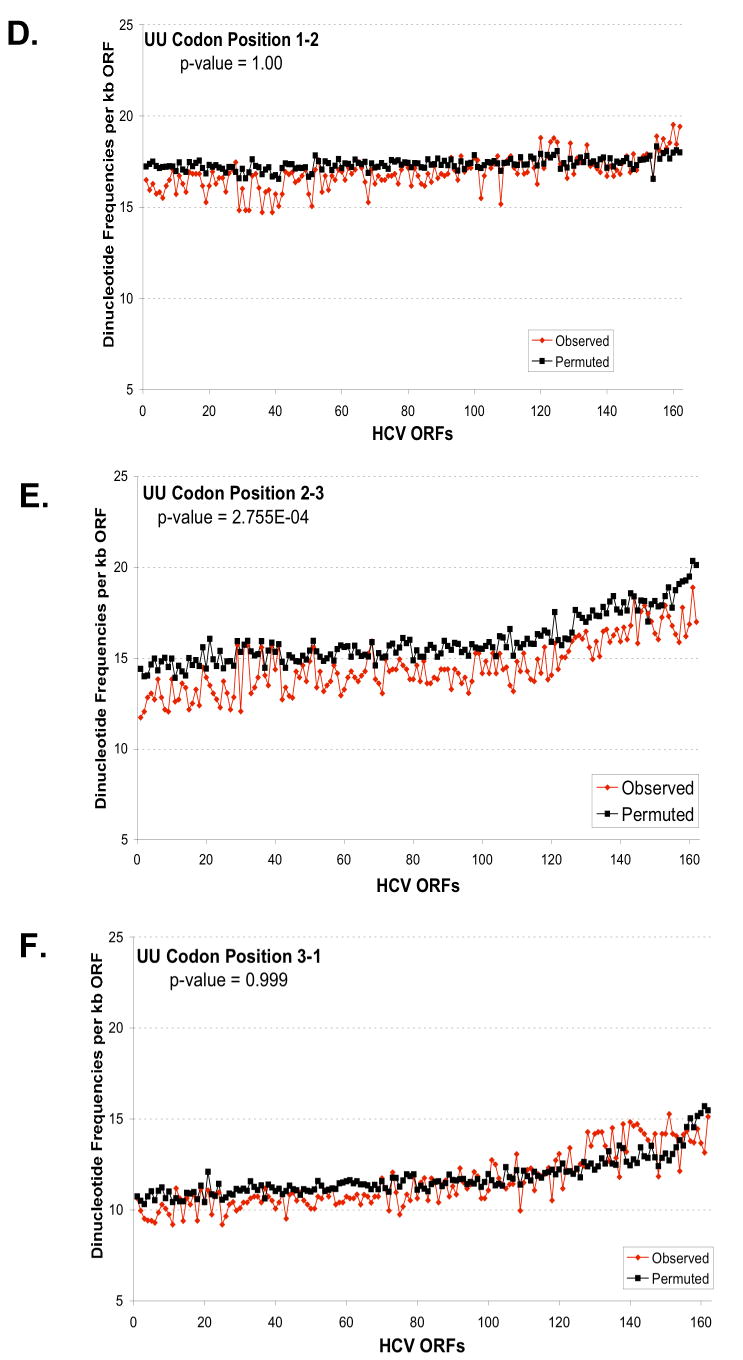
The predicted and observed UA (A-C) and UU (D-F) dinucleotide frequencies were plotted versus codon position: codon position 1-2 (A, D); codon position 2-3 (B, E); and codon position 3-1 (C, F). The frequencies per kb of ORF (y-axis) were plotted versus the order of strains established in Figure 5 (x-axis). Predicted dinucleotide frequencies were obtained using permuted sequences as described in Methods (Method 3). The aggregate results for all strains for each codon position were analysed to evaluate the null hypothesis that the predicted and observed frequencies are the same. The p-values from this test are displayed in each figure. For UA, the observed frequency is significantly different than the predicted for all positions (p-value < .01). While for UU, the observed frequency is only significantly different for position 2-3 (p-value < .01).
Figure 3. Observed to predicted dinucleotide frequencies in composite dataset.
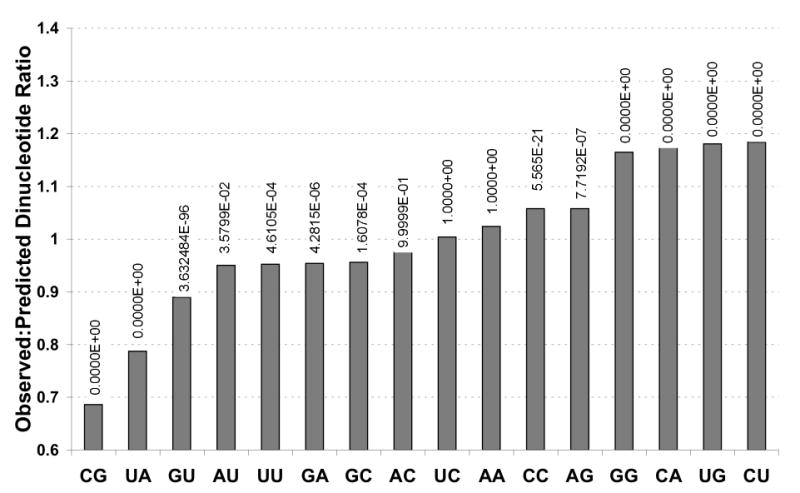
A statistical analysis was performed to test whether the observed frequency of each dinucleotide was equal to the predicted frequency (for all ORFs simultaneously). The p-values for each dinucleotide are indicated in the graph (0 means that it is less than 10-300). For all dinucleotides except “AC”, “UC”, “AA”, the observed frequencies are significantly lower (“CG”, “UA”, “GU”, “AU”, “UU”, “GA”, “GC”) or higher than expected (“CC”, “AG”, “GG”, “CA”, “UG”, “CU”) at the 0.05 level.
Statistical analyses
We tested whether the observed dinucleotide frequencies were significantly different from the predicted dinucleotide frequencies derived from permuted sequences using the prop.test function in the statistical software R (R Development Core Team, 2006). We tested the dinucleotide frequencies separately by codon positions (Figure 6) and also averaged over all codon positions (Figure 3). The prop.test was applied to all ORFs simultaneously. This test assumes that the ORFs are independent. However, there are complex dependencies between the sequences that are difficult to model. Therefore, to reduce any biases introduced by these dependencies in our statistical tests, we removed all ORFs that have sequence similarity greater than 94% with another ORF. Similarity percentages were determined by aligning the complete set of sequences using the CLC FreeWorkbench. This cutoff excluded 60 ORFs and was chosen to balance the amount of sequence similarity with the number of ORFs that were removed from the analysis. The results did not change qualitatively if we altered the cutoff.
We tested for genotype effect for the data presented in Figure 5 using a regression framework that predicts the observed to predicted dinucleotide ratio from genotype. The ratio is used instead of the observed values displayed in Figure 5 to account for any ORF-specific dinucleotide biases. We obtained a p-value from the F-test which evaluates whether there is a significant genotype effect on the ratio. Computations and permutations were performed in the statistical software package R (R, 2006).
Figure 5. Variable frequencies of UA & UU dinucleotides within HCV RNA ORFs.
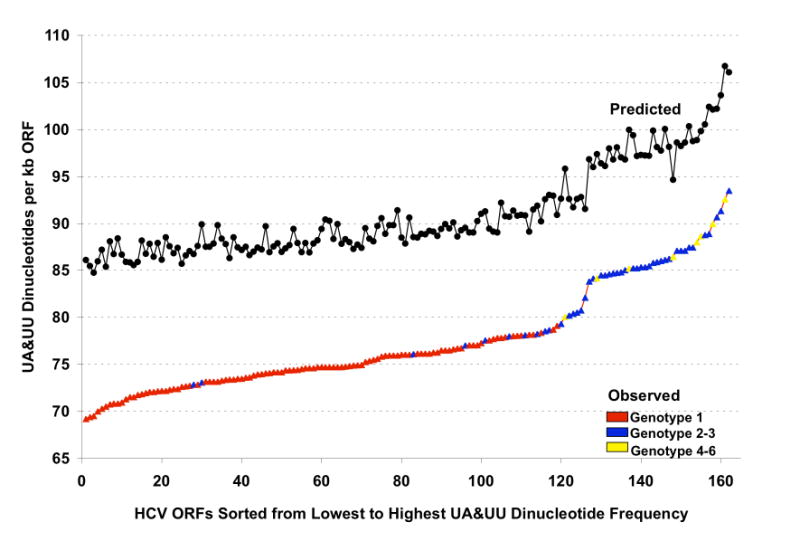
The predicted and observed UA and UU dinucleotides frequencies for 162 HCV RNA ORFs were plotted from lowest to highest observed frequencies (isolates identified in Supplemental Data Tables 1S & 2S). Predicted UA & UU frequencies were derived using Method 1 (Black circles). For observed UA & UU dinucleotide frequencies, points on the line are colored based on the genotype of each HCV RNA ORF: red, genotype 1; blue, genotype 2 or 3; yellow, genotype 4-6. Note that HCV genotype 2 and 3 strains tend to have substantially more UA and UU dinucleotides than HCV genotype 1 strains.
HCV RNA transcripts
HCV 1a and HCV 2a RNA transcripts were made by T7 transcription from cDNA clones as previously described (Han and Barton, 2002). Briefly, p90/HCVFLlong pU, an infectious clone of HCV 1a (Kolykhalov et al., 1997), and pJ6CF, an infectious clone of HCV 2a (Yanagi et al., 1999), were linearized with Mlu I and used as templates in T7 transcription reactions (Epicentre, Madison, WI). α-32P[CTP] was included in reactions to make radiolabeled RNA. HCV RNAs were precipitated with ammonium acetate, dissolved in water, and quantified by OD260nm.
HCV RNA cleavage by activation of endogenous 2′-5′ OAS and endogenous RNase L in cytoplasmic extracts of HeLa cells
HCV RNA (10 nM) was incubated in reactions containing cytoplasmic extracts from HeLa cells as previously described (Han and Barton, 2002). These reactions faithfully mimic the physiologic conditions of infected cells as evidenced by their ability to support all of the biosynthetic steps of poliovirus RNA translation and replication (Barton et al., 1995; Barton and Flanegan, 1993; Barton et al., 1996; Murray and Barton, 2003), however, while HCV RNA translates within these reactions it does not replicate (data not shown). Poly I:C, a synthetic dsRNA, was added to the reactions at the indicated concentrations to activate endogenous 2′-5′ OAS (as in (Han and Barton, 2002)). Poly I:C activation of 2′-5′ OAS results in the production of 2-5A which subsequently activates RNase L. Reaction products were purified by phenol:chloroform extraction and ethanol precipitation before fractionation by electrophoresis in 1% agarose. Radiolabeled RNAs were detected and quantified by phosphorimaging.
HCV RNA cleavage in reactions containing purified 2-5A and RNase L
2-5A and RNase L were purified as previously described (Han et al., 2004; Rusch et al., 2001; Silverman et al., 2000). HCV RNA (100 nM) was incubated at 30°C in 50 ul reactions containing 25 mM Tris-HCl, pH 7.4, 100 mM KCl, 10 mM MgCl2, 50 uM ATP, 7 mM 2-mercaptoethanol and the indicated concentrations of 2-5A and RNase L. Reaction products were purified, spiked with tRNA carrier, and fractionated by electrophoresis in 1% agarose. HCV RNA and HCV RNA fragments were stained with ethidium bromide and detected by UV light. Densitometry was used to quantify the amounts of intact HCV RNA.
Results
HCV RNA ORF dinucleotide frequencies
RNase L can manifest antiviral activity via two mechanisms: by promoting apoptosis and by cleaving viral RNA (Zhou et al., 1997). If RNase L were to cleave HCV RNA during infection it would likely exert selective pressure on the sequence of HCV RNA, leading to lower frequencies of single-stranded UA and UU dinucleotides. Enomoto et al. sequenced viral RNA from three patients infected with interferon-resistant HCV 1b strains (Enomoto et al., 1995). UA and UU dinucleotides were less frequent than predicted in the ORFs of these interferon-resistant virus (Fig. 1 A-C, Patients 1-3). There were ∼10% fewer UU dinucleotides than predicted and more than 20% fewer UA dinucleotides than predicted for each of these HCV RNAs (Fig. 1A-C, observed to predicted dinucleotide ratios were determined using the base composition of the ORF as described in Materials and Methods, Method 1). As previously reported for all single-stranded RNA viruses (Rima and McFerran, 1997), CpG dinucleotides were also less frequent than predicted for each of these HCV RNAs (Fig. 1A-C). Many dinucleotides were present at near predicted frequencies and several dinucleotides were present more frequently than predicted (Fig. 1).
Permuted HCV ORF sequences provide an alternative method for examining the expected dinucleotide frequencies in HCV ORFs (Figure 2A-C, Patients 1-3). Using permuted sequence controls, UA and UU dinucleotides were less frequent than predicted in the ORFs of HCV1b interferon-resistant virus (Fig. 2, observed to predicted dinucleotide ratios were determined by comparing the HCV RNA ORF with 100 permuted ORFs as described in Methods, Method 3). Thus, using two approaches, UA and UU dinucleotides were less frequent than predicted in the ORFs of HCV 1b interferon-resistant virus from pateints 1-3 [observed:predicted dinucleotide frequencies based on the base composition of ORF (Fig. 1, Method 1) and observed:predicted dinucleotide frequencies based on the average dinucleotide frequencies of 100 permuted RNA sequences derived from each HCV ORF (Fig. 2, Method 3)].
To expand upon these observations we analyzed the dinucleotide frequencies of 162 HCV RNA ORFs from the Los Alamos National Labs HCV Database (Supplemental Data Tables 1S & 2S). Sequences were selected to be unique and isolates from non-human sources were excluded. The collected sequences were sorted by genotype and subtype. 11 HCV 1a sequences were analyzed along with 95 HCV 1b sequences, 3 HCV 1c sequences, 15 HCV 2a sequences. 23 HCV 2b sequences, 4 HCV 3a sequences, 2 HCV 6b sequences and 1 HCV 2c, 3k, 4a, 5a, 6a, 6g, 6h, 6k sequence respectively (Supplemental Data Tables 1S & 2S). UA and UU dinucleotides were less frequent than predicted for each of the 162 HCV RNA ORFs (Supplemental Data Tables 1S & 2S, Observed to Predicted Ratios less than 1.0). Across all ORFs, dinucleotide frequencies for UA (p-value < 10-300) and UU (p-value = 4.6×10-4) were significantly smaller than the predicted frequencies obtained from the permuted sequences (Fig. 3). As previously reported for all single-stranded RNA viruses (Rima and McFerran, 1997), CpG dinucleotides were also significantly less frequent than predicted (p-value < 10-300) across the 162 HCV RNA ORFs (Supplemental Data Tables 1 & 2 and Fig. 3).
Relative to the other dinucleotides, UA and UU dinucleotides were among the least abundant dinucleotides in HCV RNA ORFs (Fig. 4). Notably, UA and UU dinucleotides were also among the least abundant dinucleotides in the (-) strand complement of the ORF (Fig. 4, note that AA and AU in the ORF would represent UU and UA in the complementary strand). Thus, relatively low UA and UU dinucleotide frequencies were a common feature of each HCV RNA ORF, consistent with the predicted selective pressure of RNase L.
Figure 4. Dinucleotide frequencies in HCV RNA ORFs.
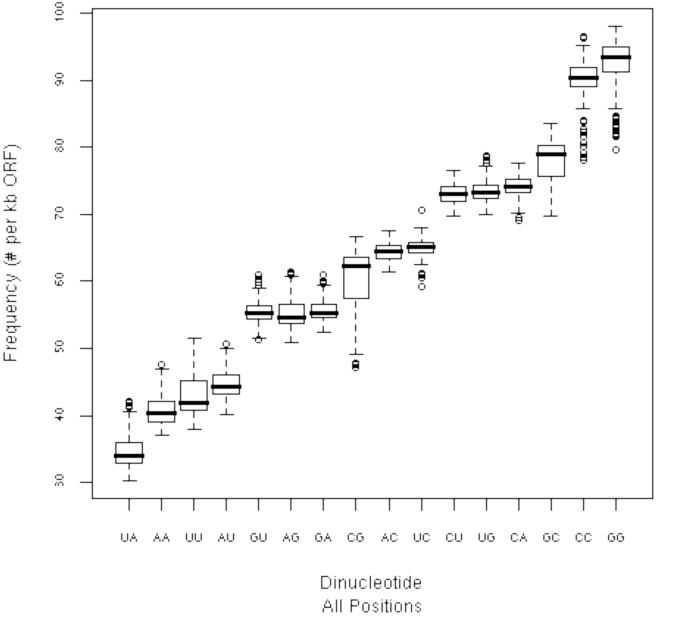
Box-plots for the observed dinucleotide frequencies in the 162 HCV RNA ORFs (isolates identified in Supplemental Data Tables 1S & 2S) were plotted. The solid line indicates the median value, the box shows the range where 25% to 75% of the data fall (called the inter-quartile range IQR). The “whiskers” show points that are within 1.5 × IQR and finally, the dots indicate outliers.
To explore the relationship between HCV genotype and UA/UU dinucleotide frequency we sorted the HCV RNA ORF sequences from lowest to highest observed frequency of UA & UU (Fig. 5). Cumulative UA + UU dinucleotide frequencies ranged in a continuum from 69 to 93 per kb of HCV RNA ORF. Notably, most HCV genotype 1 strains had fewer UA and UU dinucleotides per kb of ORF than the other HCV genotypes (Fig. 5, HCV genotype 1 sequences in red, HCV 2 and 3 sequences in blue, other HCV genotypes in yellow). We found that there was a significant genotype effect on the ratio of observed UA/UU counts compared to the predicted counts (p-value = 1.0×10-09, see Methods). UA dinucleotides were significantly less frequent than predicted at all three codon positions (Fig. 6A-C), while UU dinucleotides were significantly under-represented only at wobble positions (Fig. 6D-F). Comparable data were obtained using the base composition of the codon position as described in Methods (Method 2, Supplemental Data Fig. 1S). The redundancy of codons for specific amino acids makes it relatively easy to accomodate selection against specific bases in the wobble position of codons.
HCV RNA cleavage by RNase L
Despite different frequencies of UA and UU dinucleotides both HCV 1a and HCV 2a RNAs were cleaved efficiently in vitro (Fig. 7). HCV RNAs were susceptible to cleavage by RNase L in both cytoplasmic extracts (which contain endogenous 2′-5′ OASes and endogenous RNase L) and in reactions containing purified RNase L (Han and Barton, 2002; Han et al., 2004). The reactions containing cytoplasmic extracts are advantageous because the conditions are more physiologic while the reactions containing purified RNase L are advantageous because there are no confounding variables. We used both conditions to compare the sensitivity of HCV 1a RNA and HCV 2a RNA to cleavage by RNase L. HCV 1a RNA has 73 UA + UU dinucleotides per kb of ORF while HCV 2a RNA has 81 UA + UU dinucleotides per kb of ORF.
Figure 7. HCV 1a and HCV 2a RNA cleavage by RNase L.




A. 32P-labeled HCV 1a and HCV 2a RNAs were incubated along with the indicated amounts of poly I:C at 30°C in reactions containing cytoplasmic extracts from HeLa cells with endogenous 2′-5′ OAS and RNase L as described in Methods. RNAs from the reactions were separated by electrophoresis in 1% agarose and radiolabeled HCV RNAs were detected and quantified by phosphorimaging. B-D. HCV 1a and HCV 2a RNAs (100 nM) were incubated in reactions containing purified 2-5A and purified RNase L as described in Methods. B. HCV 1a and HCV 2a RNAs (100 nM) in reactions with 12 nM RNase L and variable concentrations of 2-5A as indicated. 30°C for 30 min. C. HCV 1a and HCV 2a RNAs (100 nM) in reactions with 0.5 nM 2-5A and variable concentrations of RNase L as indicated. 30°C for 30 min. D. HCV 1a and HCV 2a RNAs (100 nM) in reactions with 12 nM RNase L and 0.5 nM 2-5A as indicated. 30°C for 0 to 10 min as indicated.
HCV mRNA can activate 2′-5′ OAS above particular threshold concentrations (> 20 nM HCV mRNA ); however, specific HCV RNA elements involved in the activation of 2′-5′ OAS are not yet fully defined (Han and Barton, 2002). Therefore, we chose to use 10 nM HCV RNA, a concentration below the threshold associated with the activation of 2′-5′ OAS, in conjunction with increasing amounts of defined dsRNA [poly(I:C)] to activate 2′-5′ OAS and thereby activate RNase L in reactions containing cytoplasmic extracts (Fig. 7A). HCV RNA remained intact in reactions without dsRNA (Fig. 7A, lanes 1 and 8). At concentrations greater than 3 μg per ml poly(I:C) led to the activation of RNase L and the degradation of HCV RNA (Fig. 7A, lanes 3-7 and 10-14). Both HCV 1a and 2a RNAs were cleaved into fragments ∼ 200 to 2000 bases in length (Fig. 7A, lanes 7 and 14). Similar results were obtained in reactions containing purified RNase L (Fig. 7B-7D). In the absence of 2-5A, 12 nM of RNase L was unable to cleave HCV RNA (Fig. 7B, lanes 1 and 7). Increasing amounts of 2-5A up to 10 nM led to increased degradation of HCV RNAs by purified RNase L (Fig. 7B, lanes 2-6 and 8-12). Likewise, in the absence of RNase L, 0.5 nM 2-5A had no effect on HCV RNA (Fig. 7C, lanes 1 and 7). Increasing amounts of RNase L up to 20 nM led to increased degradation of HCV RNA (Fig. 7C, lanes 2-6 and 8-12). In reactions containing 12 nM RNase L and 0.5 nM 2-5A, HCV RNA was cleaved into progressively smaller fragments over a 10 minute time course (Fig. 7D). The amounts of intact HCV 1a and 2a RNAs decreased with indistinguishable kinetics (Fig. 7D). Thus, reduced frequencies of UA and UU dinucleotides within HCV 1a relative to those in HCV 2a RNA did not prevent cleavage by RNase L.
Discussion
Our analyses are consistent with the possibility that RNase L exerts evolutionary pressure on the sequence of HCV RNA ORFs. Furthermore, variably reduced frequencies of UA and UU dinucleotides in HCV RNA ORFs, as shown in Figure 5, could contribute to the variable efficacy of interferon therapy.
Various selective forces can influence the base and dinucleotide composition of RNA virus ORFs: mutational bias of viral RNA-dependent RNA polymerases, host and tissue specific codon bias, and host antiviral responses (Harris and Liddament, 2004; Hartwig et al., 2006; Jenkins and Holmes, 2003; Plotkin et al., 2004). The elevated expression of 2′-5′ OAS in HCV-infected livers (Thimme et al., 2002), in conjunction with the expression of RNase L in liver (Zhou et al., 2005), is consistent with the possibility that RNase L could be active in HCV-infected livers. HCV RNA ORFs are relatively GC rich (range from 54.7 to 59.4 % GC) and every isolate of HCV RNA examined had fewer UA and UU dinucleotides than predicted should be present (Supplemental Data Tables 1 & 2), consistent with the selective pressure of RNase L (Han et al., 2004). UA dinucleotides are infrequent in most ORFs (Beutler et al., 1989), including those of positive-strand RNA viruses (Rima and McFerran, 1997); however, the paucity of both UA and UU dinucleotides in HCV RNAs (Figs. 1-3) is consistent with the selective pressure of an endoribonuclease like RNase L.
It is extremely uncommon for positive-strand RNA viruses to possess both fewer UA and fewer UU dinucleotides than expected by their base composition (Rima and McFerran, 1997). Only 2 out of 46 representative positive-stranded RNA viruses from the Picorna-, Flavi-, Toga-, Corona-, Calici-, and Astroviridae families had notably decreased UA and UU dinucleotide frequencies; HCV and the genetically related hepatitis G virus (HGV) (Rima and McFerran, 1997). 20 to 30% of HCV infections are associated with HGV co-infection because both viruses are transmitted in contaminated blood (Alter et al., 1997). Interferon therapy in HCV/HGV co-infected patients revealed that persistent HGV infections, like chronic HCV infections, are often resistant to interferon therapy (Kao et al., 1998; McHutchison et al., 1997; Tanaka et al., 1999; Yu et al., 2001). Thus, the paucity of both UA and UU dinucleotides in HCV and HGV ORFs is a unique feature relative to other positive-strand RNA viruses and is consistent with the predicted selective pressure of RNase L. ADAR, interferon-regulated dsRNA-activated adenosine deaminase, could select against UA dinucleotides; however, ADAR functions predominantly in the nucleus and targets the adenosine of UA dinucleotides within dsRNA (Casey, 2002; DeCerbo and Carmichael, 2005; Hartwig et al., 2006; Sato et al., 2001).
RNase L is one of several antiviral proteins potentially responsible for diminished viral loads during interferon therapy. Hyper-variation at wobble position UA and UU dinucleotides within the quasispecies of HCV RNAs during interferon therapy is consistent with the selective pressure of RNase L [Fig. 10 in (Han and Barton, 2002)] as are reduced frequencies of UA and UU dinucleotides at wobble postions of HCV ORFs (Figs. 6B&E). Because HCV genotype 1 RNAs have fewer UA and UU dinucleotides per kb of ORF than other HCV genotypes (Fig. 5) and HCV genotype 1 infections are more resistant to interferon therapy than genotypes 2 and 3 (Manns et al., 2001; McHutchison et al., 1998) it seems reasonable to speculate that RNase L could play an important role in the variable efficacy of interferon therapy. Nonetheless, in vitro, HCV 1a and HCV 2a RNAs were equally sensitive to cleavage by RNase L (Fig. 7). These results partially contradict previously published results where HCV 2a mRNA appeared to be more readily cleaved by RNase L (Han and Barton, 2002). Thus, if variably reduced frequencies of UA and UU dinucleotides make HCV RNAs partially resistant to cleavage by RNase L, in vitro assays appear too blunt to detect such differences.
Although the RNA sequences of HCV ORFs could vary because of codon redundancy, there are evolutionary constraints even at wobble positions of codons (Chamary et al., 2006). Small portions of the HCV ORF encode phylogenetically conserved RNA structures needed for replication (You et al., 2004). In addition, Simmonds et al. described genome-ordered RNA structure (GORS) in HCV ORFs (Simmonds et al., 2004). A significant feature of GORS is lower than expected evolution at wobble positions of codons. Another feature of GORS is the increased stability of seemingly random RNA structures across HCV ORFs. UA and UU dinucleotides cannot be cleaved when sequestered within dsRNA portions of RNA structures and many of the UA and UU dinucleotides within HCV RNA ORFs are resistant to cleavage by RNase L due to their presence in dsRNA portions of the ORF (Han et al., 2004). Thus, perhaps RNase L is an important selective force underlying GORS.
Supplementary Material
Acknowledgments
We thank Charlie Rice and Jens Buhk for cDNAs encoding HCV RNA. This work was supported by the American Cancer Society (RSG-02-063-01-MBC to DJB) and the National Institutes of Health (NCI grant CA-44059 to RHS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alter HJ, Nakatsuji Y, Melpolder J, Wages J, Wesley R, Shih JW, Kim JP. The incidence of transfusion-associated hepatitis G virus infection and its relation to liver disease. N Engl J Med. 1997;336(11):747–54. doi: 10.1056/NEJM199703133361102. [DOI] [PubMed] [Google Scholar]
- Alter MJ. Epidemiology of hepatitis C. Hepatology. 1997;26(3) 1:62S–65S. doi: 10.1002/hep.510260711. [DOI] [PubMed] [Google Scholar]
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144(10):705–14. doi: 10.7326/0003-4819-144-10-200605160-00004. [DOI] [PubMed] [Google Scholar]
- Barton DJ, Black EP, Flanegan JB. Complete replication of poliovirus in vitro: preinitiation RNA replication complexes require soluble cellular factors for the synthesis of VPg-linked RNA. J Virol. 1995;69(9):5516–27. doi: 10.1128/jvi.69.9.5516-5527.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton DJ, Flanegan JB. Coupled translation and replication of poliovirus RNA in vitro: synthesis of functional 3D polymerase and infectious virus. J Virol. 1993;67(2):822–31. doi: 10.1128/jvi.67.2.822-831.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton DJ, Morasco BJ, Flanegan JB. Assays for poliovirus polymerase, 3D(Pol), and authentic RNA replication in HeLa S10 extracts. Methods Enzymol. 1996;275:35–57. doi: 10.1016/s0076-6879(96)75005-x. [DOI] [PubMed] [Google Scholar]
- Beutler E, Gelbart T, Han JH, Koziol JA, Beutler B. Evolution of the genome and the genetic code: selection at the dinucleotide level by methylation and polyribonucleotide cleavage. Proc Natl Acad Sci U S A. 1989;86(1):192–6. doi: 10.1073/pnas.86.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JL. RNA editing in hepatitis delta virus genotype III requires a branched double-hairpin RNA structure. J Virol. 2002;76(15):7385–97. doi: 10.1128/JVI.76.15.7385-7397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary JV, Parmley JL, Hurst LD. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 2006;7(2):98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- Davis GL, Wong JB, McHutchison JG, Manns MP, Harvey J, Albrecht J. Early virologic response to treatment with peginterferon alfa-2b plus ribavirin in patients with chronic hepatitis C. Hepatology. 2003;38(3):645–52. doi: 10.1053/jhep.2003.50364. [DOI] [PubMed] [Google Scholar]
- DeCerbo J, Carmichael GG. Retention and repression: fates of hyperedited RNAs in the nucleus. Curr Opin Cell Biol. 2005;17(3):302–8. doi: 10.1016/j.ceb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Dong B, Niwa M, Walter P, Silverman RH. Basis for regulated RNA cleavage by functional analysis of RNase L and Ire1p. Rna. 2001;7(3):361–73. doi: 10.1017/s1355838201002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Izumi N, Marumo F, Sato C. Comparison of full-length sequences of interferon-sensitive and resistant hepatitis C virus 1b. Sensitivity to interferon is conferred by amino acid substitutions in the NS5A region. J Clin Invest. 1995;96(1):224–30. doi: 10.1172/JCI118025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JD, Seeger C. Cardif: a protein central to innate immunity is inactivated by the HCV NS3 serine protease. Hepatology. 2006;43(3):615–7. doi: 10.1002/hep.21096. [DOI] [PubMed] [Google Scholar]
- Foy E, Li K, Sumpter R, Jr, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102(8):2986–91. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale M, Jr, Blakely CM, Kwieciszewski B, Tan SL, Dossett M, Tang NM, Korth MJ, Polyak SJ, Gretch DR, Katze MG. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol Cell Biol. 1998;18(9):5208–18. doi: 10.1128/mcb.18.9.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale M, Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436(7053):939–45. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- Guo JT, Bichko VV, Seeger C. Effect of alpha interferon on the hepatitis C virus replicon. J Virol. 2001;75(18):8516–23. doi: 10.1128/JVI.75.18.8516-8523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JT, Zhu Q, Seeger C. Cytopathic and noncytopathic interferon responses in cells expressing hepatitis C virus subgenomic replicons. J Virol. 2003;77(20):10769–79. doi: 10.1128/JVI.77.20.10769-10779.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JQ, Barton DJ. Activation and evasion of the antiviral 2′-5′ oligoadenylate synthetase/ribonuclease L pathway by hepatitis C virus mRNA. Rna. 2002;8(4):512–25. doi: 10.1017/s1355838202020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JQ, Townsend HL, Jha BK, Paranjape J, Silverman RH, Barton DJ. A phylogenetically conserved RNA structure in the poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J Virol. 2007;81(11) doi: 10.1128/JVI.01857-06. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JQ, Wroblewski G, Xu Z, Silverman RH, Barton DJ. Sensitivity of hepatitis C virus RNA to the antiviral enzyme ribonuclease L is determined by a subset of efficient cleavage sites. J Interferon Cytokine Res. 2004;24(11):664–76. doi: 10.1089/jir.2004.24.664. [DOI] [PubMed] [Google Scholar]
- Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4(11):868–77. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- Hartwig D, Schutte C, Warnecke J, Dorn I, Hennig H, Kirchner H, Schlenke P. The large form of ADAR 1 is responsible for enhanced hepatitis delta virus RNA editing in interferon-alpha-stimulated host cells. J Viral Hepat. 2006;13(3):150–7. doi: 10.1111/j.1365-2893.2005.00663.x. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–7. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Jenkins GM, Holmes EC. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003;92(1):1–7. doi: 10.1016/s0168-1702(02)00309-x. [DOI] [PubMed] [Google Scholar]
- Johnson CL, Gale M., Jr CARD games between virus and host get a new player. Trends Immunol. 2006;27(1):1–4. doi: 10.1016/j.it.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Kao JH, Lai MY, Chen W, Chen PJ, Chen DS. Efficacy of ribavirin plus interferon alpha on viraemia of GB virus-C/hepatitis G virus: comparison with interferon alpha alone. J Gastroenterol Hepatol. 1998;13(12):1249–53. [PubMed] [Google Scholar]
- Kolykhalov AA, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 1997;277(5325):570–4. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- Kuiken C, Yusim K, Boykin L, Richardson R. The Los Alamos hepatitis C sequence database. Bioinformatics. 2005;21(3):379–84. doi: 10.1093/bioinformatics/bth485. [DOI] [PubMed] [Google Scholar]
- Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, Carney DS, Wang T, Ishida H, Yoneyama M, Fujita T, Saito T, Lee WM, Hagedorn CH, Lau DT, Weinman SA, Lemon SM, Gale M., Jr Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103(15):6001–6. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958–65. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- McHutchison JG, Hoofnagle JH. Therapy of Chronic Hepatitis C. In: Liang TJ, Hoofnagle JH, editors. Hepatitis C. Academic Press; San Diego: 2000. pp. 203–240. [Google Scholar]
- McHutchison JG, Gordon SC, Schiff ER, Shiffman ML, Lee WM, Rustgi VK, Goodman ZD, Ling MH, Cort S, Albrecht JK. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N Engl J Med. 1998;339(21):1485–92. doi: 10.1056/NEJM199811193392101. [DOI] [PubMed] [Google Scholar]
- McHutchison JG, Nainan OV, Alter MJ, Sedghi-Vaziri A, Detmer J, Collins M, Kolberg J. Hepatitis C and G co-infection: response to interferon therapy and quantitative changes in serum HGV-RNA. Hepatology. 1997;26(5):1322–7. doi: 10.1002/hep.510260534. [DOI] [PubMed] [Google Scholar]
- McHutchison JG, Shad JA, Gordon SC, Morgan TR, Ling MH, Garaud JJ, Albrecht JK, Dienstag JL. Predicting response to initial therapy with interferon plus ribavirin in chronic hepatitis C using serum HCV RNA results during therapy. J Viral Hepat. 2001;8(6):414–20. doi: 10.1046/j.1365-2893.2001.00312.x. [DOI] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–72. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- Murray KE, Barton DJ. Poliovirus CRE-dependent VPg uridylylation is required for positive-strand RNA synthesis but not for negative-strand RNA synthesis. J Virol. 2003;77(8):4739–50. doi: 10.1128/JVI.77.8.4739-4750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavio N, Taylor DR, Lai MM. Detection of a novel unglycosylated form of hepatitis C virus E2 envelope protein that is located in the cytosol and interacts with PKR. J Virol. 2002;76(3):1265–72. doi: 10.1128/JVI.76.3.1265-1272.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314(5801):997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Player MR, Torrence PF. The 2-5A system: modulation of viral and cellular processes through acceleration of RNA degradation. Pharmacol Ther. 1998;78(2):55–113. doi: 10.1016/S0163-7258(97)00167-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Robins H, Levine AJ. Tissue-specific codon usage and the expression of human genes. Proc Natl Acad Sci U S A. 2004;101(34):12588–91. doi: 10.1073/pnas.0404957101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R Foundation for Statistical Computing; Vienna, Austria: 2006. R: A language and environment for statistical computing. URL http://www.R-project.org. [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–7. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rima BK, McFerran NV. Dinucleotide and stop codon frequencies in single-stranded RNA viruses. J Gen Virol. 1997;78(Pt 11):2859–70. doi: 10.1099/0022-1317-78-11-2859. [DOI] [PubMed] [Google Scholar]
- Rusch L, Dong B, Silverman RH. Monitoring activation of ribonuclease L by 2′,5′-oligoadenylates using purified recombinant enzyme and intact malignant glioma cells. Methods Enzymol. 2001;342:10–20. doi: 10.1016/s0076-6879(01)42531-6. [DOI] [PubMed] [Google Scholar]
- Sarrazin C, Berg T, Lee JH, Ruster B, Kronenberger B, Roth WK, Zeuzem S. Mutations in the protein kinase-binding domain of the NS5A protein in patients infected with hepatitis C virus type 1a are associated with treatment response. J Infect Dis. 2000;181(2):432–41. doi: 10.1086/315263. [DOI] [PubMed] [Google Scholar]
- Sato S, Wong SK, Lazinski DW. Hepatitis delta virus minimal substrates competent for editing by ADAR1 and ADAR2. J Virol. 2001;75(18):8547–55. doi: 10.1128/JVI.75.18.8547-8555.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman RH, Dong B, Maitra RK, Player MR, Torrence PF. Selective RNA cleavage by isolated RNase L activated with 2-5A antisense chimeric oligonucleotides. Methods Enzymol. 2000;313:522–33. doi: 10.1016/s0076-6879(00)13033-2. [DOI] [PubMed] [Google Scholar]
- Simmonds P. Hepatitis C virus genotypes. In: Liang TJ, Hoofnagle JH, editors. Hepatitis C. Academic Press; San Diego: 2000. pp. 53–70. [Google Scholar]
- Simmonds P. Genetic diversity and evolution of hepatitis C virus--15 years on. J Gen Virol. 2004;85(Pt 11):3173–88. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- Simmonds P, Smith DB. Structural constraints on RNA virus evolution. J Virol. 1999;73(7):5787–94. doi: 10.1128/jvi.73.7.5787-5794.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P, Tuplin A, Evans DJ. Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: Implications for virus evolution and host persistence. Rna. 2004;10(9):1337–51. doi: 10.1261/rna.7640104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DB, Simmonds P. Characteristics of nucleotide substitution in the hepatitis C virus genome: constraints on sequence change in coding regions at both ends of the genome. J Mol Evol. 1997;45(3):238–46. doi: 10.1007/pl00006226. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Nakanishi M, Kusakabe Y, Goto Y, Kitade Y, Nakamura KT. Structural basis for recognition of 2′,5′-linked oligoadenylates by human ribonuclease L. Embo J. 2004;23(20):3929–38. doi: 10.1038/sj.emboj.7600420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Hess G, Schlueter V, Zdunek D, Tanaka S, Kohara M. Correlation of interferon treatment response with GBV-C/HGV genomic RNA and anti-envelope 2 protein antibody. J Med Virol. 1999;57(4):370–5. doi: 10.1002/(sici)1096-9071(199904)57:4<370::aid-jmv8>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285(5424):107–10. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- Taylor DR, Tian B, Romano PR, Hinnebusch AG, Lai MM, Mathews MB. Hepatitis C virus envelope protein E2 does not inhibit PKR by simple competition with autophosphorylation sites in the RNA-binding domain. J Virol. 2001;75(3):1265–73. doi: 10.1128/JVI.75.3.1265-1273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrault NA, Pawlotsky JM, McHutchison J, Anderson F, Krajden M, Gordon S, Zitron I, Perrillo R, Gish R, Holodniy M, Friesenhahn M. Clinical utility of viral load measurements in individuals with chronic hepatitis C infection on antiviral therapy. J Viral Hepat. 2005;12(5):465–72. doi: 10.1111/j.1365-2893.2005.00615.x. [DOI] [PubMed] [Google Scholar]
- Thimme R, Bukh J, Spangenberg HC, Wieland S, Pemberton J, Steiger C, Govindarajan S, Purcell RH, Chisari FV. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci U S A. 2002;99(24):15661–8. doi: 10.1073/pnas.202608299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimme R, Lohmann V, Weber F. A target on the move: innate and adaptive immune escape strategies of hepatitis C virus. Antiviral Res. 2006;69(3):129–41. doi: 10.1016/j.antiviral.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Nagayama K, Enomoto N, Itakura J, Tanabe Y, Sato C, Izumi N, Watanabe M. Amino acid substitutions in PKR-eIF2 phosphorylation homology domain (PePHD) of hepatitis C virus E2 protein in genotype 2a/2b and 1b in Japan and interferon efficacy. Hepatol Res. 2003;26(4):268–274. doi: 10.1016/s1386-6346(03)00164-5. [DOI] [PubMed] [Google Scholar]
- Yanagi M, Purcell RH, Emerson SU, Bukh J. Hepatitis C virus: an infectious molecular clone of a second major genotype (2a) and lack of viability of intertypic 1a and 2a chimeras. Virology. 1999;262(1):250–63. doi: 10.1006/viro.1999.9889. [DOI] [PubMed] [Google Scholar]
- Yang SS, Lai MY, Chen DS, Chen GH, Kao JH. Mutations in the NS5A and E2-PePHD regions of hepatitis C virus genotype 1b and response to combination therapy of interferon plus ribavirin. Liver Int. 2003;23(6):426–33. doi: 10.1111/j.1478-3231.2003.00875.x. [DOI] [PubMed] [Google Scholar]
- You S, Stump DD, Branch AD, Rice CM. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J Virol. 2004;78(3):1352–66. doi: 10.1128/JVI.78.3.1352-1366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ML, Chuang WL, Dai CY, Chen SC, Lin ZY, Hsieh MY, Tsai JF, Wang LY, Chang WY. GB virus C/hepatitis G virus infection in chronic hepatitis C patients with and without interferon-alpha therapy. Antiviral Res. 2001;52(3):241–9. doi: 10.1016/s0166-3542(01)00165-6. [DOI] [PubMed] [Google Scholar]
- Zhou A, Molinaro RJ, Malathi K, Silverman RH. Mapping of the human RNASEL promoter and expression in cancer and normal cells. J Interferon Cytokine Res. 2005;25(10):595–603. doi: 10.1089/jir.2005.25.595. [DOI] [PubMed] [Google Scholar]
- Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, Silverman RH. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. Embo J. 1997;16(21):6355–63. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.