

Abstract
Using various methods, e.g., spectrophotometry, circular dichroism, and isothermal titration calorimetry, the interaction of poncean S (PS) with human serum albumin (HSA) was characterized at pH 1.81, 3.56, and 7.40 using the spectral correction technique, and Langmuir and Temkin isothermal models. The consistency among results concerning, e.g., binding number, binding energy, and type of binding, showed that ion pair electrostatic attraction fixed the position of PS in HSA and subsequently induced a combination of multiple noncovalent bonds such as H-bonds, hydrophobic interactions, and van der Waals forces. Ion pair attraction and H-bonds produced a stable PS-HSA complex and led to a marked change in the secondary structure of HSA in acidic media. The PS-HSA binding pattern and the process of change in HSA conformation were also investigated. The potentially toxic effect of PS on the transport function of HSA in a normal physiological environment was analyzed. This work provides a useful experimental strategy for studying the interaction of organic substances with biomacromolecules, helping us to understand the activity or mechanism of toxicity of an organic compound.
Introduction
Human serum albumin (HSA) is the major protein component of blood plasma but is also found in the interstitial fluid of body tissues. In mammals, albumin is synthesized by the liver and has a half-life of 19 days in the circulation (1,2). It is the major contributor to the oncotic pressure of blood plasma (3). Albumin is also reported to be chiefly responsible for the maintenance of blood pH (4). Serum albumin has been one of the most extensively studied proteins for the past 40 years: its primary structure (a single-chain polypeptide of 585 residues) has been known for a long time, and its tertiary structure was determined a few years ago by x-ray crystallography (5). Its secondary structure comprises 67% helix (six turns) and 17 disulfide bridges (6), including three structurally similar α-helical domains (I–III), each divided into subdomains A and B (7). HSA is called a multifunctional plasma carrier protein because of its ability to bind an unusually broad spectrum of ligands. These include inorganic cations, organic anions, various drugs, amino acids, and, perhaps most important, physiologically available insoluble endogenous compounds such as bilirubin (3), fatty acids, and bile acids. Binding to HSA facilitates their transport throughout the circulation (9).
Structural studies have mapped the locations of the fatty acid binding sites throughout the protein (10,11). Like the endogenous ligands, many exogenous compounds also bind to HSA (12), for example, commonly used drugs with acidic or electronegative features, e.g., warfarin (13), camptothecins (14), and inorganic polymers such as polyoxometalates (15,16). The primary drug binding sites on the protein have been mapped by structural studies (17,18). Recently, studies have been conducted on the binding of organic contaminants or toxins to HSA, e.g., arazine (19), ochratoxin (20), methyl parathion (21), and arsenic (22). These compounds often cause conformational changes in the protein, e.g., decrease in α-helix and increase of β-sheet content. These studies showed that the protein binds ligands selectively and covalently: few ligand molecules bind, with a high heat of reaction. In fact, the binding of any substance is likely to affect the activity of the protein, either enhancing it (23) with potential medical significance or inhibiting it (24) if an organic contaminant or toxin is involved.
Azo pigments are a structurally diverse and widely distributed group of synthetic compounds that include environmental pollutants. Representatives of this group, poncean S (PS) and its homologs discovered by Baurm (25), are used as food additives and sometimes as cosmetic pigments and biological stains, usually very effectively. PS can enter the gastrointestinal tract via food intake or permeate into the blood via skin absorption. As an azo compound, it can be reduced by azo reductases to produce aromatic amines, some of which are known carcinogens (26). Long-time use has negative effects on human and animal liver, possibly resulting in degenerative pathological changes and cirrhosis (27). pH varies widely among normal human body fluids, for example, a pH of <2 in gastric fluid, pH 4–7 in sweat, pH 4.5 in vaginal fluid, pH 5–7 in urine, and ∼pH 7.4 in blood. Any compound that binds to a protein, enzyme, gene, or other biomacromolecule will affect its structure and function to a greater or lesser extent. For example, a drug binding to HSA in the blood may be transported to the focus of disease for therapeutic effect, but binding of a toxin may impede the transport of endogenous substances.
Ligand binding to macromolecules has previously been studied by methods such as equilibrium dialysis (28), electrochemistry (29), calorimetery (30), spectrophotometry (31), and fluorescence spectroscopy (32). In this work, isothermal titration calorimetry (ITC), ultraviolet (UV), and circular dichroism (CD) were used to characterize the binding of PS to HSA at pH 1.81, 3.56, and 7.40, and the spectral correction technique (33), and the Langmuir and Temkin isothermal models were used to elucidate the mechanism of interaction. The interaction of PS with HSA was investigated and compared in detail not only at room temperature but also under normal physiological conditions: 0.8%–0.9% electrolyte, 37°C. The various methods yielded highly consistent results concerning, e.g., the saturation binding number of PS, the binding energy of the reaction, and the bonds involved. Ion pair electrostatic attraction (34) fixes the position of PS in HSA and subsequently induces the formation of a combination of other noncovalent bonds: H-bonds (35,36), hydrophobic interactions, and van der Waals forces. The type and site of binding and the noncovalent bonds involved were provisionally identified. Possible conformational change of HSA in the presence of PS was also identified.
Materials and methods
Instruments and materials
The absorption spectra of PS and protein solutions were recorded with a Model Lambda-25 spectrophotometer (Perkin-Elmer, Foster City, CA) equipped with a thermostatic cell holder linked to a Model TS-030 water-circulated thermostatic oven (Yiheng Sci. Technol., Shanghai, China). The spectrophotometer was computer-controlled using UV WinLab software (Version 2.85.04). The ITC experiments were carried out on a Model MSC-ITC system (MicroCal, Studio City, CA) with measurement software. A Model J-715 CD spectropolarimeter (Jasco Instruments, Tokyo, Japan) with secondary structure estimation-standard analysis measurement software (715 /No. B014460524, Jasco) was used to determine protein conformation. A Model DK-8D electrothermic multiporous constant temperature (Shanghai Yiheng Technol., Shanghai, China) was used in the temperature experiment. Solution pHs were measured with a Model pHS-25 acidity meter (Shanghai Precise Sci. Instrum., Shanghai, China).
HSA (250 mg, purity 96%–99%, Sigma Reagents, St. Louis, MO) was dissolved in 250 ml deionized water as a stock solution (1.00 mg/ml HSA). Solutions (0.010 mg/ml HSA) were prepared daily by diluting the stock and were stored at 2°C–8°C. PS stock solution (0.750 mmol/l) was prepared by dissolving 76 mg of PS (purity 75%, Shanghai Chemical Reagents, Chin. Med. Group, Shanghai, China) in 100 ml deionized water. Solutions of 0.075 mmol/l PS were prepared daily by diluting the stock. A series of Britton-Robinson (B-R) buffers, pH 1.81, 2.43, 2.97, 3.56, 4.19, 4.97, 5.43, 5.71, 6.38, 6.77, and 7.40, were prepared to adjust the acidities of solutions. A 2.0 mol/l NaCl solution was prepared in deionized water to adjust the ionic strengths of solutions to investigate the effects of electrolyte on noncovalent binding. Both 0.01 mol/l EDTA and pH 3.6 acetic-acetate buffer were prepared and used only in protein assays.
Photometric determination of the PS-HSA interaction
All studies were carried out in a 10.0 ml calibrated flask containing a known volume of HSA solution, 2.0 ml of B-R buffer (pH 1.81, 3.56, or 7.40), and a known volume of 0.750 mmol/l PS. The solution was diluted to 10.0 ml with deionized water and mixed thoroughly. After the reaction had proceeded for 5 min, the absorbances Aλ2 and Aλ1 of the HSA-PS solutions and and of the reagent blank (without HSA) were measured at 568 nm (λ2) and 495 nm (λ1) against water. The parameters Ac, f, and γ of each solution were calculated. The Langmuir and Temkin isothermal models were applied to fit plots γ−1 vs. and γ vs. ln(CL). Finally, N, K, and ΔQ were calculated.
Thermodynamic characterization of the PS-HSA interaction by ITC
ITC experiments were carried out as follows. The PS solution (0.600 mmol/l in pH 1.81 and 3.56 B-R buffers or 0.300 mmol/l in pH 7.40 B-R buffer) was injected ∼40 times in 5 μl increments at 3 min intervals into the isothermal cell containing HSA (1.6 μmol/l in pH 1.81 and 3.56 B-R buffers or 8.0 μmol/l in pH 7.40 B-R buffer). The cell temperature remained at 25°C or 37°C. Heats of dilution of PS, obtained separately by injecting into the buffer, were used to correct the data. The corrected heats were divided by the number of moles injected and analyzed using the Origin software (version 7.0) supplied by the manufacturer. The titration curve was fitted by a nonlinear least squares method and N, Kb, ΔH, and ΔS were determined.
CD measurement of HSA conformation in the presence of PS
Buffer (1 ml, pH 1.81, 3.56, or 7.40) was mixed with 0.030 mg/ml HSA in three flasks; 0, 0.015 or 0.060 mmol/l PS was added. The solutions were diluted to 10.0 ml with deionized water. Simultaneously, a reagent blank without PS was prepared. Before measurement, all the solutions were diluted from 1.00 to 3.00 ml with deionized water. Each sample was allowed to equilibrate for 15 min, then injected into a 0.1 cm light path cell, and the mean residue ellipticity (MRE) of HSA was measured between 200 and 250 nm. From the MRE curves, the relative contents of secondary structure forms of HSA—α-helix, β-pleated sheet, β-turn, and random coil—were calculated in all the solutions.
Protein assay
Light-absorption ratio variation (LARVA) (37) was used to assay protein with very high sensitivity. A known volume of a sample containing <70 μg of protein was added to a 10 ml flask along with 1 ml 10 mmol/l EDTA and 1 ml of pH 3.6 acetate buffer. After mixing, 3.0 μmol/l PS was added and the solution was diluted to 10.0 ml with deionized water. After 5 min, the absorbances (A495 and A568) of the solution were measured at 495 and 568 nm against water. Using the same method, a reagent blank without protein was prepared, and the absorbances ( and ) were measured. The absorbance ratio difference (ΔAr) of the PS-HSA solution was calculated by the relationship
| (1) |
ΔAr is linearly related to the protein concentration (CM0) as follows:
| (2) |
where p and q are constants obtained by linear regression of plots of ΔAr vs. CM0 from the series of standard HSA solutions. The variable p as the sensitivity factor is inversely proportional to the initial concentration (CL0) of ligand (37): the less L added, the higher the sensitivity obtained. However, too low a PS concentration will cause an obvious measurement error because of background instrument noise. To optimize the addition, PS was added in three concentrations (1.5, 3.0, and 4.5 μmol/l) to the series of standard HSA solutions and the ΔAr of each was calculated. By considering both the low limit of detection (LOD) and the linearity, an optimal PS concentration was selected.
Results and discussion
Effect of pH on the PS-HSA interaction
Fig. 1 illustrates the color change of PS solution in the presence of HSA and also shows the chemical structure of PS. PS is an orange anionic azo compound in aqueous solution 1, and it reacts with HSA to form a red product in solution 2. The absorption spectrum of the product shows a marked red shift. HSA-PS solutions were measured in various pH media, and their absorption spectra are shown in Fig. 2 A. The interval between the positive peak and the negative trough increases with increasing acidity of solution. From curves 1–4, the peak valley interval changes little when the pH is below 3.56. Given the dissociation constants (KR) of the side groups (R) of basic and acidic amino acid residues (AARs) (10.53 for Lys, 6.00 for His, 12.48 for Arg, 3.65 for Asp, and 4.25 for Glu), no R groups will have a negative charge when the pH is <3.65, whereas the Rs of the basic AARs will be positively charged.
Figure 1.
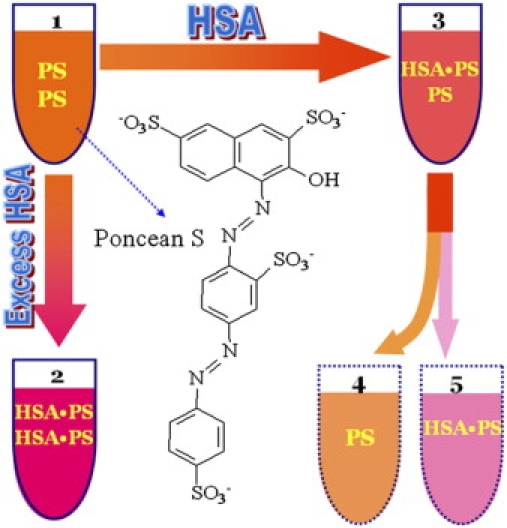
Cartoon illustrating the color change in the HSA-PS reaction: (1) PS solution; (2) HSA-PS solution containing excess HSA in which free PS approached zero; (3) As 2, but containing micro amounts of HSA. Solution 3 consists of solutions 4 and 5, which were actually nonmeasurable.
Figure 2.
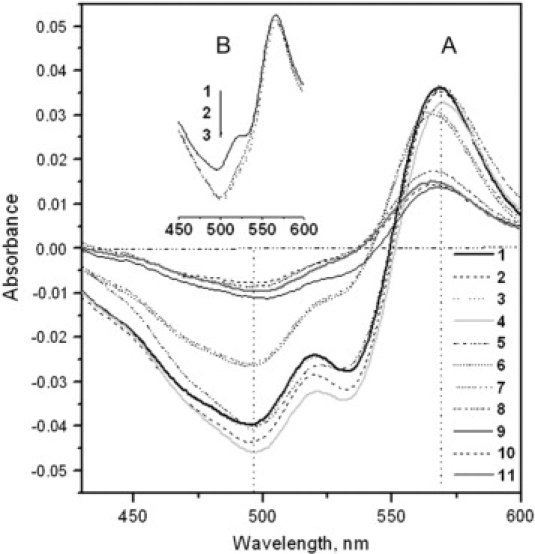
(A) Absorption spectra, measured against the reagent blank, of HSA-PS solutions containing 7.5 μmol/l PS and 50.0 mg/l HSA at pH values (1–11): 1.81, 2.43, 2.97, 3.56, 4.19, 4.97, 5.43, 5.71, 6.38, 6.77, and 7.40. (B) Absorption spectra of the solutions containing 7.5 μmol/l PS and 0.80 mg/ml HSA at pH 1.81 (1), 3.56 (2), and 7.40 (3) against water. There was no free PS because the solutions contained excess HSA.
PS anions will enter the HSA cleft with no charge repulsion. However, there are 98 negatively charged acidic AARs in HSA, together with 98 positively charged basic AARs when the pH is >4.25. Entry of PS will therefore be opposed by charge repulsion. Therefore, from curves 6–11, the binding of PS to HSA becomes progressively weaker. The two troughs in each curve indicate that PS-HSA binding is heterogeneous at any pH, i.e., there is a mixture of complexes linked via two, three, and four sulfonate groups because the PS concentration is not high enough. To compare the interactions of PS with HSA in various pH media including that of normal blood, i.e., pH 7.40, three buffer solutions, pH 1.81, 3.56, and 7.40, were used. From curves 1, 4, and 11 in A, 568 (λ2) and 495 nm (λ1) were the wavelengths that most clearly indicated the binding process and were used in subsequent work.
From the absorption spectra shown in Fig. 2 B, it is apparent that formation of the PS-HSA complex resulted in red shifts: from 497 to 565 nm (curve 1) at pH 1.81 and from 500 to 566 nm (curves 2 and 3) at pH 3.56 and 7.40. Interestingly, a secondary trough appears at 528 nm only in curve 1. A possible reason is that the unfolding of HSA at pH 1.81 increases the distances among positively charged AARs. The dissociation constants (Ka) of PS (L) were also determined spectrophotometrically (38), and the results indicated that pKa is between 2 and 3 for H2L3− and more than 9 for HL4−. Therefore, H2L3− and HL4− were both present in the pH 1.81 solution, leading to three and four ionic bonds between HSA and each PS molecule. Thus, PS-HSA binding is heterogeneous at pH 1.81 even if there is sufficient PS in solution to achieve saturation.
Photometric characterization of the PS-HSA interaction
The interaction of PS (L) with HSA (M) is summarized as follows:Eqilibrium CL = CL0 − NCM0(Aλ1,Aλ2) CM → 0 CM0(AC)
From the color change in Fig. 1, it is clear that to determine the absorbance of the PS-HSA complex in solution 3, interference by excess PS in solution 4 must be eliminated. The spectral correction technique (33) proposed previously is a useful method for this; it involves measuring solutions 1, 2, and 3. The effective fraction (f) of PS bound to HSA and the molar ratio (γ) of PS bound to HSA are calculated by the relationships (39)
| (3) |
and
| (4) |
where
| (5) |
Returning to the reaction equation above, CM0 and CL0 are the initial molar concentrations of HSA and PS; Ac indicates the real absorbance of the HSA-PS complex in solution 3 at 568 nm, which cannot be measured directly. Aλ2 and Aλ1 are the absorbances of the HSA-PS solution 3 measured at 568 and 495 nm, respectively, against water; α and β are correction constants and were calculated by measuring solutions 2 and 1 shown in Fig. 1. The γ value will approach the saturation number (N) of PS bound with the increase of PS in a HSA solution.
The absorbance ratios of the HSA-PS solutions were measured at 495 and 568 nm (Fig. 3 A). The A495/A568 ratios in three pH media all decrease with increasing HSA concentration and approach a constant value of 1.6 when the molar concentration of HSA is more than 1.2 times that of PS. This indicates that more and more PS molecules became bound to HSA until no excess PS was free in solution, i.e., solution 2 in Fig. 1. Thus, the constant minimum should be α of the binding product; the β values of PS in various media correspond to the A568/A495 ratio in the absence of HSA, which are located at the beginnings of curves 1–3. By measuring a series of PS solutions containing known concentrations of HSA, Ac, f, and γ were calculated using Eqs. 4–6. The variation in γ with PS is shown in Fig. 3 B; the value increases with increasing PS concentration. Moreover, the values approach the following maxima: 30 when the PS/HSA molar ratio is more than 30 (from curve 1), 25 when the ratio is more than 25 (from curve 2), and 1.7 when the ratio is more than 5 (from curve 3).
Figure 3.
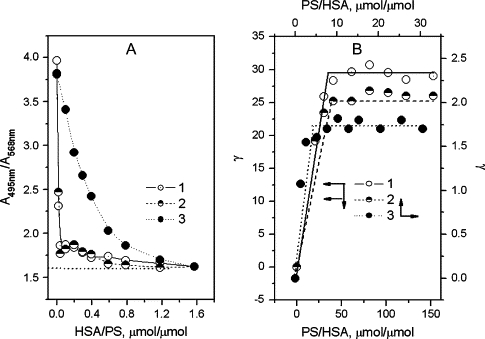
(A) Variation of the absorbance ratio (A495/A568) of solutions containing 7.5 μmol/l PS and 0−0.80 mg/ml HSA. (B) Variation of γ in solutions containing 50.0 mg/l HSA and variable PS. (1) pH 1.81, (2) pH 3.56, and (3) pH 7.40.
These results indicate that the binding of PS to HSA is saturable. When more PS is added, the fraction of excess PS steadily increases, but γ does not change. Thus, N of PS molecules bound per molecule of HSA are ∼30 at pH 1.81, ∼25 at pH 3.56, and ∼1.7 at pH 7.40. These values will be examined further in the following experiments. As described above, all the basic AARs in HSA form positively charged R groups in pH solution of <3.65, whereas all acidic AARs will not dissociate. Thus, PS anions bind easily to the HSA surface until saturation is reached. In contrast, negative R groups of the acidic AARs rejecting the concentration of PS ions increases when pH is >4.25. Therefore, N of PS at pH 7.40 is much less than those in acidic media. The Langmuir and Temkin isothermal models (40) below were used to fit the experimental data.
| (6) |
and
| (7) |
where CL is the equilibrium concentration of PS to be calculated by CL = (1 − f)CL0, K is the adsorption constant, and ΔQ the adsorption energy (J/mol) of a saturating concentration of PS. The term a is a regression constant, T the temperature in degrees Kelvin, and R the gas constant, 8.314 J mol−1 K−1. The results are given in Fig. 4. The binding of PS to HSA fitted both models when the number of PS molecules per HSA molecule was less than the maximal binding number. Therefore, the interaction of PS with HSA corresponds to chemical monolayer adsorption. From the intercepts and gradients of the regression lines in Fig. 4 A, both N and K of PS may be calculated (Table 1). The N values were consistent with those obtained from Fig. 3. ΔQ of PS was calculated from the line gradients in Fig. 4 B, as shown in Table 1. The increase in K and ΔQ values with increase in pH shows that PS binds more stably in neutral solution. A possible reason is that a low PS binding number resulted in more binding sites on HSA. In addition, the binding of PS is exothermic because of the negative heat of adsorption.
Figure 4.
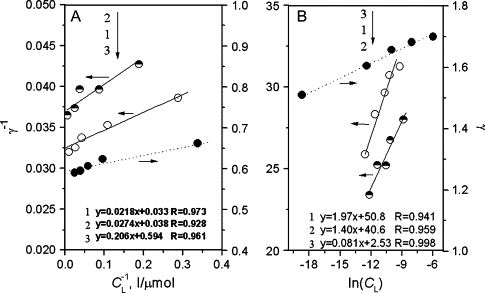
Plots of (A) γ−1 vs. and (B) γ vs. ln(CL) from solutions containing 50.0 mg/l HSA and variable PS. (1) pH 1.81, (2) pH 3.56, and (3) pH 7.40.
Table 1.
Determination of the thermodynamic parameters of the HSA-PS binding reaction at pH 1.81, 3.56, and 7.40 at 25°C
| pH | N* | K*, ×106 M−1 | ΔQ†, kcal/mol | N‡ | Kb‡, ×106 M−1 | ΔH‡, kcal/mol | ΔS‡, (cal/mol)K−1 | ΔG§, kcal/mol |
|---|---|---|---|---|---|---|---|---|
| 1.81 | 31 | 1.51 | −9.33 | 29.4 | 5.93 | −7.95 | 4.34 | −9.23 |
| 3.56 | 26 | 1.39 | −11.0 | 23.3 | 1.12 | −9.02 | −2.59 | −8.25 |
| 7.40 | 1.7 | 2.88 | −12.4 | 1.56 | 0.78 | −10.9 | −9.58 | −8.04 |
| 7.40¶ | / | / | / | 1.43 | 0.30 | −11.2 | −11.2 | −7.73 |
By the Langmuir adsorption isothermal.
By the Temkin adsorption model.
By ITC.
Calculated by Eq. 8.
At 37°C.
ITC characterization of the PS-HSA interaction
To understand the mechanism of HSA-PS binding and to assess the effect of environmental conditions, e.g., acidity, on its specificity and stability, detailed thermodynamic data are needed. ITC measurements provide information on thermodynamic quantities such as enthalpy and heat capacity changes during the molecular interaction directly from the heat produced by the reaction and have been used to study, for example, protein-ligand interactions (41), DNA triplex formation (42), and human immunodeficiency virus (HIV) protease activity (43). Fig. 5 (X-1) (X: A–D) depicts a typical isothermal titration profile obtained by injecting PS into the ITC cell containing HSA. An exothermic heat pulse is detected after each injection; its magnitude progressively decreases until a plateau is reached corresponding to the heat of dilution of the peptide species in the buffer and indicating saturation. The heat evolved at each injection was corrected for the heat of dilution, which was determined separately by injecting the PS solution into the buffer and divided by the number of moles injected. The resulting values were plotted as a function of the PS/HSA molar ratio and fitted to a sigmoid curve by a nonlinear least squares method (curves X-2 in Fig. 5). Values for the equilibrium binding constant (Kb), binding number (N), enthalpy change (ΔH), and entropy change (ΔS) of the PS-HSA reaction at various pHs were obtained from curves X-2 and calculated by the Gibbs free energy (ΔG) equation:
| (8) |
Figure 5.
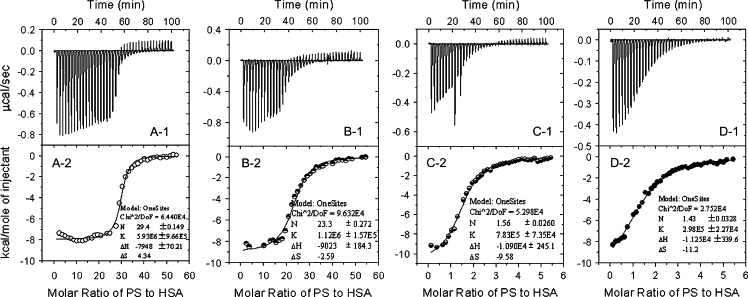
X-1 (X = A, B, C, and D): ITC titration profile of PS-HSA binding at (A) pH 1.81, (B) pH 3.56, and (C and D) pH 7.40. The temperature was 25°C (A–C) or 37°C (D). Each pulse corresponds to a 5-μl injection of (A and B) 0.600 mmol/l PS or (C and D) 0.300 mmol/l PS into the ITC cell (1.4685 ml) containing (A and B) 1.6 μmol/l HSA or (C and D) 8.0 μmol/l HSA. X-2. The area of each peak in X-1 was integrated and corrected for the heat of dilution, which was estimated by a separate experiment by injecting the PS into the B-R buffer. The corrected heat was divided by the moles of injectant, and values were plotted as a function of the PS/HSA molar ratio. The titration curve was fitted by a nonlinear least squares method.
The thermodynamic parameters derived from these curves are summarized in Table 1. Comparison with the photometric method (above) shows that the three measurements yield similar N values at three pHs. ΔH and ΔQ both become increasingly negative with increasing pH at 25°C, and ΔQ approaches the ΔH value at the same pH. Therefore, the Temkin isothermal model can be used to explain the binding mechanism. Because ΔH is much less than 60 kcal/mol (44), the PS-HSA interaction is noncovalent: it involves H-bonds, ion pair attraction, hydrophobic interactions, and van der Waals forces. The Kb values indicate that the complex becomes more stable with increasing acidity of solution. This can be explained by the tendency of ΔG to decrease with increasing pH (Table 1 and Fig. 6). Comparison of the ΔG, ΔH, and −TΔS values suggests that the PS-HSA interaction is amphipathic and H-bonds and ion pair binding are both major contributors, i.e., the interaction of PS with HSA depends on a combination of ion pair attraction and H-bonds. Moreover, the heat released during the PS-HSA reaction increases with increasing pH, i.e., the ion pair attraction and H-bonds between PS and HSA are weakened in more acidic solution. This indicates that the number of H-bonds formed by PS is lower in more acidic solutions.
Figure 6.
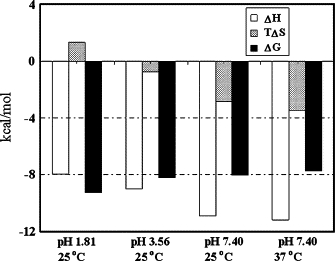
Comparative distribution of ΔH (left column) and TΔS (middle column) in ΔG (right column) at pH 1.81 (25°C), 3.56 (25°C), and 7.40 (25°C and 37°C).
As described above, the unfolding of HSA at pH 1.81 increases the distance among positively charged AARs. In contrast, ΔS becomes more negative with increasing pH. This indicates that PS binding destroyed the internal hydrophobic interactions in HSA at pH values above 3.56, replacing them with ion pair attraction and H-bonds. The chemical structure of PS shows that all the aryls are enclosed by hydrophilic groups (−SO3—, −N=N−, and −OH), and PS has no lipophilic substituent, e.g., the halogen or alkyl group. Thus, hydrophobic interactions between PS and HSA were probably weaker than the internal ones in HSA. On the other hand, PS binding could have induced folding of HSA, decreasing the entropy. In addition, comparison of the thermodynamic parameters of the solution between 37°C and 25°C at pH 7.40 shows that −ΔH becomes more positive but ΔS becomes more negative (Table 1 and Fig. 6). This indicates that ion pair attraction and H-bonds were strengthened, but hydrophobic interaction was weakened at normal physiological pH.
Fixing of the position of PS by ion pair attraction induces the formation of a combination of multiple bonds
Electrostatic forces have an important role in the binding of ligands to proteins (16), especially in acidic media. However, the relationship between the number of anionic groups on a ligand and the number of basic AARs on a protein has not yet been determined. From ϕ = N/Nb, the effective binding rate (ϕ) of PS to HSA can be calculated, where Nb is the total number of protonated basic AARs of HSA (Lys, His, and Arg). The dissociation constants of the R groups of Lys, His, and Arg indicate that they are all positively charged when the pH is <6. However, only the R groups of Lys and Arg residues are protonated in neutral medium. Thus, Nb is 98 in acidic solution and 79 in neutral solution.
In a more acidic solution, the peptide chain of a protein unfolds (16), and there is little steric hindrance. If the binding of PS to HSA depended only on ion pair attraction, ϕ should approach 100%. From the N values obtained above, ϕ of PS was calculated to be 31% at pH 1.81, 27% at pH 3.56, and 2.2% at pH 7.40. Because each PS molecule has four sulfonate groups, it can bind to at most four basic AARs, so all the basic AARs in HSA could bind to PS at pH 1.81 and 3.56. However, most of the PS molecules bound to the peptide chain via three sulfonates, i.e., three ionic bonds, at pH 1.81, whereas most of them bound via four sulfonates at pH 3.56. HSA tends to fold at pH 7.40, so steric hindrance increases, leading to a marked decrease of ϕ even though PS may still bind via four ionic bonds. From the number of bonds formed by PS, the PS-HSA binding energy is low at pH 1.81, as confirmed by the ΔH values above. In conclusion, ion pair attraction could have induced PS to enter the cleft of HSA, i.e., electrostatic attraction plays a position-fixing role in the interaction of the small ligand with the protein.
Once the PS molecules are positioned in HSA, other noncovalent bonds—H-bonds, hydrophobic interactions, and van der Waals forces—could act in combination. PS contains other potential binding groups in addition to the negatively charged −SO3— groups, e.g., −N=N− and −OH, which could form H-bonds with polar side groups of AARs in the peptide chain, but there will be little hydrophobic interaction between PS and HSA, as described above. An illustration of PS bound to the peptide chain in acidic solution is shown in Fig. 7. Thus, the combined action of noncovalent bonds will bind the small ligand firmly. As an example, the side group of K195 in the IIA domain and those of K446 and H464 in the IIIA domain would fix PS by ion pair bonds, and then the side groups of S193, Q198, and E465 would bind the PS by H-bonds. In a neutral solution, pH 7.40, the negative R groups of Asp and Glu, which are distributed throughout the peptide chain, will exclude the entry of PS anions even though the positive Rs of Lys and Arg residues attract them. Possibly only two spatial positions can be occupied by the PS anion, i.e., between K212–R218 and R348–K351, K402–R410 and R428–K432, which have no negative R groups around them. This is consistent with the number of PS molecules bound at pH 7.40.
Figure 7.
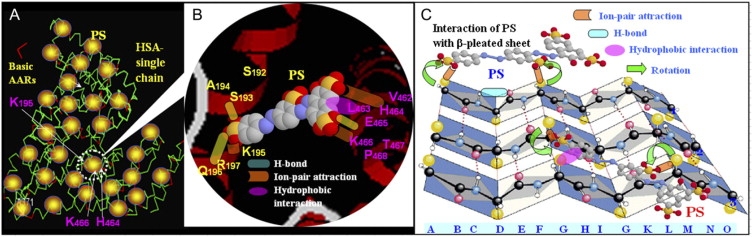
Cartoon illustrating the possible binding sites of PS in HSA and the corresponding bonds. (A) HSA single chain; red line represents positively charged amino acid residues. (B) As an example, PS is inserted between two α-helices (one from S192 to R197 in the IIA domain, and the other in green from V462 to P468 in the IIIA domain), replacing the original internal noncovalent bonds and forming ion pair bonds (green concave-convex rectangle), H-bonds (yellow rounded rectangle), and possibly hydrophobic interactions (red ellipse). (C) Cartoon illustrating how the secondary structure of HSA changed from β-sheet to helix (PS binding with the side groups (Rs) on lines 2 and 3) and turn (PS binding with the Rs on line 1 only) in PS solution. Lines 1, 2, and 3: peptide chain, and Rank A-P: AAR sequence.
Effects of electrolyte, temperature, and in situ pH change
The stability of a noncovalent complex is always affected by properties of the liquid environment in the body such as pH, ionic strength, and temperature (45,46). The effect of electrolytes on the γ value of PS in the HSA-PS interaction is shown in Fig. 8 A. With increasing electrolyte concentration, γ decreases in various pH media and the change from curve 3 was most evident at pH 7.40. Its value in 1.0 mol/l electrolyte is 70% that in the absence of electrolyte at pH 1.81 and pH 3.56, but only 25% at pH 7.40. This is due to the Debye-Huckel screening, where the Debye length is inversely proportional to the square root of the ionic strength of solution (47). Human blood normally contains ∼0.15 mol/l electrolyte. From all curves in A, γ is only slightly different in such a solution from γ in the absence of electrolyte. This implies that the ions in body fluids would not affect PS binding. When the ion concentration is increased to >0.5 mol/l, the marked adsorption of PS suggests that such conditions could be favorable for transmission of endogenous substances and drugs.
Figure 8.
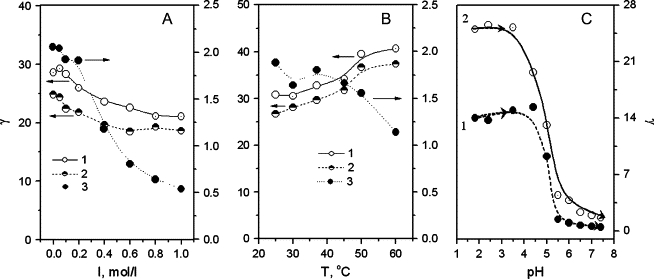
Effects of (A) electrolyte, (B) temperature, and (C) pH on γ of solutions. Both A and B contained 0.030 mmol/l PS and 50.0 mg/l HSA (1 and 2) or 200 mg/l HSA (3) at (1) pH 1.81, (2) pH 3.56, and (3) pH 7.40. (C) The pH of the solution (25 ml) was adjusted in situ step-by-step with 1 mol/l and 0.1 mol/l NaOH, where solution 1 contained 60.0 mg/l HSA and 0.015 mmol/l PS and solution 2 contained 60.0 mg/l HSA and 0.060 mmol/l PS.
At higher temperature, the protein cleft expands, increasing the distance between the adjacent peptide chains. Thus, the effective number of ligand binding sites is reduced, often causing desorption of ligand from the protein (48). However, the interaction of PS with HSA seems contradictory. At pH 7.4, γ decreases from curve 3 in Fig. 8 B, especially when the temperature is >40°C. At 37°C, γ decreases to ∼1.7 from ∼1.9 at room temperature. In contrast, curves 1 and 2 indicate that γ increases slightly with warming. A PS-HSA complex with a high binding number would form aggregates so that the PS would be physically adsorbed on the particle surfaces.
The slightness of the effects of ionic strength <0.2 mol/l and temperature <40°C indicates that PS binds strongly to HSA although the interaction is noncovalent. Thus, the penetration of PS-like organic structures into the blood could inhibit the endogenous transport of drugs. To investigate the stability of the product with the highest PS binding number at pH 1.81, a PS-HSA solution prepared at pH 1.81 was measured and then the pH was adjusted in situ step-by-step with 1 mol/l and 0.1 mol/l NaOH from pH 1.81 to 7.40. γ was calculated at each pH by Eq. 4, and its variation is shown in Fig. 8 C. The γ values in two such solutions, containing saturating (solution 1) and semisaturating (solution 2) concentrations of PS, approach those in Fig. 3 B obtained from PS-HSA solutions prepared independently at pH 3.56 and 7.40. Moreover, γ decreases markedly when the pH exceeds the isoelectric point of HSA. This is attributed to the unfolding of HSA in acidic media and the protonation of basic AARs, which is less at pH values above 4.7. In addition, the PS-HSA complex formed at pH 1.81 can be destroyed by low pH, as indicated by the trend in γ, i.e., binding to HSA is reversible and has not caused permanent denaturation.
Effect of PS on the secondary structure of HSA
The specific conformation of a protein with a particular function results from covalent and noncovalent interactions among its amino acid residues. When an organic compound such as a pollutant, drug, or toxin is added to a protein solution, the internal noncovalent interactions of the peptide chain may be altered and possibly destroyed, changing the original conformation. In particular, strong binding between a protein and an organic ligand may cause a permanent and irreversible change in the conformation and loss of the original function. CD spectrometry is often used to evaluate the secondary structure of a protein, i.e., the contents of β-pleated sheet, β-turn, α-helix, and random coil. The molar ellipticity CD curves of the PS-HSA solutions in various pH media are shown in Fig. 9, and the analytical results are summarized in Table 2.
Figure 9.
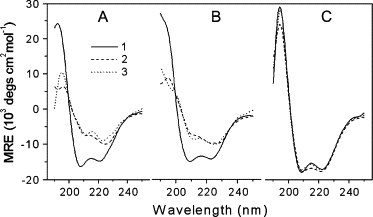
The molar ellipticity CD curves for HSA (0.030 mg/ml) solutions at (A) pH 1.81, (B) pH 3.56, and (C) pH 7.40, containing PS: (1) 0, (2) 0.015, and (3) 0.060 mmol/l in both A and B and (1) 0, (2) 0.56, and (3) 2.25 μmol/l in C.
Table 2.
Variation of the secondary conformational factors of HSA in the presence of PS in various pH media
| Rate, % |
||||
|---|---|---|---|---|
| pH | Factor | no PS | 0.015 mmol/l | 0.060 mmol/l PS |
| 1.81 | α-helix | 29.3 | 35.6 | 39.1 |
| β-sheet | 35.2 | 0 | 0 | |
| Turn | 4.3 | 30.1 | 32 | |
| Random coil | 31.2 | 34.3 | 29 | |
| 3.56 | α-helix | 37.8 | 42.4 | 45 |
| β-sheet | 28.6 | 0 | 0 | |
| Turn | 5.8 | 30.7 | 31.5 | |
| Random coil | 27.8 | 26.9 | 23.5 | |
| 7.40 | α-helix | 32.2 | 34.1* | 31.5† |
| β-sheet | 26 | 16.5* | 27.2† | |
| Turn | 16.4 | 20* | 15.5† | |
| Random coil | 25.4 | 29.4* | 25.8† | |
All of the solutions contained 0.030 mg/ml HSA.
Contained 0.56 μmol/l PS.
2.25 μmol/l PS.
The β-sheet content of HSA decreased to 0 at pH 1.81 and 3.56 with less than saturating concentrations of PS (15 μmol/l), where ∼80% of all available binding sites in HSA were bound by PS. The disappearance of β-sheet resulted in a marked increase in β-turn content, for example, from 4.3% at pH 1.81 and 5.8% at pH 3.56 to 30.1% and 30.7%. Moreover, the helix content increased by ∼20%. Comparison between saturating and subsaturating concentrations of PS produced no obvious change in secondary structure contents though 4 times the molarity of PS was added. Thus, a small amount of PS leads to a marked change of protein conformation in acidic media. Fig. 7 C illustrates how the binding may affect changes from β-pleated sheet to helix and turn. On a single peptide chain (chain 1), PS could bridge the side groups (Rs) CA1 (rank A and line 1) and CG1 by ion pair attraction.
Thus, both the A–C and E–G sector sheets would rotate inversely around CD1 to split the original H-bonds between chains 1 and 2, leading to formation of an H-bond between C=O (E1) and NH (C1). Thus, the pleated sheet is changed into a turn. If both Rs were located on chains 2 and 3, such as CG2 and CM3, respectively, ion pair attraction would draw both of them close to the two available −SO3— groups of PS by inverse rotation. Because of the perturbation caused by PS, the original H-bonds between chains 2 and 3 would be destroyed and a new H-bond between −NH (I3) and −OH of PS formed. Therefore, the β-sheet was changed into helix. In contrast, all the secondary conformational factors changed little at pH 7.40 even with saturating PS concentrations. This is presumably because there is too little binding of PS at pH 7.40 to affect the conformation of HSA markedly. As a deduction, the binding of PS to proteins or enzymes in acidic environments, e.g., stomach and skin secretions, might produce obvious toxicity. In normal blood, the tight binding of PS to HSA would inhibit the transmission of endogenous substances and drugs, though the secondary conformation of HSA would not be markedly altered.
Application of the interaction to protein assay
Calibration graphs and LOD
The characterization of the binding reaction and the effects of electrolyte and pH described above indicate that the interaction between PS and HSA is influenced by a range of experimental conditions such as ionic strength <0.2 mol/l, pH between 2 and 4, and temperature <40°C. Thus, it may be used for protein assay by LARVA (37), which has more than 10 times the analytical sensitivity of ordinary spectrophotometry. If EDTA is added to sequester most metal ions, this binding reaction is very selective. Three standard series of HSA were prepared, and the regression equations are given in Table 3. The LOD of HSA, defined as 3 times the standard deviation of 10 replicated blanks, was also calculated and is given in Table 3: the less PS added, the higher the sensitivity obtained. However, too low a PS concentration could cause an obvious measurement error because of background instrument noise. The LODs of series 2 and 3 are the lowest, but series 2 gives the best linearity. Thus, 3.0 μmol/l PS was added to determine the protein concentrations in samples such as food and blood. The corresponding LOD of protein is 0.2 mg/l.
Table 3.
The linear regression equations at pH 3.6 for HSA assay and LOD of HSA
| Series | PS, μmol/l | Linear scope of HSA, mg/l | Regression equation | R* | σ† | LOD‡, mg/l |
|---|---|---|---|---|---|---|
| 1 | 1.5 | 0–3.0 | ΔAr = 0.0491CM0 + 0.011 | 0.9867 | 0.0060 | 0.4 |
| 2 | 3.0 | 0–7.0 | ΔAr = 0.0354CM0 + 0.005 | 0.9980 | 0.0025 | 0.2 |
| 3 | 4.5 | 0–10.0 | ΔAr = 0.0237CM0 - 0.004 | 0.9969 | 0.0016 | 0.2 |
Linear correlation coefficient.
Standard deviation of 10 replicated reagent blanks.
LOD = 3σ/p (p-slope of line).
Effect of foreign substances
EDTA was used to sequester most of the metals that might occur in the samples. The results indicate that none of the following species affected the direct determination of 5.0 mg/l HSA (error less than ±5%) with PS: 100 mg/l lysine, glutamine, alanine; 100 mg/l K+, Cl−; 5.0 mg/l Ca2+, glucose; 2 mg/l Mg2+, vitamin C; 1 mg/l Zn2+, Fe2+, Fe3+; and 0.2 mg/l Cu2+. Therefore, the recommended method was selective and suitable for quantitative analysis of water-soluble proteins.
Analyses of samples
Five samples—quail egg white, milk, human blood, chicken serum, and eel serum—were determined. Both quail egg white and milk may be diluted and then colored directly. The other samples were treated according to the following procedures: 5 ml blood extracted from human or animal veins was centrifuged at 3000 rpm for 10 min and 1.00 ml of the supernatant was diluted to 100 ml. After mixing, protein was assayed. The results are given in Table 4. They agreed with the values obtained by classical Coomassie brilliant blue colorimetry (49). This indicates that the proposed method is accurate and reliable for practical analysis.
Table 4.
Determination of proteins in samples with PS at pH 3.60
| Sample* | HSA added, μg | Proteins found†, μg | Recovery, % | Protein content in sample, mg/ml |
|---|---|---|---|---|
| Quail egg white | 0 | 38.7 ± 1.7 | 38.7 | |
| 10 | 50.7, 48.4 | 120.1, 97.5 | ||
| Fresh milk | 0 | 58.3 ± 0.6 | 58.3 | |
| 10 | 69.8, 67.8 | 115, 95.2 | ||
| Personal serum | 0 | 20.1 ± 1.5 | 20.1 | |
| 10 | 31.2, 30.6 | 111, 105 | ||
| Chicken serum | 0 | 37.1 ± 1.4 | 37.1 | |
| 20 | 56.8, 57.7 | 98.7, 103.2 | ||
| Eel serum | 0 | 11.9 ± 2.4 | 11.9 | |
| 10 | 20.9, 22.3 | 90.0, 104.2 |
0.100 mL of the diluted solution of a sample was added.
Average of four replicated determinations in a 10-ml colorimetric flask.
Conclusions
Knowledge of biomolecular interactions such as protein-protein (50) and protein-ligand binding, enzyme catalysis, and inhibition is important for our understanding of cellular processes including signal transduction, gene regulation, and enzyme reactions (51). Such knowledge significantly improves our understanding of biological systems. More and more studies of protein-ligand interactions have led to increasingly widespread interest (52–54), where conventional molecular spectrometric methods such as fluorescent probe, UV, and CD are most widely used. Recently, binding mechanisms have been studied intensively using x-ray crystallography, NMR, ITC, and surface plasmon resonance biosensors (55,56), which are powerful analytical tools in enzymology, rational drug design, and toxicology. Ligand binding is often associated with denaturation of macromolecules (folding or unfolding).
Some modes of binding are exothermic (such as electrostatic interaction) and others are endothermic (such as unfolding). Thus, the primary interactions are usually electrostatic (16). The formation of covalent complexes with specific residues in a protein is being studied increasingly in, for example, aspects of DNA repair (57), identification of enzyme active sites (58), pharmaceutical development (59), and heavy metal toxicity (60). However, noncovalent interactions are more numerous in cells. Although a noncovalent bond is often weaker than a covalent bond, the combination of many noncovalent bonds will produce a stronger association. This work proposed that electrostatic interaction induced a combination of noncovalent bonds—H-bonds, hydrophobic interactions, and van der Waals forces—which was illustrated. In contrast to the binding of metal complexes and pesticides (21), noncovalent binding of organic compounds is unselective.
By characterizing the interaction of PS with HSA using various instrumentations and isothermal models, highly consistent results were obtained concerning, e.g., binding number, binding energy, and type of binding. PS formed a stable complex with HSA, and the combination of noncovalent bonds caused a marked change in the secondary conformation of HSA in acidic media. This is the first change process of this kind to be illustrated. The potentially toxic effect of PS was analyzed by determining PS-HSA binding under normal physiological conditions. The mechanism of interaction with the protein is very complicated. Although the crystal structures of a number of proteins have been analyzed and possible binding regions identified (2), it has not been possible to distinguish all the intermediate forms or to determine single noncovalent bond energies accurately. This work provides a useful experimental strategy for studying the interaction of organic substances with biomacromolecules. It helps us to understand the activity or mechanism of toxicity of an organic compound.
Acknowledgments
We thank Dr. Shao-Feng Luo of Univ. Sci. Technol. China for his help with ITC measurement and Dr. Xue-Ling Ao for her assistance in the use of the computer programs and data analysis.
We thank the Natural Science Foundation of China (Grant No. 20477030) and the Shanghai Fundamental Research Project (Grant No. 04JC14072) for financially supporting this work.
Footnotes
Editor: Jonathan B. Chaires.
References
- 1.Carter D.C., Chang B., Ho J.X., Keeling K., Krishnasami Z. Preliminary crystallographic studies of four crystal forms of serum albumin. Eur. J. Biochem. 1994;226:1049–1052. doi: 10.1111/j.1432-1033.1994.01049.x. [DOI] [PubMed] [Google Scholar]
- 2.He X.M., Carter D.C. Atomic structure and chemistry of human serum albumin. Nature. 1992;358:209–215. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 3.Shakai N., Garlick R.L., Bunn H.F. Nonenzymatic glycosylation of human serum albumin alters its conformation and function. J. Biol. Chem. 1984;259:3812–3817. [PubMed] [Google Scholar]
- 4.Carter D.C., Ho J.X. Structure of serum albumin. Adv. Protein Chem. 1994;45:153–203. doi: 10.1016/s0065-3233(08)60640-3. [DOI] [PubMed] [Google Scholar]
- 5.Brown J.R., Shockely P. In: Jost P.C., Griffith O.H., editors. Vol. 1. Wiley; New York: 1982. pp. 26–28. (Lipid-Protein Interaction). [Google Scholar]
- 6.Curry S., Mandelkow H., Brick P., Franks N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 1998;5:827–835. doi: 10.1038/1869. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharya A.A., Curry S., Franks N.P. Binding of the general anesthetics propofol and halothane to human serum albumin. J. Biol. Chem. 2000;275:38731–38738. doi: 10.1074/jbc.M005460200. [DOI] [PubMed] [Google Scholar]
- 8.Shakai N., Garlick R.L., Bunn H.F. Nonenzymatic glycosylation of human serum albumin alters its conformation and function. J. Biol. Chem. 1984;259:3812–3817. [PubMed] [Google Scholar]
- 9.Kragh-Hansen U. Molecular aspects of ligand binding to serum albumin. Pharmacol. Rev. 1981;33:17–53. [PubMed] [Google Scholar]
- 10.Bhattacharys A.A., Grüne T., Curry S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 2000;303:721–732. doi: 10.1006/jmbi.2000.4158. [DOI] [PubMed] [Google Scholar]
- 11.Zunszain P.A., Ghuman J., Komatsu T., Tsuchida E., Curry S. Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct. Biol. 2003;3:6. doi: 10.1186/1472-6807-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghuman J., Zunszain P.A., Petitpas I., Bhattacharya A.A., Otagiri M., Curry S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005;353:38–52. doi: 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 13.Petitpas I., Bhattacharya A.A., Twine S., East M., Curry S. Crystal structure analysis of warfarin binding to human serum albumin. J. Biol. Chem. 2001;276:22804–22809. doi: 10.1074/jbc.M100575200. [DOI] [PubMed] [Google Scholar]
- 14.Burk T.G., Mi Z. The structural basis of camptothecin interactions with human serum albumin: impact on drug stability. J. Med. Chem. 1994;37:40–46. doi: 10.1021/jm00027a005. [DOI] [PubMed] [Google Scholar]
- 15.Rhule J.T., Hill C.L., Judd D.A., Schinazi R.F. Polyoxometalates in medicine. Chem. Rev. 1998;98:327–358. doi: 10.1021/cr960396q. [DOI] [PubMed] [Google Scholar]
- 16.Ajloo D., Behnam H., Saboury A.A., Mohamadi-Zonoz F., Ranjbar B., Moosavi-Movahedi A.A., Hasani A., Alizadeh K., Gharanfoli M., Amani M. Thermodynamic and structural studies on the human serum albumin in the presence of a polyoxometalate. Bull. Korean Chem. Soc. 2007;28:733–736. [Google Scholar]
- 17.Ashbrook J.D., Spector A.A., Fletcher J.E. Medium chain fatty acid binding to human plasma albumin. J. Biol. Chem. 1972;247:7030–7042. [PubMed] [Google Scholar]
- 18.Sudlow G., Birkett D.J., Wade D.N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975;11:824–832. [PubMed] [Google Scholar]
- 19.Purcell M., Neault J.F., Malonga H., Arakawa H., Carpentier R., Tajmir-Riahi H.A. Interaction of atrazine and 2, 4-D with human serum albumin studied by gel and capillary electrophoresis, and FTIR spectroscopy. Biochim. Biophys. Acta. 2001;1548:129–138. doi: 10.1016/s0167-4838(01)00229-1. [DOI] [PubMed] [Google Scholar]
- 20.Berger V., Gabriel A.F., Sergent T., Trouet A., Larondelle Y., Schneider Y.J. Interaction of ochratoxin A with human intestinal Caco-2 cells: possible implication of a multidrug resistance-associated protein (MRP2) Toxicol. Lett. 2003;140:465–476. doi: 10.1016/s0378-4274(03)00043-2. [DOI] [PubMed] [Google Scholar]
- 21.Silva D., Cortez C.M., Cunhua-Bastos J., Louro S.R. Methyl parathion interaction with human and bovine serum albumin. Toxicol. Lett. 2004;147:53–61. doi: 10.1016/j.toxlet.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 22.Uddin S.J., Shilpi J.A., Murshid G.M.M., Rahman A.A., Sarder M.M., Alam M.A. Determination of the binding sites of arsenic on bovine serum albumin using warfarin (site-I specific probe) and diazepam (site-II specific probe) J. Biol. Sci. 2004;4:609–612. [Google Scholar]
- 23.Marolia K.Z., D'Souza S.F. Enhancement in the lysozyme activity of the hen egg white foam matrix by cross-linking in the presence of N-acetyl glucosamine. J. Biochem. Biophys. Methods. 1999;39:115–117. doi: 10.1016/s0165-022x(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 24.Pitzurra L., Marconi P., Bistoni F., Blasi E. Selective inhibition of cytokine-induced lysozyme activity by tetanus toxin in the GG2EE macrophage cell line. Infect. Immun. 1989;57:2452–2456. doi: 10.1128/iai.57.8.2452-2456.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ministry of Food. 1954. Food Standards Committee Report on Colouring Matters Recommendations Relating to the Use of Coloring Matters in Foods. H. M. S. O., London.
- 26.Skipper P.L., Tannenbaum S.R. Molecular dosimetry of aromatic amines in human populations. Environ. Health Perspect. 1994;102:17–21. doi: 10.1289/ehp.94102s617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Y.P., Zheng P., Tang J.S. Characterization of synthetic azo colorant in foodstuffs. Phys. Test. Chem. Anal. B. 2002;38:456–461. [Google Scholar]
- 28.Housaindokht M.R., Bahrololoom M., Tarighatpoor S., Moosavi-Movahedi A.A. An approach based on diffusion to study ligand-macromolecule interaction. Acta Biochim. Pol. 2002;49:703–707. [PubMed] [Google Scholar]
- 29.Bathaie S.Z., Moosavi-Movahedi A.A., Saboury A.A. Energetic and binding properties of DNA upon interaction with dodecyl trimethylammonium bromide. Nucleic Acids Res. 1999;27:1001–1005. doi: 10.1093/nar/27.4.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saboury A.A. Application of a new method for data analysis of isothermal titration calorimetry in the interaction between human serum albumin and Ni2+ J. Chem. Thermodyn. 2003;35:1975–1981. [Google Scholar]
- 31.Aspuru E.O.D., Zaton A.M. Usefulness of difference spectroscopy in the study of the binding of uracil derivatives to human serum albumin. J. Biochem. Biophys. Methods. 1993;27:87–94. doi: 10.1016/0165-022x(93)90052-p. [DOI] [PubMed] [Google Scholar]
- 32.Bordbar A.K., Sohrabi N., Tangestaninejad S. Study of interaction of potassium dodecatangestato cobaltate(iii) with bovine serum albumin using fluorescence spectroscopy. Phys. Chem. Liq. 2004;42:127–133. [Google Scholar]
- 33.Gao H.W., Jiang J., Yu L.Q. Study on spectrometric probe of protein solution with p-iodochlorophosphonazo as adsorbate by MPASC technique. Analyst. 2001;126:528–533. doi: 10.1039/b006760l. [DOI] [PubMed] [Google Scholar]
- 34.Dominiak P.M., Volkov A., Li X., Messerschmidt M., Coppens P. Theoretical databank of transferable aspherical atoms and its application to electrostatic interaction energy calculations of macromolecules. J. Chem. Theory Comput. 2007;3:232–247. doi: 10.1021/ct6001994. [DOI] [PubMed] [Google Scholar]
- 35.Kortemme T., Morozov A.V., Baker D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J. Mol. Biol. 2003;326:1239–1259. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 36.Gao H.W., Zhao J.F. Langmuir aggregation of thionin on sodium dodecyl sulfate and application. J. Anal. Chem. 2003;58:322–327. [Google Scholar]
- 37.Gao H.W., Wang C.L., Jia J.Y., Zhang Y.L. Continuous flow analysis combined with light absorption ratio variation spectrophotometry for determination of copper at ng/ml level in natural water. Anal. Sci. 2007;23:655–659. doi: 10.2116/analsci.23.655. [DOI] [PubMed] [Google Scholar]
- 38.Cong P.S., Zhu Z.L., Li T.H. The determination of ionization constants of polyprotic by using evolving factor analysis and pH-spectrophotometric data. Chem. J. Chin. Univ.-Chin. 1994;15:1292–1296. [Google Scholar]
- 39.Li L., Gao H.W., Ren J.R., Chen L., Li Y.C., Zhao J.F., Zhao H.P., Yuan Y. Binding of Sudan II and IV to lecithin liposomes and E. coli membranes: insights into the toxicity of hydrophobic azo dyes. BMC Struct. Biol. 2007;7:16. doi: 10.1186/1472-6807-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai S.Y., Lin S.C., Suen S.Y., Hsu W.H. Effect of number of poly(His) tags on the adsorption of engineered proteins on immobilized metal affinity chromatography adsorbents. Process Biochem. 2006;41:2058–2067. [Google Scholar]
- 41.Cooper A., McAlpine A., Stockley P.G. Calorimetric studies of the energetics of protein-DNA interactions in the E. coli methionine repressor (MetJ) system. FEBS Lett. 1994;348:41–45. doi: 10.1016/0014-5793(94)00579-6. [DOI] [PubMed] [Google Scholar]
- 42.Kamiya M., Torigoe H., Shindo H., Sarai A. Temperature dependence and sequence specificity of DNA triplex formation: an analysis using isothermal titration calorimetry. J. Am. Chem. Soc. 1996;118:4532–4538. [Google Scholar]
- 43.Luque I., Todd M.J., Gomez J., Semo N., Freire E. Molecular basis of resistance to HIV-1 protease inhibition: a plausible hypothesis. Biochemistry. 1998;37:5791–5797. doi: 10.1021/bi9802521. [DOI] [PubMed] [Google Scholar]
- 44.Yang M. Molecular recognition of DNA targeting small molecule drugs. J. Beijing Med. Univ. 1998;30:97–99. [Google Scholar]
- 45.Malkov V., Voloshin O., Soyfer V.N., Frank-Kamenetskii M.D. Cation and sequence effects on stability of intermolecular pyrimidine-purine-purine triplex. Nucleic Acids Res. 1993;21:585–591. doi: 10.1093/nar/21.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singleton S.F., Dervan P.B. Temperature dependence of the energetics of oligonucleotide-directed triple-helix formation at a single DNA site. J. Am. Chem. Soc. 1994;116:10376–10382. [Google Scholar]
- 47.Quinn S.J., Kifor O., Trivedi S., Diaz R., Vassilev P., Brown E. Sodium and ionic strength sensing by the calcium receptor. J. Biol. Chem. 1998;273:19579–19586. doi: 10.1074/jbc.273.31.19579. [DOI] [PubMed] [Google Scholar]
- 48.Gao H.W., Yang J.X., Jiang J. MPASC technique: a novel method for studying spectrometric probe of proteins with picramine CA as reactant. Supramol. Chem. 2002;14:315–321. [Google Scholar]
- 49.Lopez J.M., Imperial S., Valderrama R., Navarro S. An improved Bradford protein assay for collagen proteins. Clin. Chim. Acta. 1993;220:91–100. doi: 10.1016/0009-8981(93)90009-s. [DOI] [PubMed] [Google Scholar]
- 50.Almogren A., Furtado P.B., Sun Z., Perkins S.J., Kerr M.A. Purification, properties and extended solution structure of the complex formed between human immunoglobulin a1 and human serum albumin by scattering and ultracentrifugation. J. Mol. Biol. 2006;356:413–431. doi: 10.1016/j.jmb.2005.11.060. [DOI] [PubMed] [Google Scholar]
- 51.Kraut D.A., Carroll K.S., Herschlag D. Challenges in enzyme mechanism and energetics. Annu. Rev. Biochem. 2003;72:517–571. doi: 10.1146/annurev.biochem.72.121801.161617. [DOI] [PubMed] [Google Scholar]
- 52.Strombergsson H., Kryshtafovych A., Prusis P., Fidelis K., Wikberg J.E.S., Komorowski J., Hvidsten T.R. Generalized modeling of enzyme-ligand interactions using proteochemometrics and local protein substructures. Proteins. 2006;65::568–579. doi: 10.1002/prot.21163. [DOI] [PubMed] [Google Scholar]
- 53.Ciulli A., Abell C. Biophysical tools to monitor enzyme-ligand interactions of enzymes involved in vitamin biosynthesis. Biochem. Soc. Trans. 2005;33:767–771. doi: 10.1042/BST0330767. [DOI] [PubMed] [Google Scholar]
- 54.Lodarczyk J.W., Galitonov G.S., Kierdaszuk B. Identification of the tautomeric form of formycin A in its complex with Escherichia coli purine nucleoside phosphorylase based on the effect of enzyme-ligand binding on fluorescence and phosphorescence. Eur. Biophys. J. 2004;33:377–385. doi: 10.1007/s00249-003-0369-9. [DOI] [PubMed] [Google Scholar]
- 55.Eichmuller C., Tollinger M., Krautler B., Konrat R. Mapping the ligand binding site at protein side-chains in protein-ligand complexes through NOE difference spectroscopy. J. Biomol. NMR. 2001;20:195–202. doi: 10.1023/a:1011299009214. [DOI] [PubMed] [Google Scholar]
- 56.Jones R.B., Gordus A., Krall J.A., MacBeath G. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature. 2006;439:168–174. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 57.Liao Z.Y., Thibaut L., Jobson A., Pommier Y. Inhibition of human tyrosyl-DNA phosphodiesterase by aminoglycoside antibiotics and ribosome inhibitors. Mol. Pharmacol. 2006;70:366–372. doi: 10.1124/mol.105.021865. [DOI] [PubMed] [Google Scholar]
- 58.Brazeau B.J., Johnson B.J., Wilmot C.M. Copper-containing amine oxidases. Biogenesis and catalysis; a structural perspective. Arch. Biochem. Biophys. 2004;428:22–31. doi: 10.1016/j.abb.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 59.Yang X.X., Hu Z.P., Chan S.Y., Zhou S.F. Monitor drug-protein interaction. Clin. Chim. Acta. 2006;361:9–29. doi: 10.1016/j.cca.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 60.Giaginis C., Gatzidou E., Theocharis S. DNA repair systems as targets of cadmium toxicity. Toxicol. Appl. Pharmacol. 2006;213:282–290. doi: 10.1016/j.taap.2006.03.008. [DOI] [PubMed] [Google Scholar]