

Abstract
Scaffolding proteins act as chaperones for the assembly of numerous viruses, including most double-stranded DNA bacteriophages. In bacteriophage P2, an internal scaffolding protein, gpO, is required for the assembly of correctly formed viral capsids. Bacteriophage P4 is a satellite phage that has acquired the ability to take control of the P2 genome and use the P2 capsid protein gpN to assemble a capsid that is smaller than the normal P2 capsid. This size determination is dependent on the P4 external scaffolding protein Sid. Although Sid is sufficient to form morphologically correct P4-size capsids, the P2 internal scaffolding protein gpO is required for the formation of viable capsids of both P2 and P4. In most bacteriophages, the scaffolding protein is either proteolytically degraded or exits intact from the capsid after assembly. In the P2/P4 system, however, gpO is cleaved to an N-terminal fragment, O*, that remains inside the mature capsid after DNA packaging. We previously showed that gpO exhibits autoproteolytic activity, which is abolished by removal of the first 25 amino acids. Co-expression of gpN with this N-terminally truncated version of gpO leads to the production of immature P2 procapsid shells. Here, we use protein analysis and mass spectroscopy to show that P2 and P4 virions as well as procapsids isolated from viral infections contain O* and that cleavage occurs between residues 141 and 142 of gpO. By co-expression of gpN with truncated gpO proteins, we show that O* binds to gpN and retains the proteolytic activity of gpO and that the C-terminal 90 residues of gpO (residues 195–284) are sufficient to promote the formation of P2-size procapsids. Using mass spectrometry we have also identified the head completion protein gpL in the virions.
Keywords: virus assembly, capsid maturation, mass spectrometry
Introduction
Scaffolding proteins are proteins that are required for the productive progress of an assembly process but are not present in the final assembly product, acting like molecular chaperones for the proteins undergoing assembly. Scaffolding proteins are almost universal among the complex, double-stranded (ds) DNA bacteriophages and common among eukaryotic dsDNA viruses as well (Dokland, 1999; Fane and Prevelige, 2003).
The dsDNA bacteriophage P2 consists of a T=7 icosahedral capsid composed of 415 copies of gpN-derived capsid protein connected to a complex, contractile tail via a connector or portal, which serves as the entry and exit point for the DNA (Bertani and Six, 1988; Dokland et al., 1992). P2 encodes gpO, a 31.7 kDa internal scaffolding protein that is essential for correct assembly of the procapsid and hence for the production of viable phage (Lengyel et al., 1973). Maturation of the P2 procapsid involves packaging of the DNA, removal of the scaffolding protein and expansion of the capsid. Maturation is accompanied by cleavage of gpN, gpO and the connector protein gpQ, to their mature forms, N*, O* and Q*, respectively (Lengyel et al., 1973; Rishovd and Lindqvist, 1992)(Table 1). An essential “head completion protein”, gpL (18.8 kDa) is added in the later stages of assembly (Lengyel et al., 1973; Pruss and Calendar, 1978).
Table 1.
P2 and P4 structural proteins detected by SDS-PAGE and mass spectrometry.
| Gene | Protein | Function | Sequence | Calculated massa (Da) | Apparent massb (kDa) | Measured massc (Da) | |
|---|---|---|---|---|---|---|---|
| P2 | P4 | ||||||
| T | gpT | Tail length | 1–805 | 86,522.7 | 85d | n.d. | n.d. |
| H | gpH | Tail fiber | 1–669 | 71,504.2 | 75d | n.d. | n.d. |
| FI | gpFI | Tail sheath | 1–396 | 43,143.7 | 43 | n.d. | n.d. |
| N | gpN | Capsid protein | 1–357 | 40,246.5 | n.d. | n.d. | n.d. |
| N* | 32–357 | 36,672.5 | 38 | 36,673.8 | 36,673.7 | ||
| Q | gpQ | Portal protein | 1–344 | 39,114.6 | n.d. | n.d. | n.d |
| Q* | 27–344 | 36,380.3 | 36 | n.d. | n.d | ||
| FII | gpFII | Tail core | 2–172 | 18,937.5 | 22 | 18,938.1f | n.d |
| L | gpL | Head completion | 3–169 | 18,767.4 | 17e | 18,767.7 | n.d |
| O | gpO | Scaffolding protein | 1–284 | 31,445.5 | 33 | n.d. | n.d |
| O* | 2–141 | 15,514.7 | 17 | 15,514.9f | 15,514.4 | ||
| sid | Sid | External scaffold in P4 | 1–244 | 27,126.5 | 32 | n.d | 27,127.3 |
Calculated from the known sequence;
by SDS-PAGE;
by ESI-TOF mass spectrometry;
gpT and gpH are outside the linear range of the SDS-PAGE size determination;
gpL overlaps with gpFII by SDS-PAGE;
mass spectra are shown in Fig. 3. n.d. = not detected
Bacteriophage P4 is a genetically distinct replicon that lacks genes coding for most structural proteins, but has acquired the ability to exploit P2 proteins to assemble its own capsid (Christie and Calendar, 1990; Lindqvist et al., 1993; Six, 1975). However, the capsid assembled under control of P4 is smaller (T=4) than the normal T=7 P2 capsid (Dokland et al., 1992). This size determination is dependent on the P4-encoded 27.3 kDa gene product Sid (Shore et al., 1978), which forms an external scaffold around the P4 procapsid (Dokland et al., 2002; Marvik et al., 1995; Wang et al., 2000). The gpN processing pattern in P4 differs from that in P2, containing significant amounts of the larger cleavage products h1 and h2 (Rishovd and Lindqvist, 1992).
Morphologically correct P4 capsids can be formed by the co-expression of Sid and gpN alone, showing that gpO is not required for assembly in the presence of Sid (Dokland et al., 2002). However, functional gpO is required for the formation of viable P4 phage (Six, 1975), indicating that gpO has additional functions, such as incorporation of the gpQ portal, processing of structural proteins and subsequent capsid maturation.
Regulation of bacteriophage scaffolding protein levels may be important for correct capsid assembly in vivo. In bacteriophage P22, for example, excessive amounts of the gp8 scaffolding protein leads to an increase in aberrant assembly (Parent et al., 2006), and the gp8 mRNA exerts negative feedback on its own translation (Wyckoff and Casjens, 1985). It has been suggested that a similar mechanism plays a role in the regulation of gpO levels in P2 (Larsen, 1994). However, unlike P22 gp8, which is recycled and reused for further assembly (King and Casjens, 1974), gpO is proteolytically cleaved to O*, an N-terminal fragment with apparent MW of 17 kDa (Lengyel et al., 1973; Rishovd and Lindqvist, 1992). We previously found that gpO possesses autoproteolytic activity and that full-length gpO is rapidly degraded upon overexpression in E. coli (Wang et al., 2006). An MBP-gpO fusion protein that included maltose binding protein (MBP) at the N-terminus was cleaved to a shorter fragment, presumed to be MBP-O*, about 40 min post induction of expression (Wang et al., 2006). Thus, it seems likely that gpO autoproteolytic activity may also provide a mechanism for scaffolding protein level regulation in vivo. Co-expression of full-length gpO and gpN led to the formation of aberrant shells and increased levels of N* (Wang et al., 2006), implicating gpO in gpN processing (Goldstein et al., 1974; Lengyel et al., 1973). Removal of the first 25 amino acids of gpO abolished the protease activity; however, this truncated protein, O(26–284) or OΔ25, was able to promote the formation of P2 procapsid-like particles upon co-expression with gpN (Wang et al., 2006), demonstrating that gpO protease activity and gpN cleavage is not required for assembly. Indeed, cleavage of gpN occurs after assembly during an infection (Marvik et al., 1994a). This truncated gpO protein is also incorporated into P4-like procapsids upon co-expression with Sid and gpN (Wang et al., 2006).
Removal of the scaffolding is required to make room for the DNA. In P22 and φ29, the respective scaffolding proteins exit intact (King et al., 1976; Morais et al., 2003), but in most phages, including P2/P4 as well as T4 (Leiman et al., 2003) and lambda (Ray and Murialdo, 1974), the scaffolding protein is removed through proteolytic cleavage. What is unusual about P2 compared to these systems is the O* cleavage product remains within the mature capsid following DNA packaging (Lengyel et al., 1973; Rishovd and Lindqvist, 1992). It is unknown whether O* remains bound to gpN and whether it has some function in the mature capsid, such as a role in DNA packaging or ejection, or whether its presence in the mature capsids is purely incidental.
The purpose of the present study is to analyze the functional domain organization of gpO and the role of the domains in assembly and capsid maturation of P2 and P4. Firstly, we have used biochemical analysis combined with mass spectroscopy (MS) to analyze the protein composition of P2 and P4 virions and procapsids isolated from wild-type phage lysates and find that all particles contain processed O* protein. We also detect the head completion protein gpL in P2 and P4 capsids for the first time. Secondly, we have used MS analysis to identify the exact maturation cleavage site of gpO between residues 141 and 142. Finally, we find that O* (residues 1–141) and the more highly expressed N-terminally truncated version O(26–141) are unable to direct the assembly of correctly formed P2 procapsids upon co-expression with gpN, although O(26–141) does bind to gpN. In contrast, the complementary C-terminal half, O(142–284) and the even shorter fragment O(195–284) both support the assembly of P2 procapsids upon co-expression with gpN. These data suggest that gpO is divided into two distinct domains: an N-terminal gpN-binding protease domain that is involved in targeting of gpO to the shells, processing of gpN and regulation of gpO levels, and a C-terminal scaffolding domain that interacts transiently with gpN during capsid assembly.
Results and discussion
Purification and characterization of P2 and P4
P2 phage was grown as previously described (Kahn et al., 1991) and purified by isopycnic density centrifugation on a CsCl density gradient (average density 1.5 g/ml). Under these conditions, P2 virions form a clearly visible band in the lower part of the gradient; however, we also observed a fainter “top” band at a density of approximately 1.3 g/ml, consistent with previously described P2 procapsids that were isolated on CsCl gradients at a density of 1.27 g/ml (Bowden and Modrich, 1985).
As expected, negative stain EM showed that the lower band contained predominantly intact, DNA-filled virions with an average capsid diameter of 65 nm (Fig. 1A). The top band consisted predominantly of empty shells with smaller diameter (average 56 nm) than the mature capsids and a characteristic serrated, circular outline typical of P2 and other bacteriophage procapsids (Bowden and Modrich, 1985; Marvik et al., 1994a) and similar in appearance to particles produced by co-expression of gpN and O(26-284) (Wang et al., 2006). By comparison, virions that have lost their DNA or spontaneously expanded procapsids have a larger, more angular appearance (Fig. 1B). The procapsid fraction also contained loose tails that co-purified with empty shells in the CsCl gradients.
Figure 1.

Negative stain electron micrographs of P2 virions (A), P2 procapsids from the CsCl top fraction (B), P4 virions (C) and P4 procapsids (top fraction, D). A few larger shells are visible in the procapsid fractions in B and D (white arrows). A GroEL particle is indicated in B (black arrowhead). Scale bar, 100 nm.
P4 was grown on the P2 lysogen E. coli strain C-1895 and purified on a CsCl gradient as previously described (Kahn et al., 1991). This sample also formed two bands on CsCl gradients. As expected, the main band contained primarily complete P4 phage with an average diameter of 49 nm (Fig. 1C). The top band contained predominately P4 procapsids (diameter 40 nm; Fig. 1D), similar to the capsids isolated on glycerol gradients from P4 infections (Barrett et al., 1976) and P4-like procapsids produced by co-expression of gpN and Sid with or without O(26-284) (Dokland et al., 2002; Marvik et al., 1994a; Wang et al., 2006). An additional, small band near the bottom of the gradient was sometimes observed, which was found to contain full capsids, without tails attached (not shown).
Biochemical characterization of P2 and P4 capsids
The CsCl gradient fractions from both P2 and P4 were subjected to SDS-PAGE analysis (Fig. 2A). For the P2 virion fraction, a series of protein bands were visible that by comparison with previous studies (Geisselsoder et al., 1982; Lengyel et al., 1973; Rishovd et al., 1994), could be identified as (in order from the largest): tail length determination protein gpT (predicted molecular weight 86.5 kDa), tail fiber protein gpH (71.5 kDa), gpFI (tail sheath; 43.1 kDa), N* (major capsid protein; 36.7 kDa), Q* (portal protein; 36.4 kDa), gpFII (tail core; 18.9 kDa) and the internal scaffolding fragment O* (previously known as h7) at an apparent MW of 17 kDa (Table 1). Western blot analysis with a polyclonal rabbit anti-gpO antibody (Marvik et al., 1994a; Rishovd et al., 1994) confirmed the identity of the 17 kDa band as O* (Fig. 2B). However, the signal from the anti-gpO antibody was not strong, as this antibody recognizes relatively weakly the N-terminal part of gpO (Marvik et al., 1994a).
Figure 2.

(A) SDS-PAGE analysis of P4 virions (lane 1), P4 procapsids (lane 2), P2 virions (lane3) and P2 procapsids (lane 4) after transfer to nitrocellulose and staining with colloidal gold. M, marker; sizes (kDa) indicated. (B) Western blot of P2 virion proteins separated by SDS-PAGE using a rabbit polyclonal anti-gpO antibody (Marvik et al, 1994), identifying the 17 kDa band as O*. Note that the protein markers used in (A) and (B) are different, giving rise to a discrepancy in the apparent MW of O*.
The P2 procapsid-containing top fraction showed a similar pattern to the P2 virion fraction (Fig. 2A), including the presence of O*, not previously observed in purified P2 procapsids. The presence of tail proteins (gpFI, gpFII and gpH) in this fraction is due to loose tails that sediment at essentially the same density as the empty procapsids in the CsCl gradients. The P2 procapsid fraction also contained a band at 60 kDa, assumed to be GroEL, as previously described (Marvik et al., 1994b), and which can be observed in the micrographs as sevenfold symmetric “flowers”. Another band that can be seen at around 52 kDa (Fig. 2A) is likely to be the same one that was previously observed by Marvik et al. (1994b). Although it was sequenced, it did not match any protein in the database at the time. A search through the current database reveals that the sequence corresponds to the E. coli enzyme lipoamide dehydrogenase (Lpd). The P2 procapsids also displayed a weak band at around 32 kDa. While it could not be conclusively identified, it is likely to correspond to residual amounts of full-length gpO.
As previously described, P4 virions are comprised of essentially the same P2-derived proteins as P2 virions (Barrett et al., 1976; Geisselsoder et al., 1982; Rishovd et al., 1994). Indeed, SDS-PAGE analysis of the P4 virions showed bands at positions corresponding to gpT, gpH, gpFI, N*, Q* gpFII and O* as well as small amounts of a 32 kDa band, most likely corresponding to full-length gpO (Fig. 2A). The top fraction from the CsCl gradient, which contained P4 procapsids and loose tails (Fig. 1D), showed protein bands at positions corresponding to gpFI, N*, gpFII and O*, as well as a strong band at apparent molecular weight of 31 kDa that corresponds to the external scaffolding protein Sid (actual size 27.3 kDa; Fig. 2A). The presence of Sid clearly identifies this fraction as P4 procapsids, as Sid is not found in P4 virions (Barrett et al., 1976). The Psu decoration protein was not observed, however, as it overlaps with the gpFII band (Barrett et al., 1976; Isaksen et al., 1993).
Unprocessed gpO was previously found in cell extracts from P2 and P4 infections at time points prior to lysis (Marvik et al., 1994a). In contrast, we find very little full-length gpO incorporated into procapsids purified from P2 and P4 wild-type phage lysates (Fig. 2A). Similarly, the P2 procapsids and virions both contained only mature, processed N* and no full-length gpN capsid protein (Fig. 2A). The P4 procapsids do contain some full-length gpN and/or larger gpN cleavage products, as previously described (Fig. 2A)(Rishovd and Lindqvist, 1992). Presumably, the majority of the procapsids isolated from wild-type phage lysates represents a mature form of the procapsids, ready for DNA packaging and capsid expansion. We previously found that P4 procapsids could be artificially expanded by cleavage of gpN by trypsin (Wang et al., 2003). In contrast, the present results suggest that protein cleavage and capsid maturation and expansion are disconnected processes in vivo. The role of gpN (and gpO) cleavage in vivo is therefore to prepare the capsid for expansion rather than being the direct cause of it. Most likely, DNA packaging itself is needed to complete maturation and capsid expansion.
An involvement of gpO in the processing of gpN (and other structural proteins) in P2 was previously established (Goldstein et al., 1974; Lengyel et al., 1973), and gpN and gpO cleavage was found to be co-dependent during a P2 infection. Indeed, the proteolytic activity of gpO and the presence of elevated levels of N* during co-expression of gpN and full-length gpO (Wang et al., 2006) strongly suggests that gpO is the protease for gpN and possibly for gpQ as well. This dual role of scaffold and protease is also found in the UL26 protein of herpes simplex virus (Kennard et al., 1995). However, the fact that gpO has autoproteolytic activity in the absence of gpN (Wang et al., 2006) suggests that at least in this overexpression system, gpO protease activity is not dependent on gpN.
Determination of the O* cleavage site
Rishovd et al. (1994) showed by N-terminal protein sequencing that O* shares the same N-terminus as gpO (starting on residue 2, after removal of the N-terminal Met residue). However, the exact location of the C-terminal cleavage site was not identified. In order to determine the exact O* cleavage site we subjected the O*-containing P2 virions to electrospray ionization mass spectrometry (ESI-MS) analysis after disruption in 4M urea and 0.5% formic acid (Fig. 3).
Figure 3.

Determination of masses of O* and tail core proteins. Mass spectrum of the early eluting peak from C4 reverse phase column, which contains P2’s O* and tail core (gpFII) proteins. O* peaks are prefixed by A, gpFII by B, and the number next to the letter stands for the charge state of the corresponding peak.
Four major protein species were detected in P2 phage by this method (Table 1). A peak at 36,673.83 ± 0.19 Da corresponds to N*, and the 18,938.07 ± 0.31 Da peak is equivalent to the tail core protein gpFII after removal of the N-terminal Met residue (Fig. 3). A third peak, at a mass of 15,514.94 ± 0.12 Da (Fig. 3), was interpreted as residues 2–141 of gpO, with a calculated mass of 15,514.7 Da, taking into consideration the earlier protein sequencing data. This measured mass is significantly lower than the apparent mass of 17 kDa by SDS-PAGE (Lengyel et al., 1973). The fourth peak at measured mass 18,767.74±1.28 Da was found to correspond to residues 3–169 of gpL (gpL has two N-terminal Met residues; Table 1). This essential protein, known as a “head completion protein”, has never previously been detected in virions, presumably due to overlap with the gpFII band (Lengyel et al., 1973; Pruss and Calendar, 1978). Its role and location in the head is unknown, but the fact that it could be detected by ESI-MS suggests that it is present in substantial quantities.
The mass assignments were confirmed by in-gel digestion protein bands cut out from the SDS-PAGE gels, followed by MALDI-TOF mass spectrometry analysis of the resulting peptides. Tryptic peptides from the O* band with theoretical masses within 0.02 Da of the observed peaks are indicated in Fig. 4A. All the peptides are located in the N-terminal region of gpO, within the boundaries of residues 2–141 (Fig. 4C). No peptides were detected corresponding to residues 120–142, most likely due to the acidic nature of this sequence. Finally, tandem mass spectrometry using laser-induced dissociation confirmed that the mass of 1,110.71 Da was indeed correctly assigned to the N-terminal peptide AKKVSKFFR (residues 2–10; Fig. 4B, C). Thus, these results demonstrate conclusively that O* corresponds to residues 2–141 of gpO (Fig. 4C). The identity of the gpFII band was likewise sequence confirmed, as was gpL, which was detected in the same SDS-PAGE band as gpFII at apparent MW of 22 kDa.
Figure 4.

Identification of O* protein by mass spectrometry. (A) MALDI TOF mass spectrum of the in-gel tryptic digest of O* band from SDS-PAGE. Peaks within 0.02 Da of the theoretical mass of whole length gpO are indicated by arrows and the corresponding peptide sequences are shown. (B) Tandem mass spectrometry of 1110.71 Da peptide. The resulting spectrum corresponds to the N-terminal peptide (residues 2–10) of gpO, AKKVSKFFR. y and b ions are indicated by arrows. (C) The resulting map of gpO protein. Black, sequence of O* protein inferred from mass spectrometric data; gray, sequence of gpO not present in O*; bold, regions of gpO for which peptides were identified; underlined, peptide sequence confirmed by tandem mass spectrometry.
MS analysis of P4 procapsids yielded peaks at 36,673.68 ± 0.72 Da, 27,127.32 ± 2.37 Da and 15,514.41±0.26 Da, corresponding to N*, Sid and O*, respectively (Table 1). The measured mass of O* is the same as that in P2, showing that there is no difference in scaffolding processing between the two viruses, as is also the case for gpN and gpQ (Rishovd and Lindqvist, 1992). This was confirmed by MALDI-TOF and tandem MS.
Expression and assembly of truncated gpO
With knowledge of the exact cleavage site of gpO we carried out further experiments to elucidate the function of O* and its complement sequence O(142–284). Which part of gpO is involved in gpN binding and capsid assembly? Does O* have a function in the mature virus? To address these questions, the sequence corresponding to O* (gpO residues 1–141) was cloned into the pET16 vector and expressed in E. coli by IPTG induction (Fig. 5) As in the case of full-length gpO, no visible expression of O* was observed above background on SDS-PAGE gels by Coomassie staining (Fig. 6A). Upon co-expression of O* with gpN, neither protein was visibly expressed (Fig. 6A), consistent with previous experiments with full-length gpO (Wang et al., 2006). However, when the SDS-PAGE was subjected to Western blotting with the anti-gpO antibody, a signal was observed at a size of < 6kDa, suggesting that O* had been cleaved into smaller fragments (Fig. 6B). These results suggest that O* retains the proteolytic activity of gpO and that this activity is responsible for the poor expression of O* and gpN, either specifically or through a more general cell toxicity as we previously noted for full-length gpO (Wang et al., 2006). [Additional negative regulation at the translational level cannot be excluded (Larsen, 1994; Wyckoff and Casjens, 1985).] During a phage infection the (proteolytically active) O* protein would be sequestered inside the assembled shells and presumably unable to exert any further cleavage on soluble gpN in the cytoplasm. It is unknown whether this or any other activity of O* is required once gpN cleavage and capsid expansion has occurred. However, the binding of gpO and gpN may represent an important control mechanism to prevent an excess of both scaffolding and protease activity during an infection.
Figure 5.
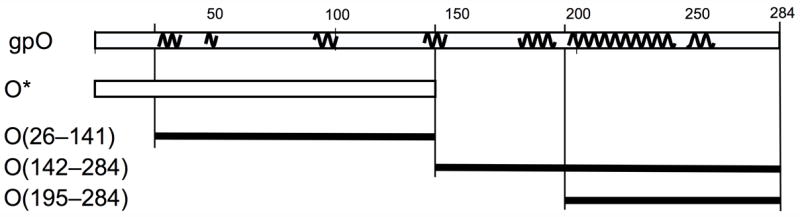
gpO expression constructs. The top line represents the complete gpO sequence (284) amino acids. The squiggly lines indicate predicted α-helical stretches (using the PredictProtein server). The following lines show the truncated clones O* (residues 1–141), O(26–141), O(142–284) and O(195–284).
Figure 6.
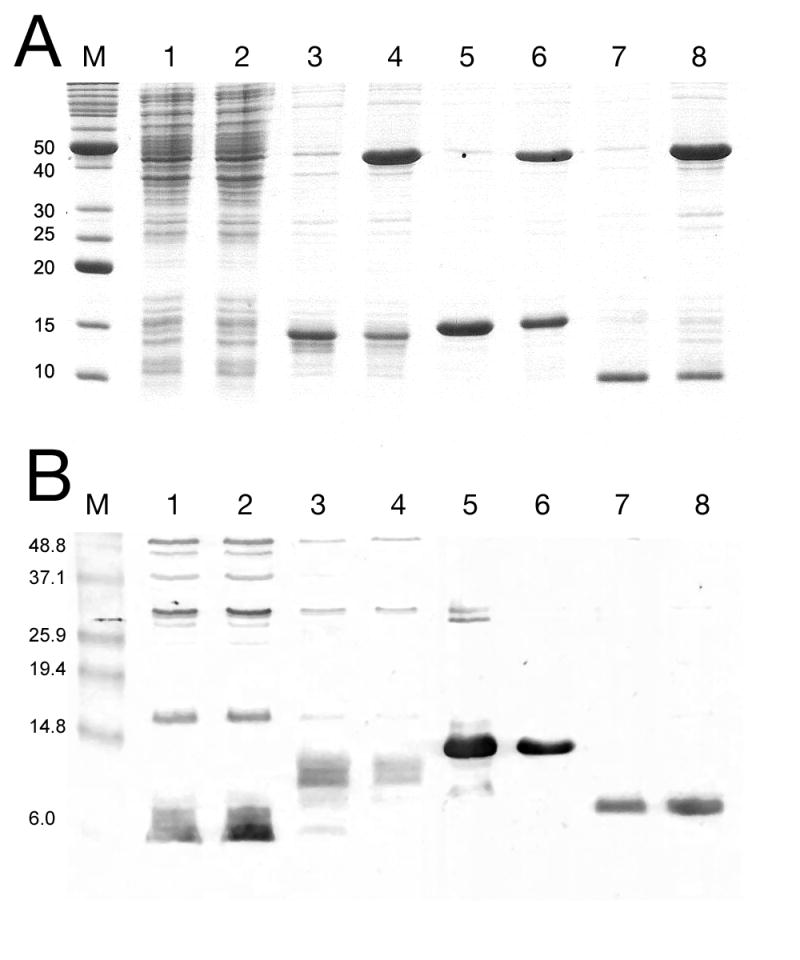
SDS-PAGE analysis of protein expression from truncated gpO clones, by Coomassie staining (A) or immunological detection with an anti-gpO antibody (B). Lane 1, O* alone; lane 2, O* and gpN; lane 3, O(26–141); lane 4, O(26–141)+gpN; lane 5, O(142–284); lane 6, O(142–284)+gpN; lane 7, O(195–284); and lane 8, O(195–284)+gpN. The truncated gpO proteins are indicated by arrows. M, markers, MW (kDa) as indicated. A few non-specific bands are seen on the immunoblot for the O* and O(26–142) expressions. Also note that the antibody is reportedly less sensitive to the N-terminal half of the protein than to the C-terminal half (Marvik et al., 1994a).
In contrast, a removal of the first 25 amino acids from O*, resulting in clone O(26–141) (Fig. 5) led to high level, stable expression from the same vector under the same conditions, either alone or together with gpN (Fig. 6A). A strong signal was observed for O(26–141) by Western blotting with anti-gpO antibody (Fig. 6B). This was expected, since previous observations had implicated the first 25 amino acids in the protease activity of gpO (Wang et al., 2006). The gpO sequence includes a Cys residue at position 18, which could potentially form part of a catalytic triad for a putative cysteine protease activity. Co-expression of O(26–141) and gpN led to the formation of protein complexes that sedimented in a broad band on 10–40% sucrose gradients (Fig. 7A). Negative stain EM revealed that this band contained mostly aberrant shells, with a few closed shells of different sizes (Fig. 8A). These results show that although the N-terminal half gpO exhibits gpN-binding activity, the C-terminal part is required for assembly fidelity.
Figure 7.

Coomassie stained SDS-PAGE separated proteins from co-expressions with truncated gpO. (A) Particles produced in O(26–141)+gpN expression, separated on a 10–40% sucrose gradient. The arrow indicates the direction of sedimentation; P, pellet from gradient; M, marker, MW (kDa) indicated. (B) Expression of O(142–284)+gpN. Lane P, 120,000×g pellet; lane G, band from 10–40% sucrose gradient. (C) Expression of O(195–284)+gpN. Supernatant (S) and pellet (P) after centrifugation at 100,000×g for 1 hr. The gpN and truncated gpO protein bands in (B) and (C) are indicated by N and OΔ, respectively. MW markers (M) as in (A).
Figure 8.

Negative stain electron microscopy of particles formed by co-expression of O(26–141) and gpN (A), O(142–284)+gpN (B) and O(195–284)+gpN (C) and purified on 10–40% sucrose gradients. Scale bar, 100 nm.
The C-terminal, complementary part of the gpO sequence, O(142–284), was also stably expressed at high level (Fig. 6). Co-expression of O(142–284) with gpN led to the formation of shells that were pelleted at 120,000×g and formed a band on 10–40% sucrose gradients (Fig. 7B). By negative stain EM, these particles consisted of mostly well-formed, P2 procapsid-like particles (Fig. 8B). This result shows that only the C-terminal half of gpO is required for assembly, while the O* fragment is dispensable for this purpose. Furthermore, whereas proper virus assembly in vivo may require careful modulation of gpO levels through cleavage and removal of the C-terminal assembly domain, such regulation does not seem to be essential in the expression system, since the stably and highly expressed O(142–284) produces large amounts of well-formed procapsids. On the other hand, even the 10% aberrant shells observed in the co-expression system might be detrimental to phage viability in vivo.
The C-terminal half of gpO includes a long predicted α-helical region between residues 195 and 257, with a predicted loop at residues 241–245 (Fig. 5). This suggested that the protein might contain a helix-loop-helix motif similar to that found in the C-terminal half of the gp8 scaffolding protein of bacteriophage P22 (Parker et al., 1998). To test whether this predicted α-helical region was sufficient for assembly, the construct O(195–284) was generated (Fig. 5). This 90 amino acid fragment was also highly expressed (Figs 6 and 7) and was able to direct the formation of P2 procapsids upon co-expression with gpN, forming particles that were indistinguishable from those produced in the O(142–284)N co-expression (Fig. 8C). Interestingly, most of the O(195–284) protein remained in the supernatant after pelleting the assembled particles at 120,000×g (Fig. 7C), suggesting that control of capsid fidelity by gpO requires only a transient interaction between the C-terminal domain of gpO and gpN. The N-terminal protease domain remains tightly bound to gpN and is presumably responsible for targeting the scaffolding protein to the capsid.
In conclusion, we have shown that the gpO can be divided into two distinct and independent functional domains: an N-terminal gpN-binding domain that is sufficient for proteolytic activity, and a C-terminal scaffolding domain that is sufficient to promote capsid assembly. Our data suggest that a transient interaction with gpN coupled with autoproteolytic cleavage of the C-terminal domain of gpO functions to remove the potentially toxic effect of too much scaffolding domain, while tight binding of the N-terminal domain of gpO to gpN targets the proteolytic activity to the shell and prevents illicit processing from occurring in the cytoplasm. Further work on the scaffolding activity of gpO will be focused on the C-terminal half, while experiments on the N-terminal domain will focus on its role in capsid maturation and protein processing.
Experimental Methods
Growth and purification of phage
P2 phage was grown by infection of E. coli strain C-2 with P2 vir1 at 37°C in Luria Broth (LB) supplemented with 0.1% glucose, 1 mM MgCl2, and 0.25 mM CaCl2 as previously described (Kahn et al., 1991). After addition of 1.28 mM EGTA, the lysate was clarified by centrifugation at 5,000 g for 10 min. Following the addition of 0.43 M NaCl and 8% polyethylene glycol (PEG) 6,000, the virus was incubated on ice for 1 hr and centrifuged at 5,400×g for 20 min. The pellet was resuspended in phage buffer (20 mM Tris-HCl pH 7.4, 10 mM MgCl2, 1% ammonium acetate). Chloroform was added to the resuspended pellet and the solution was centrifuged at 4,000 × g for 20 min. 0.64 g CsCl was added per ml of extracted phage. The resulting solution was centrifuged at 70,000 rpm (339,000×g) for 20 hr in a Beckman NVT90 rotor. P4 vir1 del22 (Raimondi et al., 1985) was grown on the P2 lysogen E. coli strain C-1895 at 37°C in LB supplemented with 0.1% glucose, 1.6 mM MgCl2, and 0.5 mM CaCl2. Purification followed essentially the same procedure as for P2, except that 0.50 g CsCl was added per ml resuspended PEG pellet (Kahn et al., 1991). The phage- and procapsid- containing bands were collected and dialyzed against phage buffer.
Electron microscopy
Virus samples from the CsCl gradient were dialyzed against phage buffer on a 0.025μm filter for 15 minutes. A 3 μl sample was applied to Formvar/carbon-coated grids, rinsed once with the same buffer and stained with 1% uranyl acetate (Dokland and Ng, 2006). Sucrose gradient fraction samples from co-expression experiments (see below) were applied to the grid, rinsed twice with phage buffer and stained with 1% uranyl acetate. The samples were observed in an FEI Tecnai F20 electron microscope operated at 200 kV. Images were collected on a Gatan Ultrascan 4000 4 k × 4k CCD camera using a magnification of 65,500×.
SDS-PAGE and Western blot analysis
Protein purified samples were separated by SDS-PAGE and stained with Coomassie. Apparent protein sizes were calculated by plotting their migration against the known sizes of the markers. The proteins were transferred to nitrocellulose using a Bio-Rad Trans-Blot semidry cell. Total proteins were detected using the Bio-Rad colloidal gold total protein stain kit. For specific antigen detection, the blots were labeled with a rabbit polyclonal anti-O antibody raised against a β-galactosidase(1–4):(30 residue linker):gpO(25–284) fusion protein (Marvik et al., 1994a) (gift from Björn H. Lindqvist, University of Oslo), diluted 1:1,000 in phosphate-buffered saline (PBS) with 0.05% Tween 20 and 5% milk blocker, followed by a HRP-conjugated goat anti-mouse IgG heavy-chain secondary antibody (1:5,000 dilution; Southern Biotech, Birmingham, AL). The blots were developed using the Opti-4CN detection kit (Bio-Rad). For the Coomassie stained gels and the total protein blot, the Benchmark Protein Ladder (Invitrogen) was used; for the Western blot we used the Benchmark Prestained Protein Ladder (Invitrogen). The two markers run differently on the gels.
Mass spectrometry
The samples were mixed at a 1:1 (v/v) ratio with denaturation buffer consisting of 8M urea, 1% formic acid and 0.5 mg/ml protamine sulfate and injected into a C4 reverse phase column. The proteins were eluted by gradient of 1:1 mixture of acetonitrile and 2-propanol with 0.1% formic acid. The eluate was directly injected into electrospray ionization (ESI) time-of-flight (TOF) mass spectrometer (LCT, Micromass).
For obtaining sequence information, the protein bands were cut out of the SDS-PAGE gel and digested with 5 μg/ml trypsin solution (Trypsin Gold, Promega). The resulting peptides were extracted and analyzed by MALDI-TOF tandem mass spectrometry (Ultraflex III, Bruker Daltonics). Peptide fragmentation for tandem mass spectrometry was performed by laser-induced dissociation only.
Cloning and expression of gpO
Expression clones of O* (residues 2–141) and other gpO truncation constructs were generated identically by inserting the truncated O gene between the NcoI and NdeI sites of the pET16b vector (Novagen). Tandem clones of truncated O and N were produced by the addition of the full-length N gene between the NdeI and XhoI sites in the appropriate O deletion construct. For expression, all clones were grown at 37ºC in LB with 100 μg/ml ampicillin until OD600 = 0.6, induced by the addition of 0.5mM IPTG, and harvested after 2 h. The cloning was done in the recA− E. coli strain DH5α (Invitrogen) while expression was performed in BL21 (DE3) (Novagen).
Protein purification
The harvested cells were resuspended in lysis buffer (10mM Tris pH 7.4, 200mM NaCl, 1mM PMSF, 1% Triton X-100, and 0.5% deoxycholate) and frozen overnight at −20ºC. After thawing, the resuspended cells were lysed by sequential passages at 1000, 5000, 10000, and 15000 psi through an Emulsiflex EF-C3 high pressure cell disruptor (Avestin, Inc., Ottawa). The lysates were clarified by centrifugation at 11,800×g for 30 minutes. Virus-like particles were collected by centrifuging the clarified lysates at 40,000 rpm (114,000×g )for 1 hr in a Beckman Type 60Ti rotor. The pellets were resuspended in procapsid buffer (50mM Tris-HCl pH 8.0, 100mM NaCl, 10mM MgCl2), loaded onto 10–40% sucrose gradients in the same buffer and centrifuged for 2 hrs at 30,000 rpm (110,000×g) in a Beckman SW 41 rotor. One ml gradient fractions were collected manually. Total lysates, pellets and sucrose gradients fractions were analyzed by SDS-PAGE. Pooled fractions were pelleted by centrifugation for 40 min at 50,000 rpm (178,000×g) in a Beckman Type 60Ti rotor, resuspended in procapsid buffer and prepared for EM.
Acknowledgments
We acknowledge Michael Spilman and Cindy Rodenburg for assistance with various aspects of this work. PEP was supported by NIH grant GM47980.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barrett KJ, Marsh ML, Calendar R. Interactions between a satellite bacteriophage and its helper. J Mol Biol. 1976;106:683–707. doi: 10.1016/0022-2836(76)90259-x. [DOI] [PubMed] [Google Scholar]
- Bertani LE, Six E. The P2-like phages and their parasite, P4. In: Calendar R, editor. The Bacteriophages. Vol. 2. Plenum Press; New York: 1988. pp. 73–143. 2 vols. [Google Scholar]
- Bowden DW, Modrich P. In vitro maturation of circular bacteriophage P2 DNA. J Biol Chem. 1985;260:6999–7007. [PubMed] [Google Scholar]
- Christie GE, Calendar R. Interactions between satellite bacteriophage P4 and its helpers. Annu Rev Genet. 1990;24:465–490. doi: 10.1146/annurev.ge.24.120190.002341. [DOI] [PubMed] [Google Scholar]
- Dokland T. Scaffolding proteins and their role in viral assembly. Cell Mol Life Sci. 1999;56:580–603. doi: 10.1007/s000180050455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokland T, Lindqvist BH, Fuller SD. Image reconstruction from cryo-electron micrographs reveals the morphopoietic mechanism in the P2–P4 bacteriophage system. EMBO J. 1992;11:839–846. doi: 10.1002/j.1460-2075.1992.tb05121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokland T, Ng ML. Electron microscopy of biological samples. In: Dokland T, Hutmacher DW, Ng ML, Schantz JT, editors. Techniques in microscopy for biomedical applications. World Scientific Press; Singapore: 2006. [Google Scholar]
- Dokland T, Wang S, Lindqvist BH. The structure of P4 procapsids produced by coexpression of capsid and external scaffolding proteins. Virology. 2002;298:224–231. doi: 10.1006/viro.2002.1485. [DOI] [PubMed] [Google Scholar]
- Fane BA, Prevelige PE. Mechanism of scaffolding-assisted viral assembly. Adv Prot Chem. 2003;64:259–299. doi: 10.1016/s0065-3233(03)01007-6. [DOI] [PubMed] [Google Scholar]
- Geisselsoder J, Sedivy JM, Walsh RB, Goldstein R. Capsid structure of satellite phage P4 and its helper. J Ultrastruct Res. 1982;79:165–173. doi: 10.1016/s0022-5320(82)90028-4. [DOI] [PubMed] [Google Scholar]
- Goldstein R, Lengyel J, Pruss G, Barrett K, Calendar R, Six E. Head size determination and the morphogenesis of satellite phage P4. Curr Top Microbiol Immunol. 1974;68:59–75. doi: 10.1007/978-3-642-66044-3_3. [DOI] [PubMed] [Google Scholar]
- Isaksen ML, Dokland T, Lindqvist BH. Characterization of the capsid associating activity of bacteriophage P4's Psu protein. Virology. 1993;194:647–681. doi: 10.1006/viro.1993.1307. [DOI] [PubMed] [Google Scholar]
- Kahn ML, Ziermann R, Deho G, Ow DW, Sunshine MG, Calendar R. Bacteriophage P2 and P4. Methods Enzymol. 1991;204:264–280. doi: 10.1016/0076-6879(91)04013-e. [DOI] [PubMed] [Google Scholar]
- Kennard J, Rixon FJ, McDougall IM, Tatman JD, Preston VG. The 25 amino acid residues at the carboxy terminus of the herpes simplex type 1 UL26.5 protein are required for the formation of the capsid shell around the scaffold. J Gen Virol. 1995;76:1611–1621. doi: 10.1099/0022-1317-76-7-1611. [DOI] [PubMed] [Google Scholar]
- King J, Botstein D, Casjens S, Earnshaw W, Harrison SC, Lenk E. Structure and assembly of the capsid of bacteriophage P22. Phil Trans R Soc London B. 1976;276:37–49. doi: 10.1098/rstb.1976.0096. [DOI] [PubMed] [Google Scholar]
- King J, Casjens S. Catalytic head assembling protein in virus morphogenesis. Nature. 1974;251:112–119. doi: 10.1038/251112a0. [DOI] [PubMed] [Google Scholar]
- Larsen T. MS University of Oslo. Oslo; Norway: 1994. [Google Scholar]
- Leiman PG, Kanamaru S, Mesyanzhinov VV, Arisaka F, Rossmann MG. Structure and morphogenesis of bacteriophage T4. Cell Mol Life Sci. 2003;60:2356–2370. doi: 10.1007/s00018-003-3072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel JA, Goldstein RN, Marsh M, Sunshine MG, Calendar R. Bacteriophage P2 head morphogenesis: cleavage of the major capsid protein. Virology. 1973;53:1–23. doi: 10.1016/0042-6822(73)90461-3. [DOI] [PubMed] [Google Scholar]
- Lindqvist BH, Deho G, Calendar R. Mechanisms of genome propagation and helper exploitation by satellite phage P4. Microbiol Rev. 1993;57:683–702. doi: 10.1128/mr.57.3.683-702.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvik OJ, Dokland TE, Nøkling RH, Jacobsen E, Larsen T, Lindqvist BH. The capsid size-determining protein Sid forms an external scaffold on phage P4 procapsids. J Mol Biol. 1995;251:59–75. doi: 10.1006/jmbi.1995.0416. [DOI] [PubMed] [Google Scholar]
- Marvik OJ, Jacobsen E, Dokland T, Lindqvist BH. Bacteriophage P2 and P4 morphogenesis: assembly precedes proteolytic processing of the capsid proteins. Virology. 1994a;205:51–65. doi: 10.1006/viro.1994.1619. [DOI] [PubMed] [Google Scholar]
- Marvik OJ, Sharma P, Dokland T, Lindqvist BH. Bacteriophage P2 and P4 assembly: alternative scaffolding proteins regulate capsid size. Virology. 1994b;200:702–714. doi: 10.1006/viro.1994.1234. [DOI] [PubMed] [Google Scholar]
- Morais MC, Kanamaru S, Badasso MO, Koti JS, Owen BAL, McMurray CT, Anderson DL, Rossmann MG. Bacteriophage phi29 scaffolding protein gp7 before and after prohead assembly. Nat Struct Biol. 2003;10:572–576. doi: 10.1038/nsb939. [DOI] [PubMed] [Google Scholar]
- Parent KN, Zlotnick A, Teschke CM. Quantitative analysis of multi-component spherical viral assembly: scaffolding protein contributes to the global stability of phage P22 procapsids. J Mol Biol. 2006;359:1097–1106. doi: 10.1016/j.jmb.2006.03.068. [DOI] [PubMed] [Google Scholar]
- Parker MH, Casjens S, Prevelige PE. Functional domains of bacteriophage P22 scaffolding protein. J Mol Biol. 1998;281:69–79. doi: 10.1006/jmbi.1998.1917. [DOI] [PubMed] [Google Scholar]
- Pruss GJ, Calendar R. Maturation of bacteriophage P2 DNA. Virology. 1978;86:454–467. doi: 10.1016/0042-6822(78)90085-5. [DOI] [PubMed] [Google Scholar]
- Raimondi A, Donghi R, Monaguti A, Pessina A, Deho G. Analysis of spontaneous deletion mutants of satellite bacteriophage P4. J Virol. 1985;54:233–235. doi: 10.1128/jvi.54.1.233-235.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P, Murialdo H. The role of gene Nu3 in bacteriophage lambda head morphogenesis. Virology. 1974;64:247–263. doi: 10.1016/0042-6822(75)90096-3. [DOI] [PubMed] [Google Scholar]
- Rishovd S, Lindqvist BH. Bacteriophage P2 and P4 morphogenesis: protein processing and capsid size determination. Virology. 1992;187:548–554. doi: 10.1016/0042-6822(92)90457-z. [DOI] [PubMed] [Google Scholar]
- Rishovd S, Marvik OJ, Jacobsen E, Lindqvist BH. Bacteriophage P2 and P4 morphogenesis: identification and characterization of the portal protein. Virology. 1994;200:744–751. doi: 10.1006/viro.1994.1238. [DOI] [PubMed] [Google Scholar]
- Shore D, Deho G, Tsipis J, Goldstein R. Determination of capsid size by satellite bacteriophage P4. Proc Natl Acad Sci USA. 1978;75(1):400–404. doi: 10.1073/pnas.75.1.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six EW. The helper dependence of satellite bacteriophage P4: which gene functions of bacteriophage P2 are needed by P4? Virology. 1975;67:249–263. doi: 10.1016/0042-6822(75)90422-5. [DOI] [PubMed] [Google Scholar]
- Wang S, Chandramouli P, Butcher S, Dokland T. Cleavage leads to expansion of bacteriophage P4 procapsids in vitro. Virology. 2003;314:1–8. doi: 10.1016/s0042-6822(03)00375-1. [DOI] [PubMed] [Google Scholar]
- Wang S, Chang JR, Dokland T. Assembly of bacteriophage P2 and P4 procapsids with internal scaffolding protein. Virology. 2006;348:133–140. doi: 10.1016/j.virol.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Wang S, Palasingam P, Nøkling RH, Lindqvist BH, Dokland T. In vitro assembly of bacteriophage P4 procapsids from purified capsid and scaffolding proteins. Virology. 2000;275:133–144. doi: 10.1006/viro.2000.0521. [DOI] [PubMed] [Google Scholar]
- Wyckoff E, Casjens S. Autoregulation of the bacteriophage P22 scaffolding protein gene. J Virol. 1985;53:192–197. doi: 10.1128/jvi.53.1.192-197.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]