

Summary
Exon 2 of the Bcl-x gene undergoes alternative splicing in which the Bcl-xS splice variant promotes apoptosis in contrast to the anti-apoptotic splice variants, Bcl-xL. In this study, the regulation of the alternative splicing of pre-mRNA of Bcl-x was examined in response to emetine. Treatment of different types of cancer cells with emetine dihydrochloride down regulated the level of Bcl-xL mRNA with a concomitant increase in the mRNA level of Bcl-xS in a dose-and time-dependent manner. Pretreatment with calyculin A, an inhibitor of protein phosphatase-1 (PP1) and protein phosphatase 2A (PP2A) blocked emetine-induced alternative splicing in contrast to okadaic acid, a specific inhibitor of PP2A in cells, demonstrating a PP1-mediated mechanism. This is the first report on the regulation of RNA splicing of members of the Bcl-2 families in response to emetine.
Introduction
Alternative splicing occurs when the introns of a certain pre-mRNA are excised in more than one way, producing several possible mature mRNAs from one gene. Alternative pre-mRNA splicing is an essential mechanism for generating protein diversity [1–4] according to different regulatory programs. It is estimated that more than 60% of human genes undergo alternative splicing, leading to production of diversified functional isoforms [5]. Alternative splicing is precisely regulated. Aberrant splicing can lead to human disorders such as growth hormone deficiency, Frasier syndrome, Parkinson’s disease, cystic fibrosis, retinitis pigmentosa, spinal muscular atrophy, and myotonic dystrophy [6, 7].
Apoptosis, or programmed cell death, plays an important role in normal tissue equilibrium by counterbalancing the cell production and cell loss. Cancers and neurodegenerative disorders often show a defective cell death program [8], the former originating from too little apoptosis and the latter emerging from too much cell death. Deregulation of the balance between proliferation and cell death represents a protumorigenic principle in human carcinogenesis. Alternative splicing plays a critical role in the control of apoptosis. Several pre-mRNAs for cell death signals are alternatively spliced, yielding isoforms with opposing functions during programmed cell death [7].
One distinct example is the Bcl-x transcript, which is alternatively spliced in exon 2 to produce the pro-apoptotic Bcl-xS or the anti-apoptotic Bcl-xL [9]. The protein product of the larger Bcl-xL functions as a repressor of programmed cell death [10], whereas the smaller Bcl-xS encodes a protein that can accelerate cell death [11, 12]. Bcl-xL is highly expressed in several types of cancers and overexpression of Bcl-xL inhibits apoptosis and promotes resistance to chemotherapy in tumors in vivo [13]. It has been found that Bcl-xL is over-expressed in high-grade prostate cancer and associated with hormone refractory phenotype [14]. Breast cancer also has a high level of Bcl-xL that is correlated with an increased risk of metastasis [15] and disallows apoptosis, gains resistance against cytokines, alters the relationship between cells and extracellular matrix, and probably renders a mechanism for cells to adapt to a new environment [16]. In ovarian cancers, Bcl-xL expression conferred resistance to chemotherapy-induced apoptosis resulting from treatment with cisplatin, paclitaxel, topotecan, and gemcitabine [17]. On the other hand, reduction of Bcl-xL protein and/or increase of production of Bcl-xS by specific anti-sense oligonucleotide (ISIS 16009) treatment or other approaches enhanced the chemosensitivity or radiosensitivity of colon cancer cells, breast cancer cells, HepG2 hepatoblastoma cells and of other tumor cell lines [18–23]. Since Bcl-xS over-expression can induce apoptosis in tumoral cell lines [24], the ability to alter the Bcl-xL/Bcl-xS ratio thus has promising therapeutic potential for cancer treatment.
Numerous reports have demonstrated that Bcl-x alternative splicing can be regulated by small molecules such as ceramide in cancer cells [25], as well as by other biological molecules including IL-6, GM-CSF, Amphetamine and TPA [26, 27]. To identify additional small molecules that may be potentially used in cancer treatment by regulating Bcl-x splicing, we performed RT-PCR experiments in cells treated with 1,040 FDA approved drugs and compounds (NINDS custom collection, MicroSource Discovery Systems, Inc.). We found that emetine, a potent protein synthesis inhibitor in eukaryotes [28] down-regulated Bcl-xL and up-regulated Bcl-xS. We further demonstrated that this novel and newly defined emetine function is mediated by phosphorylation.
Results
Emetine Regulates the Alternative Splicing of Bcl-x Pre-mRNA
It has been hypothesized that modulation of Bcl-xL/Bcl-xS ratio by regulating alternative splicing of exon 2 (Fig. 2A) in the Bcl-x gene may have potential for cancer treatment. To identify small molecules that regulate Bcl-x splicing, we screened 1,040 FDA approved drugs and compounds (NINDS Custom Collection, MicroSource Discovery Systems, Inc.) using RT-PCR in C33A cells, a cervical cancer line. We found that emetine reduced Bcl-xL mRNA with a concomitant increase of Bcl-xS (Fig. 2B &2D). To further validate our findings, C33A cells were treated overnight with various concentration of emetine. There was a decrease in the ratio of Bcl-xL/Bcl-xS from 9.6 to 2.6, 2.2, 1.8, 2.0, and 1.5 with emetine 0.1, 0.3, 1.0, and 10.0 μM respectively (Fig. 2B). Interestingly, emetine didn’t alter splicing of tau, SMN and BACE1 genes, indicating its relative specificity on Bcl-x splicing. Emetine (C29H40N2O4), the ipecac alkaloid, is an amoebicidal agent that inhibits polypeptide chain elongation in parasites [29–31]. Emetine is a potent protein synthesis inhibitor in mammalian cells, plants and yeasts [32]. Grollman has shown that emetine and cycloheximide, another protein synthesis inhibitor, have a similar site and mode of action for inhibition of protein synthesis and his studies of the conformational, configurational and electrostatic properties of the emetine molecule suggest that both emetine and cycloheximide share certain structural properties around two nitrogen atoms that are essential for their activity (Fig. 1) [32]. Consistent with this notion, we also demonstrate that cycloheximide regulates alternative splicing of exon 2 in the Bcl-x gene (Fig. 2C & 2D). On the other hand, several other protein synthesis inhibitors such as anisomycin and puromycin in the NINDS collections of 1,040 FDA approved drugs and compounds have no or little effects on splicing of exon 2 in the Bcl-x gene, suggesting a relative specificity of emetine on Bcl-x splicing. All experiments were repeated at least three times.
Fig. 2. Emetine regulates Bcl-x splicing in C33A cells.
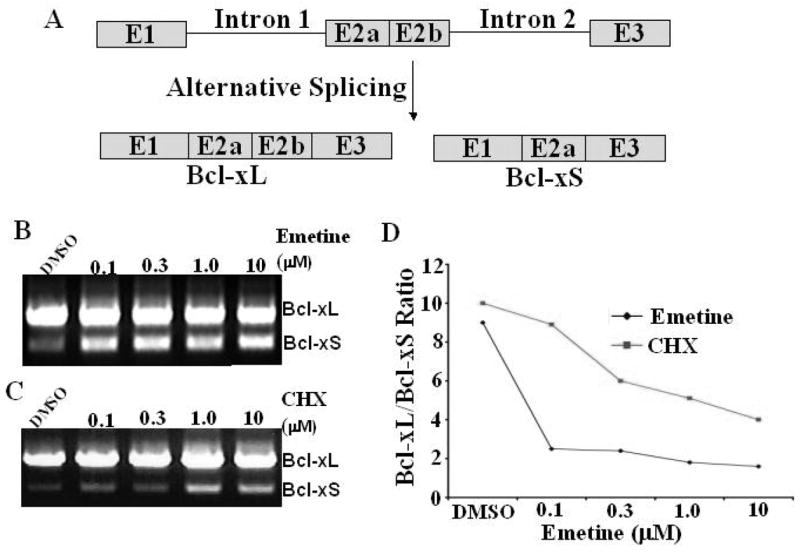
C33A cells were treated with emetine. Total RNA was extracted and analyzed by RT-PCR for the alternative splicing of Bcl-x. A. Alternative splicing of Exon 2 in the Bcl-x gene produces larger Bcl-xL and smaller Bcl-xS. B. Decrease of Bcl-xL and increase of Bcl-xS is correlated with the emetine concentration. C. Cycloheximide (CHX) also regulates exon 2 splicing in the Bcl-x gene. D. Ratio of Bcl-xL/Bcl-xS. Each experiment was repeated at least three times. Results from all experiments consistently show that emetine and cycloheximide (CHX) modulate exon 2 splicing in the Bcl-x gene. CHX: cycloheximide.
Fig. 1. Chemical Structures.
A. Emetine; B. Cycloheximide (CHX).
Time- and Dosage-dependence of emetine on Bcl-x splicing in cancer cells
To further validate if emetine regulates exon 2 splicing in the Bcl-x gene and to study if regulation of exon 2 splicing has potential relevance to cancer therapy, we examined the effects of emetine on the pre-mRNA processing of Bcl-x in several tumor cell lines. We treated cells with different time durations and/or different concentrations of emetine. Semi-quantitative RT-PCR was used to determine the effects of emetine. We found that in MCF-7 (a breast cancer cell line) (Fig. 3A & 3C) and PC3 (a prostate cancer cell line) cells (Fig. 3B &3C), regulation of Bcl-x splicing is slightly more pronounced. There was a decrease in the ratio of Bcl-xL/Bcl-xS from 8.6, 7.2, 5.1, 3.8, 3.3 and 3.0 in MCF-7 cells and 7.1, 5.0, 4.8, 4.6, 3.3, 3.1 in PC3 cells with emetine 0.1, 0.3, 1.0, 3.0 and 10.0 μM respectively (Fig. 3C). However, we see a more dramatic regulation of Bcl-x splicing in lung cancer cell line A549 cells by emetine (Fig 4). The effects of emetine on the pre-mRNA processing of Bcl-x in A549 cells were time-course and dosage-dependent. For dosage-dependent study, A549 cells were treated overnight with various concentration of emetine. There was a decrease in the ratio of Bcl-xL/Bcl-xS from 7.5 to 3.9, 2.9, 2.0, 1.5, and 1.2 with emetine 0.1, 0.3, 1.0, 3.0 and 10.0 μM respectively (Figure 4A &4C). For time-course study, A549 cells were treated with 1 μM emetine. There was a decrease in the ratio of Bcl-xL/Bcl-xS from 7.2 to 3.3, 2.8, 1.3, 1.1, and 0.9 with the duration of treatment of 6 h, 12 h, 24 h, 36 h and 48 h respectively (Fig. 4B & 4D). However, since emetine is a potent protein synthesis inhibitor, no synthesis of new protein with exon 2b skipping should be expected after emetine treatment. Consistent with this notion, we observed only a slight change of Bcl-xL/Bcl-xS ratio at the protein level in A549 cells treated with emetine (Supplemental Figure 1).
Fig. 3. Emetine regulates Bcl-x splicing in MCF-7 breast cancer cells and PC3 prostate cancer cells.
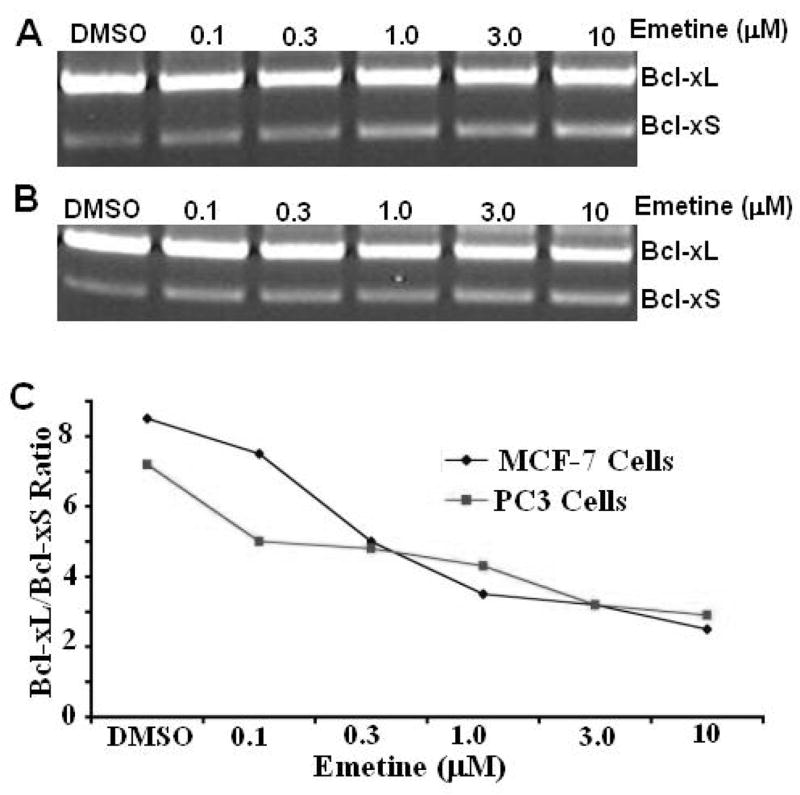
Total RNA was extracted from MCF-7 and PC3 cells that were treated with emetine. Splicing of exon 2 in the Bcl-x gene was analyzed by RT-PCR. A. RT-PCR results from MCF-7 cells treated with different concentrations of emetine. B. RT-PCR results from PC3 cells treated with different concentrations of emetine. C. Reduction of Bcl-xL and increase of Bcl-xS were observed and quantified. All experiments were repeated at least three times and consistently showed that emetine regulated exon 2 splicing of the Bcl-x gene in MCF-7 and PC3 cells.
Fig. 4. Emetine regulates Bcl-x splicing in A549 lung cancer cells.
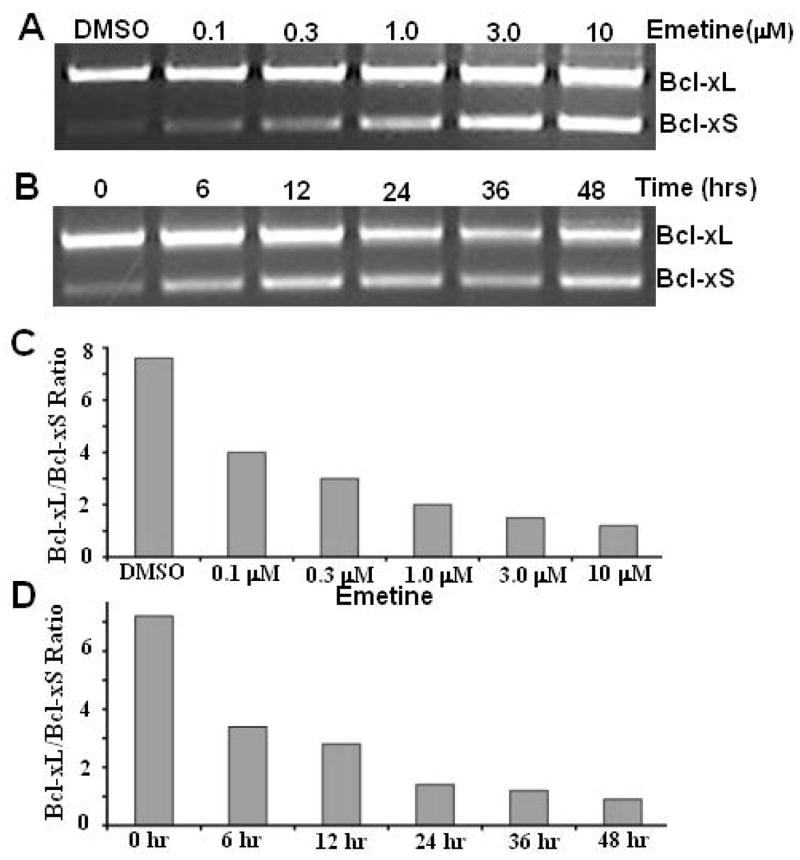
A549 cells were treated with emetine. Total RNA was extracted and analyzed by RT-PCR for the alternative splicing of Bcl-x. A & C. Reduction of Bcl-xL and increase of Bcl-xS is correlated with the emetine concentration. B & D. Cells were treated with 1 μM of emetine for different durations. RT-PCR was carried out to quantify alternative splicing of Bcl-x. All experiments were repeated at least three times.
Emetine exerts its effect on Bcl-x Splicing, via protein phosphatase-1 (PP1)
Previous studies show that ceramide affects splicing of Bcl-x in a phosphorylation dependent pathway. To examine whether emetine exerts the effects on Bcl-x splicing in a similar manner, we treated C33A and PC3 cells with phosphatase inhibitors, calyculin A and okadaic acid. We found that 5 μM of calyculin A, an inhibitor of both protein phosphatase1 (PP1) and protein phosphatase 2A (PP2A), completely blocked the emetine effects on Bcl-x alternative splicing in both C33A and PC3 cells (Fig. 5, comparing +emetine with +emetine/calyculin A). To establish whether PP1 or PP2A was the emetine-responsive protein phosphatase that regulates Bcl-x alternative splicing, C33A and PC3 cells were pretreated for 1 h with 5 μM of okadaic acid, a selective PP2A inhibitor. Pretreatment with okadaic acid had no effect on Bcl-x alternative splicing (Fig. 5, comparing +emetine with +emetine/Okadaic acid). Taken together, these results suggest that PP1 mediates the effects of emetine on the alternative splicing of Bcl-x.
Fig. 5. Calyculin A but not okadaic acid blocks effects of emetine on Bcl-x splicing.
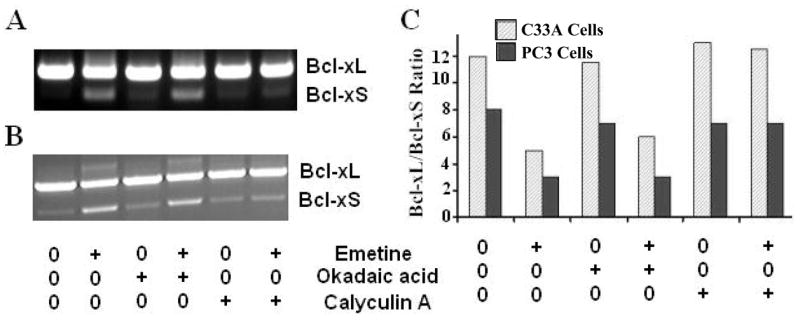
Cells were pretreated with either 5 μM of calyculin A, an inhibitor of both protein phosphatase1 (PP1) and protein phosphatase 2A (PP2A) or 5 μM of okadaic acid, a selective PP2A inhibitor and then exposed to 1.0 μM of emetine for 24 hours. RT-PCR was carried out. The results suggest that PP1 not PP2A mediates the effects of emetine on the alternative splicing of Bcl-x. A. Effects of calyculin A in C33A cells. B. Effects of calyculin A in PC3 cells. All experiments were repeated at least three times. C. Effects of calyculin A or okadaic acid on Bcl-x splicing mediated by emetine were quantified and plotted. Ratios of Bcl-xL/Bcl-xS in C33A cells: no treatment, 12.0; with emetine, 4.5; with okadaic acid, 12.0; with emetine and okadaic acid, 5.8; with calyculin A, 12.0; with emetine and calyculin A, 11.8. Ratios of Bcl-xL/Bcl-xS in PC3 cells: no treatment, 7.9; with emetine, 3.0; with okadaic acid, 7.0; with emetine and okadaic acid, 3.1; with calyculin A, 7.0; with emetine and calyculin A, 7.0. +: With emetine, okadaic acid or calyculin A. 0: Without emetine, okadaic acid or calyculin A.
Discussion
Bcl-x belongs to the Bcl-2 family and plays an important role in apoptosis. Bcl-x produces antiapoptotic Bcl-xL and proapoptotic Bcl-xS via alternative splicing of exon 2 (Fig. 2A). It has been suggested that the expression of Bcl-xL in tumor cells is one of the important indicators of chemotherapeutic efficacy because Bcl-xL protected cells from a wide variety of apoptotic stimuli and conferred a multidrug resistance phenotype [33]. In contrast, the smaller form Bcl-xS sensitizes cells to cell death inducers. These data indicate that manipulating levels of Bcl-xL and Bcl-xS proteins in tumors may provide a venue for cancer treatment with combination of chemotherapeutic agents. Recent reports demonstrated that the alternative splicing of exon 2 in the Bcl-x gene can be altered for this purpose [20]. For instance, modifying the ratio of Bcl-xL to Bcl-xS in the cells with an antisense oligonucleotide allows cells to be sensitized to undergo apoptosis in response to ultraviolet B radiation and chemotherapeutic agent treatment [34]. On the other hand, down-regulation of Bcl-xL by RNA interference was shown to suppress cell growth and induce apoptosis in human esophageal cancer cells [35]. In chemo-resistant human colon cancer cells, Bcl-xL small interfering RNA was found to suppress cell proliferation. Therefore, Bcl-xL down-regulation might provide a new target for human chemo-resistant cancer therapy [36].
Emetine is a potent inhibitor of protein synthesis in mammalian cells, plants, and yeast [37]. It has been shown that (−)-emetine is an effective chemotherapeutic agent by increasing the life-span in tumor-bearing mice [38, 39] and thus has possibility for clinical advantage [40]. To this end, emetine has been evaluated in Phase II clinical studies as a potential chemotherapeutic agent for the treatment of solid tumors [41]. However, to our knowledge, no previous study explains the mechanism responsible for the anti-tumor effect of emetine. In this report, we demonstrate that emetine regulates alternative splicing of exon 2 in the Bcl-x gene, resulting in more production of pro-apoptotic Bcl-xS with a concomitant decrease of anti-apoptotic Bcl-xL, leading to our speculation that regulating Bcl-x splicing is one of the underlying mechanisms of the anti-tumor effect of emetine. Since several other protein synthesis inhibitors including anisomycin and puromycin in the 1,040 FDA approved drugs/compounds didn’t alter the splicing of Bcl-x, we reason that inhibition of new protein synthesis is unlikely the mechanism of action for emetine on splicing. To further test this hypothesis, it is crucial in the future to investigate emetine analogs that don’t inhibit protein synthesis on the splicing of Bcl-x.
This newly defined and novel mechanism was also shown to be dependent on protein phosphatase 1 (PP1) activation. This conclusion was based on the use of the potent inhibitors of serine/threonine-protein phosphatases, okadaic acid, and calyculin A. Calyculin A, the PP1 and PP2A inhibitor, completely blocked emetine-induced alternative splicing of Bcl-x, while okadaic acid, a specific inhibitor of PP2A, had no effect on emetine-induced alternative splicing of Bcl-x (Fig 5). This therefore infers that the mechanism is dependent of PP1 activation. With the demonstration of PP1 as an emetine-activated protein phosphatase, potential PP1 substrates and mechanisms regulated by PP1 became candidate targets for emetine action. These findings are significant for several reasons. First, this mechanism of emetine-induced alternative splicing defines a novel mechanism of controlling the gene expression of pro-apoptotic factors in response to extracellular inducing agents. Second, a specific and direct mechanism mediated by an emetine-activated protein phosphatase has been established. Interestingly, the mechanistic action of emetine is similar to what has been described for ceramide, a small molecule that also induces, via alternative splicing, the expression of the pro-apoptotic splice variant Bcl-xS, with a concomitant loss in the anti-apoptotic splice variant Bcl-xL [25]. It appears that ceramide affects phosphorylation of SR proteins. Therefore, it would be important to examine if and how emetine affects splicing factors and if the ceramide is generated downstream of emetine. Moreover, it is also possible that emetine selectively affects stability of either Bcl-xL or Bcl-xS mRNA, resulting in the change of Bcl-xL/Bcl-xS ratio. This should be explored in the future. Finally, although cells with treatment of emetine at low concentrations do not undergo apoptosis, we observed that these cells are sensitized to high concentration of emetine and to other death inducers (Supplemental Fig. 2), implying that regulation of Bcl-xL/Bcl-xS ratio may indeed affect cell survival.
In summary, we demonstrated that emetine reduced the expression of the cell survival factors Bcl-xL and increased the expression of pro-apoptotic factor Bcl-xS in MCF-7 breast cancer cells, PC3 prostate cancer cells, A549 Lung cancer cells and C33A cervical carcinoma cells. Further study is needed to examine whether phosphorylation of SR proteins is regulated by emetine-activated protein phosphatase 1 (PP1) and what are the consequences of the dephosphorylation of SR-proteins. Is this effect contributing to apoptosis process? What is the critical percentage of Bcl-x splicing that determines the chemotherapeutic sensitivity? How much Bcl-xL needs to be spliced to Bcl-xS in order to allow cells to become susceptible to chemotherapy? These studies will have direct relevance to chemotherapeutic sensitivity because specific control of the alternative splicing of Bcl-x is linked to the sensitization of cells to chemotherapeutic agents, initiating a new target for anti-cancer treatment.
Significance
Emetine is a crystalline alkaloid, C29H40N2O4, derived from ipecac root. It is a potent protein synthesis inhibitor and is clinically used in the treatment of protozoan infection. Emetine has shown promise as an anti-tumor agent without bone marrow suppression. In present study, we have examined the effect of emetine on the alternative pre-mRNA processing of Bcl-x. We demonstrated that emetine down regulated the levels of Bcl-xL mRNA with a concomitant increase in the mRNA levels of Bcl-xS in a dose-and time-dependent manner. Mechanistically, emetine-induced alternative splicing was dependent on the activation of Protein Phosphatase-1 (PP1). This conclusion was based on the use of the potent inhibitors of serine/threonine-protein phosphatases, okadaic acid, and calyculin A. To our knowledge, this is the first report on the regulation of RNA splicing of members of the Bcl-2 families in response to Emetine. This significant finding may have direct relevance to chemotherapeutic sensitivity, giving rise to a new target for anti-cancer therapies. Future study is needed to confirm whether chemosensitivity is associated with increased Bcl-x splicing and whether SR proteins are dephosphorylated by PP1-mediated action of emetine.
Experimental Procedures
Compounds
All chemicals including emetine, cycloheximide, Calyculin A and Okadaic acid were purchased from Sigma.
Cell Culture
The human cervical carcinoma C33A cells were maintained in Dulbecco’s modified Eagle’s media (DMEM), supplemented with 10% (vol/vol) fetal bovine serum (FBS), L-Glutamine and penicillin-streptomycin. PC3 prostate cancer cells were cultured in RPMI media supplemented with 10% (v/v) fetal bovine serum, L-Glutamine and penicillin-streptomycin. Adenocarcinoma 549 lung cancer cells were grown in Dulbecco’s modified Eagle’s media/nutrient mixture F-12 (Ham) supplemented with 10 %(v/v) fetal bovine serum, L-Glutamine and penicillin-streptomycin. Human breast cancer cells MCF-7 and MCF-7/Adr were cultured in RPMI media supplemented with 10% (v/v) fetal bovine serum, L-Glutamine and penicillin-streptomycin. All cells were maintained at less than 80% confluence under standard incubator conditions.
Emetine Treatment
Emetine dihydrochloride hydrate (Sigma) with the stock solution concentration of 100 μM was used. Twenty-four hours prior to emetine treatment, the cells were plated in 2 ml of medium in 6-well plates at a density of 200,000 cells/well. The cells were treated with different concentration of emetine for 24 hours for dosage dependence study. For time course experiment, cells were treated with 1.0 μM of emetine for various durations.
Protein Phosphatase Inhibitors Treatment
Cells were pretreated with calyculin A or okadaic acid for 1 hour. The media were removed. Fresh regular media with emetine were added to treat cells for the duration of 24 hours. RT-PCR was then carried out to examine Bcl-x splicing.
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from cultured cells using Trizol reagent (Invitrogen) according to manufacture’s instruction. Reverse transcription was carried out with 1 μg of total RNA using Improm II reverse transcriptase (Promega) and oligo (dT) as the priming agent. After 1 hour incubation at 42°C, the reactions were terminated by heating at 70°C for 15 min. To analyze alternative splicing of exon 2 in the Bcl-x gene, an upstream 5’ primer to Bcl-x (5′-GAGGCAGGCGACGAGTTTGAA-3′) and a downstream 3′ primer (5′-TGGGAGGGTAGAGTGGATGGT-3′) were used for PCR amplification (32 cycles, 94°C,30s; 55° C,30s;72°C,1 min) with Choice Taq Blue Mastermix (Denville). PCR products were separated and analyzed on agarose gels.
Supplementary Material
Acknowledgments
Dr. Kritsanapol Boon-Unge was partly funded by the Royal Thai Government, Staff Development Scholarship. JZ was funded by NIH R03-CA119270.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Black DL. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000;103:367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- 2.Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17:100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 3.Goldstrohm AC, Greenleaf AL, Garcia-Blanco MA. Co-transcriptional splicing of pre-messenger RNAs: considerations for the mechanism of alternative splicing. Gene. 2001;277:31–47. doi: 10.1016/s0378-1119(01)00695-3. [DOI] [PubMed] [Google Scholar]
- 4.Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18:186–193. doi: 10.1016/s0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 5.Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2002;30:13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- 6.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 7.Schwerk C, Schulze-Osthoff K. Regulation of apoptosis by alternative pre-mRNA splicing. Mol Cell. 2005;19:1–13. doi: 10.1016/j.molcel.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 8.Mercatante D, Kole R. Modification of alternative splicing pathways as a potential approach to chemotherapy. Pharmacol Ther. 2000;85:237–243. doi: 10.1016/s0163-7258(99)00067-4. [DOI] [PubMed] [Google Scholar]
- 9.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 10.Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- 11.Antonsson B, Martinou JC. The Bcl-2 protein family. Exp Cell Res. 2000;256:50–57. doi: 10.1006/excr.2000.4839. [DOI] [PubMed] [Google Scholar]
- 12.Tsujimoto Y, Shimizu S. VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ. 2000;7:1174–1181. doi: 10.1038/sj.cdd.4400780. [DOI] [PubMed] [Google Scholar]
- 13.Liu R, Page C, Beidler DR, Wicha MS, Nunez G. Overexpression of Bcl-x(L) promotes chemotherapy resistance of mammary tumors in a syngeneic mouse model. Am J Pathol. 1999;155:1861–1867. doi: 10.1016/S0002-9440(10)65505-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castilla C, Congregado B, Chinchon D, Torrubia FJ, Japon MA, Saez C. Bcl-xL is overexpressed in hormone-resistant prostate cancer and promotes survival of LNCaP cells via interaction with proapoptotic Bak. Endocrinology. 2006;147:4960–4967. doi: 10.1210/en.2006-0502. [DOI] [PubMed] [Google Scholar]
- 15.Olopade OI, Adeyanju MO, Safa AR, Hagos F, Mick R, Thompson CB, Recant WM. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J Sci Am. 1997;3:230–237. [PubMed] [Google Scholar]
- 16.Fernandez Y, Espana L, Manas S, Fabra A, Sierra A. Bcl-xL promotes metastasis of breast cancer cells by induction of cytokines resistance. Cell Death Differ. 2000;7:350–359. doi: 10.1038/sj.cdd.4400662. [DOI] [PubMed] [Google Scholar]
- 17.Williams J, Lucas PC, Griffith KA, Choi M, Fogoros S, Hu YY, Liu JR. Expression of Bcl-xL in ovarian carcinoma is associated with chemoresistance and recurrent disease. Gynecol Oncol. 2005;96:287–295. doi: 10.1016/j.ygyno.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 18.Heere-Ress E, Thallinger C, Lucas T, Schlagbauer-Wadl H, Wacheck V, Monia BP, Wolff K, Pehamberger H, Jansen B. Bcl-X(L) is a chemoresistance factor in human melanoma cells that can be inhibited by antisense therapy. Int J Cancer. 2002;99:29–34. doi: 10.1002/ijc.10248. [DOI] [PubMed] [Google Scholar]
- 19.Wacheck V, Selzer E, Gunsberg P, Lucas T, Meyer H, Thallinger C, Monia BP, Jansen B. Bcl-x(L) antisense oligonucleotides radiosensitise colon cancer cells. Br J Cancer. 2003;89:1352–1357. doi: 10.1038/sj.bjc.6601254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sumantran VN, Ealovega MW, Nunez G, Clarke MF, Wicha MS. Overexpression of Bcl-XS sensitizes MCF-7 cells to chemotherapy-induced apoptosis. Cancer Res. 1995;55:2507–2510. [PubMed] [Google Scholar]
- 21.Lei XY, Zhong M, Feng LF, Zhu BY, Tang SS, Liao DF. Bcl-XL small interfering RNA enhances sensitivity of Hepg2 hepatocellular carcinoma cells to 5-fluorouracil and hydroxycamptothecin. Acta Biochim Biophys Sin (Shanghai) 2006;38:704–710. doi: 10.1111/j.1745-7270.2006.00212.x. [DOI] [PubMed] [Google Scholar]
- 22.Lei XY, Zhong M, Feng LF, Zhu BY, Tang SS, Liao DF. siRNA-mediated Bcl-2 and Bcl-xl gene silencing sensitizes human hepatoblastoma cells to chemotherapeutic drugs. Clin Exp Pharmacol Physiol. 2007;34:450–456. doi: 10.1111/j.1440-1681.2007.04593.x. [DOI] [PubMed] [Google Scholar]
- 23.Ray S, Bullock G, Nunez G, Tang C, Ibrado AM, Huang Y, Bhalla K. Enforced expression of Bcl-XS induces differentiation and sensitizes chronic myelogenous leukemia-blast crisis K562 cells to 1-beta-D-arabinofuranosylcytosine-mediated differentiation and apoptosis. Cell Growth Differ. 1996;7:1617–1623. [PubMed] [Google Scholar]
- 24.Dole MG, Clarke MF, Holman P, Benedict M, Lu J, Jasty R, Eipers P, Thompson CB, Rode C, Bloch C, Nunez, Castle VP. Bcl-xS enhances adenoviral vector-induced apoptosis in neuroblastoma cells. Cancer Res. 1996;56:5734–5740. [PubMed] [Google Scholar]
- 25.Chalfant CE, Rathman K, Pinkerman RL, Wood RE, Obeid LM, Ogretmen B, Hannun YA. De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J Biol Chem. 2002;277:12587–12595. doi: 10.1074/jbc.M112010200. [DOI] [PubMed] [Google Scholar]
- 26.Li CY, Chu JY, Yu JK, Huang XQ, Liu XJ, Shi L, Che YC, Xie JY. Regulation of alternative splicing of Bcl-x by IL-6, GM-CSF and TPA. Cell Res. 2004;14:473–479. doi: 10.1038/sj.cr.7290250. [DOI] [PubMed] [Google Scholar]
- 27.Stumm G, Schlegel J, Schafer T, Wurz C, Mennel HD, Krieg JC, Vedder H. Amphetamines induce apoptosis and regulation of bcl-x splice variants in neocortical neurons. Faseb J. 1999;13:1065–1072. doi: 10.1096/fasebj.13.9.1065. [DOI] [PubMed] [Google Scholar]
- 28.Grollman AP. Inhibitors of protein biosynthesis. V. Effects of emetine on protein and nucleic acid biosynthesis in HeLa cells. J Biol Chem. 1968;243:4089–4094. [PubMed] [Google Scholar]
- 29.Baliga BS, Cohen SA, Munro HN. Effect of cycloheximide on the reaction of puromycin with polysome-bound peptidyl-tRNA. FEBS Lett. 1970;8:249–252. doi: 10.1016/0014-5793(70)80278-2. [DOI] [PubMed] [Google Scholar]
- 30.Pestka S. Inhibitors of ribosome functions. Annu Rev Microbiol. 1971;25:487–562. doi: 10.1146/annurev.mi.25.100171.002415. [DOI] [PubMed] [Google Scholar]
- 31.Carrasco L, Jimenez A, Vazquez D. Specific inhibition of translocation by tubulosine in eukaryotic polysomes. Eur J Biochem. 1976;64:1–5. doi: 10.1111/j.1432-1033.1976.tb10268.x. [DOI] [PubMed] [Google Scholar]
- 32.Grollman AP. Structural Basis for Inhibition of Protein Synthesis by Emetine and Cycloheximide Based on an Analogy between Ipecac Alkaloids and Glutarimide Antibiotics. Proc Natl Acad Sci U S A. 1966;56:1867–1874. doi: 10.1073/pnas.56.6.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minn AJ, Rudin CM, Boise LH, Thompson CB. Expression of bcl-xL can confer a multidrug resistance phenotype. Blood. 1995;86:1903–1910. [PubMed] [Google Scholar]
- 34.Taylor JK, Zhang QQ, Wyatt JR, Dean NM. Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nat Biotechnol. 1999;17:1097–1100. doi: 10.1038/15079. [DOI] [PubMed] [Google Scholar]
- 35.Xie YE, Tang EJ, Zhang DR, Ren BX. Down-regulation of Bcl-XL by RNA interference suppresses cell growth and induces apoptosis in human esophageal cancer cells. World J Gastroenterol. 2006;12:7472–7477. doi: 10.3748/wjg.v12.i46.7472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu H, Guo W, Zhang L, Davis JJ, Teraishi F, Wu S, Cao X, Daniel J, Smythe WR, Fang B. Bcl-XL small interfering RNA suppresses the proliferation of 5-fluorouracil-resistant human colon cancer cells. Mol Cancer Ther. 2005;4:451–456. doi: 10.1158/1535-7163.MCT-04-0162. [DOI] [PubMed] [Google Scholar]
- 37.Grollman AP. Inhibitors of protein biosynthesis. II. Mode of action of anisomycin. J Biol Chem. 1967;242:3226–3233. [PubMed] [Google Scholar]
- 38.Jondorf WR, Abbott BJ, Greenberg NH, Mead JA. Increased lifespan of leukemic mice treated with drugs related to (−)-emetine. Chemotherapy. 1971;16:109–129. doi: 10.1159/000220718. [DOI] [PubMed] [Google Scholar]
- 39.Johnson RK, Jondorf WR. Some inhibitory effects of (--)-emetine on growth of Ehrlich ascites carcinoma. Biochem J. 1974;140:87–94. doi: 10.1042/bj1400087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panettiere F, Coltman CA., Jr Experience with emetine hydrochloride (NSC 33669) as an antitumor agent. Cancer. 1971;27:835–841. doi: 10.1002/1097-0142(197104)27:4<835::aid-cncr2820270413>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 41.Siddiqui S, Firat D, Olshin S. Phase II study of emetine (NSC-33669) in the treatment of solid tumors. Cancer Chemother Rep. 1973;57:423–428. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.