

Abstract
DNA-binding protein (DBP) of Autographa californica multiple nucleopolyhedrovirus (AcMNPV) was expressed as an N-terminal His6-tag fusion using a recombinant baculovirus and purified to near homogeneity. Purified DBP formed oligomers that were crosslinked by redox reagents resulting in predominantly protein dimers and tetramers. In gel retardation assays, DBP showed a high affinity for single-stranded oligonucleotides and was able to compete with another baculovirus SSB protein, LEF-3, for binding sites. DBP binding protected ssDNA against hydrolysis by a baculovirus alkaline nuclease AN/LEF-3 complex. Partial proteolysis by trypsin revealed a domain structure of DBP that is required for interaction with DNA and that can be disrupted by thermal treatment. Binding to ssDNA, but not to dsDNA, changed the pattern of proteolytic fragments of DBP indicating adjustments in protein structure upon interaction with ssDNA. DBP was capable of unwinding short DNA duplexes, and also promoted the renaturation of long complementary strands of ssDNA into duplexes. The unwinding and renaturation activities of DBP, as well as the DNA binding activity, were sensitive to sulfhydryl reagents and were inhibited by oxidation of thiol groups with diamide or by alkylation with N-ethylmaleimide. A high affinity of DBP for ssDNA and its unwinding and renaturation activities confirmed identification of DBP as a member of the SSB/recombinase family. These activities and a tight association with subnuclear structures suggests that DBP is a component of the virogenic stroma that is involved in the processing of replicative intermediates.
Keywords: baculovirus, SSB, DBP, LEF-3, replication, recombination
Introduction
Nucleopolyhedroviruses (NPVs) and granuloviruses (GVs) belong to the family Baculoviridae and contain circular double-stranded (ds) DNA genomes of 80 to 180 kb (for review see (Okano et al., 2006)). Baculoviruses are infectious for invertebrates and produce two types of virions, budded viruses (BV) that spread the infection within insects and are also infectious for cultured insect cells and occlusion-derived virions (ODV) that mediate transmission between insects. Replication of baculovirus genomes is initiated in a limited number of nuclear foci in close proximity to nuclear domain 10 (ND10) or promyelocytic leukemia protein (PML) domains (Okano et al., 1999; Mainz et al., 2002). Although a rolling circle model was proposed for baculovirus replication (Leisy and Rohrmann, 1993; Oppenheimer and Volkman, 1997), the actual mechanism of replication remains unclear. The best studied baculovirus is the Autographa californica multiple nucleopolyhedrovirus (AcMNPV) that is widely used for expression of foreign genes under control of the polyhedrin (polh) promoter. The AcMNPV genome of 134 kb encodes approximately 155 genes (Ayres et al., 1994) including seven genes essential for viral DNA replication, ie-1 (a transactivator of early gene transcription IE-1), dnapol (DNA polymerase), hel (P143, DNA helicase), lef-1 (LEF-1 - Late Expression Factor 1, DNA primase), lef-2 (primase-associated factor LEF-2), lef-3 (a single-stranded DNA-binding protein (SSB) LEF-3) (Kool et al., 1994; Lu and Miller, 1995), and lef-11 (factor LEF-11 with unknown function) (Lin and Blissard, 2002). Some other viral products including an inhibitor of apoptosis, P35, a transcriptional transactivator, PE-38, and LEF-7 are stimulatory in transient replication assays, but may be indirectly involved in virus replication (for review see (Okano et al., 2006)). The replicative protein LEF-3 is a multifunctional SSB that serves as an accessory factor for DNA polymerase (McDougal and Guarino, 1999), forms complexes with alkaline nuclease AN (Mikhailov et al., 2003) and helicase P143 (Wu and Carstens, 1998; Evans et al., 1999; Ito et al., 2004), and facilitates transport of helicase into nuclei (Chen and Carstens, 2005). AcMNPV LEF-3 has a molecular mass of 44.5 kDa and forms oligomers in solution (Evans and Rohrmann, 1997). It binds specifically to ssDNA (Hang et al., 1995) and promotes Mg2+ and ATP-independent unwinding of partial DNA duplexes (Mikhailov, 2000; Mikhailov et al., 2005). Both LEF-3 activities, DNA binding and unwinding, are highly sensitive to redox agents and are inhibited by the oxidation or alkylation of thiol groups in the protein (Mikhailov et al., 2005).Thermally unfolded LEF-3 promotes renaturation of complementary strands, and is active in strand exchange reactions (Mikhailov et al., 2006). All these properties are consistent with the involvement of LEF-3 as a principal DNA-binding factor in the replication and recombination of baculovirus genomes.
A second SSB, called DNA-binding protein (DBP) was originally discovered in cells infected with Bombyx mori NPV (BmNPV). Although unrelated in amino acid sequence, DBP is similar to LEF-3 in that it binds ssDNA, forms oligomers, and destabilizes DNA duplexes (Mikhailov et al., 1998). The functional similarity of DBP and LEF-3 raises questions concerning the different roles these two proteins play in the baculovirus infection cycle. Homologs of dbp have been identified in all baculovirus genomes sequenced, except for one infectious for a dipteran, that also lacks a homolog of lef-3 (Okano et al., 2006). The NPVs of Lymantria dispar (LdNPV) (Kuzio et al., 1999) and Ectropis obliqua (EcobNPV) (Ma et al., 2007) contain two copies of the dbp gene. AcMNPV DBP (the product of ORF 25) has a molecular mass of 36.7 kDa (316 amino acids), and is closely related to BmNPV DBP (95.9% amino acid sequence identity). DBP is expressed as an early gene product that is more abundant than LEF-3 in cells infected with BmNPV. It was not found among the structural proteins from budded or occlusion-derived virions (Okano et al., 1999). In nuclei, both LEF-3 and DBP colocalize with IE-1 in viral replication foci (Okano et al., 1999; Mainz et al., 2002; Nagamine et al., 2006), that progressively enlarge during the course of infection eventually occupying more than half of the nucleus (Okano et al., 1999). The resulting electron dense structure is called the virogenic stroma. Upon fractionation of nuclei in aqueous solutions, both LEF-3 and DBP are found in a fraction of proteins that tightly binds to chromatin (Vanarsdall et al., 2007). In cells infected with AcMNPV, DBP is essential for production of viable virions, but in contrast to LEF-3, it is not required for synthesis of viral DNA or expression of viral genes (Vanarsdall et al., 2007; Quadt et al., 2007). However in the absence of a functional dbp gene, the virogenic stroma appeared to be absent and the yield of viral DNA was substantially decreased and no full-size viral genomes were produced (Vanarsdall et al., 2007) indicating that DBP may participate in the formation of the virogenic stroma and stabilize nascent viral DNA or participate in processing of replicative intermediates.
In this report, we describe purification and biochemical analysis of AcMNPV DBP. This protein showed a high affinity for ssDNA and possessed both unwinding and renaturation activities thus confirming its initial identification as a member of a diverse family of SSBs. Similar to LEF-3, DBP formed oligomers in solution and its activities were sensitive to sulfhydryl agents. However, DBP associated more tightly than LEF-3 with subnuclear structures and appeared to function at later stages in baculovirus replication than LEF-3.
Results
Purification of AcMNPV DBP
DNA-binding protein (DBP) was originally described as an abundant early viral protein possessing a high affinity for ssDNA in BmN cells infected with BmNPV (Mikhailov et al., 1998). In order to isolate DBP of AcMNPV from Sf9 cells, we initially used the protocol adopted for BmNPV-infected BmN cells that includes isolation of cell nuclei, salt extraction of nuclear proteins, and column chromatography on ssDNA-cellulose and DEAE-Toyopearl. However, this approach resulted in low yields (data not shown). Further analysis revealed that the low yield of DBP under this purification protocol was affected by two factors. Firstly, DBP appeared to be less abundant in Sf9 cells infected with AcMNPV than in BmN cells infected with BmNPV. A quantitative Western blot analysis performed as previously described (Mikhailov et al., 1998) showed that an average Sf9 cell infected with AcMNPV contains approximately 3 × 107 molecules of DBP at 20 hpi, significantly lower than the amount of DBP (7 × 107 molecules) in the BmN cell infected with BmNPV at 14 hpi (Okano et al., 1999). Secondly, the treatment with high salt released only a minor fraction of DBP from the nuclei. A major DBP fraction remained associated with chromatin even after repeated washing with buffers containing 1.4 to 2 M NaCl (Fig. 1A). Most LEF-3, another viral DNA-binding protein that also has a high affinity for ssDNA, was extracted by 0.6 to 1 M NaCl (Fig. 1A) indicating that DBP is more tightly associated with nuclear structures than LEF-3. Despite a tight association of DBP with components of the nucleus, about half of the cellular pool of this protein was recovered in a cytoplasmic fraction of Sf9 cells infected with AcMNPV (Vanarsdall et al., 2007). Therefore in the subsequent experiments, we excluded salt extraction of DBP from nuclei because of its inefficiency and used the cytoplasmic fraction as a source for purification of DBP. Using this approach, about 30 μg of pure DBP was obtained from 500 ml of AcMNPV-infected Sf9 cells collected at 22 hpi (Fig. 1B). This protein is referred as wild type DBP (wtDBP) in this report.
Figure 1.
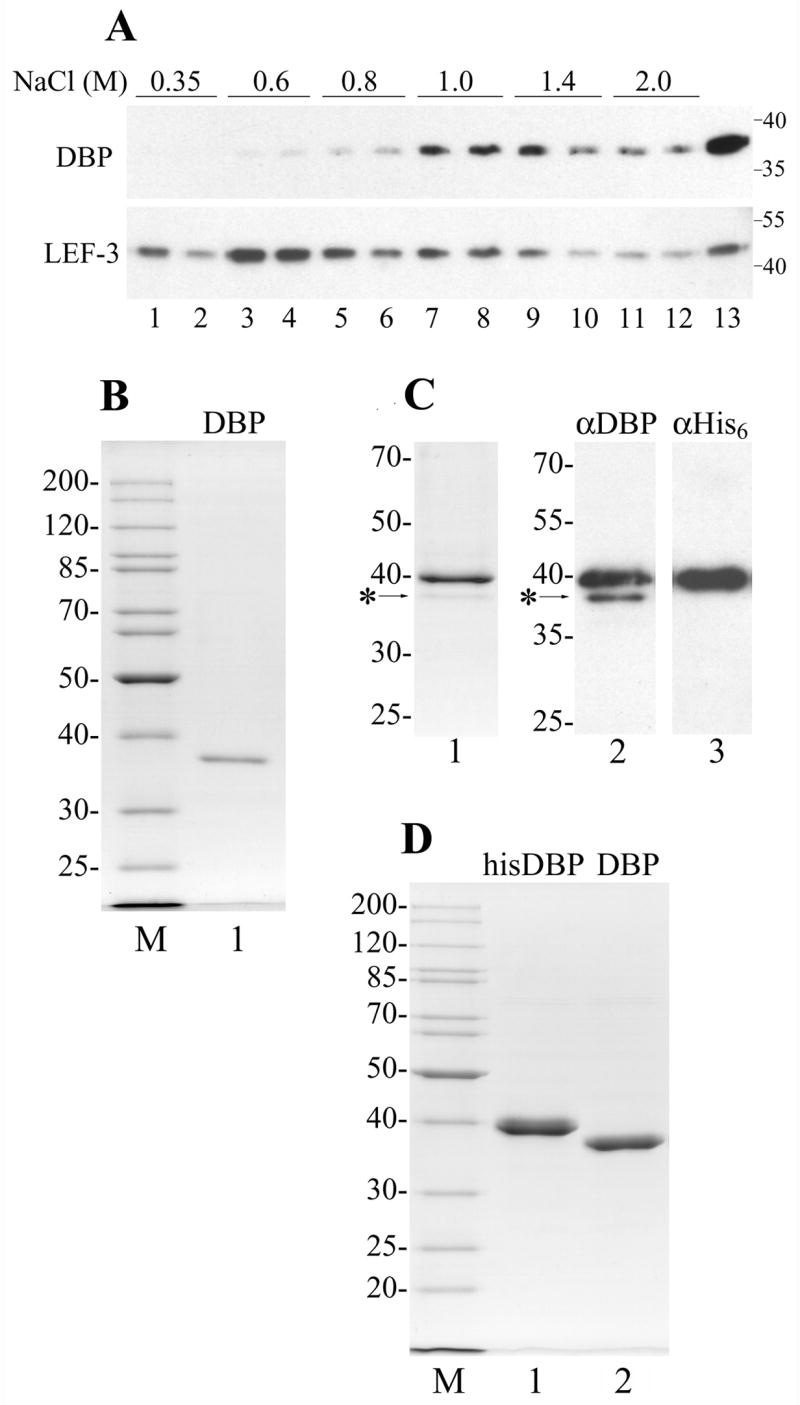
Purification of AcMNPV DBP. (A) Chromatin was obtained from 3 × 107 cells infected with AcMNPV at 20 hpi and was sequentially extracted with buffers containing NaCl as indicated. Each extraction step was repeated twice. Lane 13 shows the residue after the chromatin extraction with salt. After precipitation of insoluble material, the supernatants were subjected to SDS-11% PAGE followed by Western blotting with polyclonal antibodies to DBP and LEF-3. The molecular weight standards (in kDa) are shown on the right. (B) DBP was purified from the cytoplasm of Sf9 cells infected with wild type AcMNPV by column chromatography on ssDNA-cellulose and DEAE-Toyopearl. 230 ng were subjected to SDS-10% PAGE followed by Coomassie staining (lane 1). (C), Copurification of his-tagged DBP with wild type DBP. Lane 1, 400 ng of his-tagged DBP purified by chromatography on Ni-NTA column was subjected to SDS-11% PAGE followed by Coomassie staining. Lanes 2 and 3, 80 ng of the his-tagged DBP was subjected to SDS-10% PAGE followed by Western blotting with polyclonal antibodies to DBP (lane 2) or with monoclonal antibodies to His6 (lane 3). The position of wild type DBP is indicated by the asterisk. (D) The his-tagged DBP was purified from Sf9 cells infected with the recombinant virus vAcHisDBP by column chromatography on Ni-NTA agarose and DEAE-Toyopearl. 1.96 μg of the his-tagged DBP (lane 1) and 1.6 μg of the his-tagged DBP treated with AcTEV protease (lane 2) were subjected to SDS-11% PAGE. Lanes M in panels B and D show molecular weight standards.
In order to obtain DBP in amounts sufficient for experimentation, it was expressed as an N-terminal His6-tag fusion under control of the polh promoter in a recombinant baculovirus, vAcHisDBP. The Sf9 cells infected with vAcHisDBP were collected 3 days post infection. The lysate was applied to a Ni-NTA agarose column and the his-tagged DBP was eluted with imidazole. SDS-PAGE analysis of fractions collected from the Ni-NTA agarose revealed a major band of his-tagged DBP (39.7 kDa) and a minor band with mobility corresponding to DBP without the his-tag (36.7 kDa, marked by the asterisk in Fig. 1C). Western blot analysis confirmed that the upper major band reacts with antibodies to both DBP (αDBP) and the His6-tag (αHis6), whereas the lower minor band reacts only with αDBP (Fig. 1C, lanes 2 and 3). Because only the his-tagged proteins are specifically retained by Ni-NTA agarose, the presence of untagged DBP in the samples indicated that monomers of the his-tagged DBP and untagged DBP may interact with each other. The his-tagged DBP was expressed under the control of the very late polh promoter, whereas the untagged DBP was presumably the wild type protein expressed under control its own early promoter. This was confirmed by a decrease in the ratio of the untagged to the his-tagged DNA in the course of infection cycle. When the samples from Ni-NTA agarose were compared for cells infected with vAcHisDBP and collected at three time points, 24, 48, and 72 hpi, the ratio of the untagged to tagged DBP was maximal at 24 hpi and minimal at 72 hpi (data not shown). For further purification, the DBP samples collected from Ni-NTA agarose were subjected to chromatography on a DEAE-Toyopearl column. This procedure allows removal of contaminating proteins and separates hisDBP from the untagged protein. The his-tagged DBP eluted at NaCl concentrations of 150 to 200 mM, whereas most of the untagged DBP eluted at lower concentrations (50 to 70 mM). The elution at 50 to 70 mM NaCl from DEAE-Toyopearl resin was observed also for wtDBP from AcMNPV-infected cells (data not shown) and for BmNPV DBP (Mikhailov et al., 1998). Approximately 2.4 mg of the his-tagged DBP (hisDBP) was purified from a 100-ml culture of Sf9 cells (Fig. 1D, lane 1). A portion of the his-tagged DBP was treated with AcTEV protease to remove the N-terminal fragment of 23 amino acids that includes the His6-tag thus leaving only two extra amino acids on the N-terminus of DBP (Fig. 1D, lane 2). The hisDBP sample treated with the protease is referred to as DBP in this report, and it was used as a control in experiments with the his-tagged DBP.
DNA binding of DBP
The 5′-labeled oligonucleotides dT34 and 62 mer were used as probes for the analysis of DBP interaction with DNA by using electrophoretic mobility shift assay (EMSA). A binding site size of about 30 nt was previously estimated for the BmNPV DBP monomer (Mikhailov et al., 1998). Therefore dT34 should provide a probe for one DNA-binding site of DBP. Binding of hisDBP to dT34 generated complexes migrating very slowly in the polyacrylamide gel (Fig. 2A). In the presence of reducing agent (5 mM DTT), the half saturation of 1 nM dT34 was achieved at a hisDBP concentration of 5 × 10−8 M (Fig. 2B, line 1). This concentration represents the Kd value in the case when DBP has one DNA-binding site per protein monomer. Oxidation of hisDBP with the sulfhydryl reagent diamide markedly reduced the protein binding. The half saturation of dT34 in the presence of 10 mM diamide was observed at a DBP concentration of 1.6 × 10−6 M (Fig. 2B, line 3). Modification of cysteines in hisDBP with N-ethylmaleimide (NEM) also dramatically decreased the protein affinity for ssDNA. The half saturation of dT34 with hisDBP pretreated by 5 mM NEM and then incubated with dT34 at reducing conditions was observed at protein concentration of 3.1 × 10−6 M (Fig. 2B, line 4). The control DBP sample from which the his-tag was removed showed an affinity for dT34 similar to that of the his-tagged DBP at different redox conditions. The half saturation of dT34 by the control DBP was observed at 5.4 × 10−8 M at the reducing conditions (Fig. 2B, line 2), at 1.6 × 10−6 M at the oxidizing conditions, and at 4.7 × 10−6 M for protein alkylated with NEM (data not shown). These results suggested that reduced cysteines in DBP are essential for the efficient interaction of this protein with DNA and that the N-terminal His6-tag sequence does not interfere with DBP binding to DNA.
Figure 2.
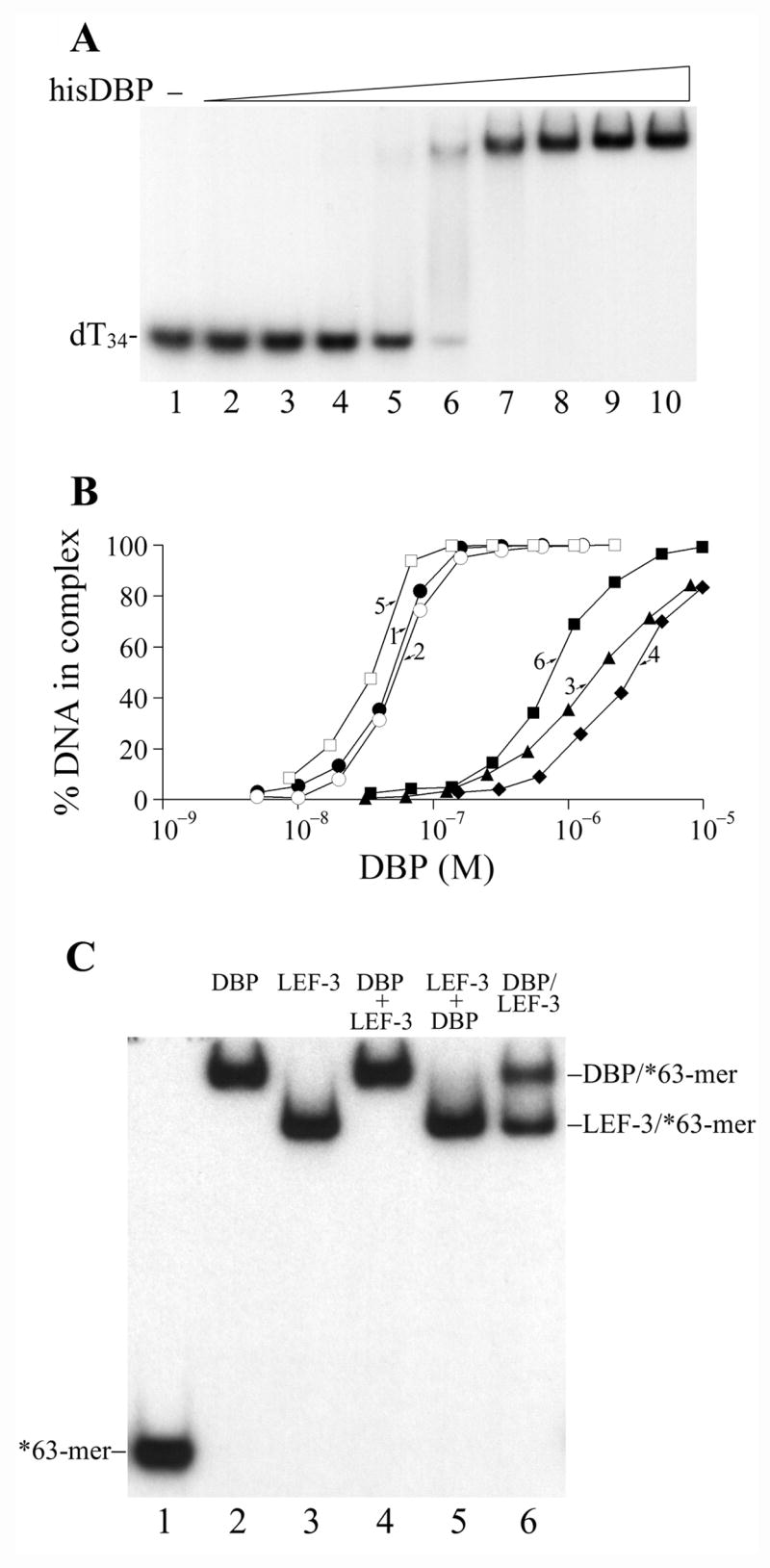
Electrophoretic mobility shift assay of DBP binding to oligonucleotides. (A) The binding assay was carried out in 10-μl reaction mixtures containing 1 nM of 32P-labeled dT34, 30 mM Tris-HCl, pH 7.5, 50 mM NaCl, 5 mM DTT, 12.5% glycerol, 0.5% Nonidet P-40, and 100 μg/ml BSA. The His6-tagged DBP was added in the following concentrations: control, no DBP (lane 1), 5 nM (lane 2), 10 nM (lane 3), 20 nM (lane 4), 40 nM (lane 5), 80 nM (lane 6), 160 nM (lane 7), 320 nM (lane 8), 640 nM (lane 8), and 1280 nM (lane 10). The mixtures were incubated for 15 min at 22ºC and then analyzed by EMSA. (B) DBP binding to 32P-labeled dT34 and 62-mer (each of 1 nM) analyzed by EMSA. Line 1, dT34, his-tagged DBP. The gel shown in panel A was used for quantification. Line 2, dT34, DBP with the his-tag removed by AcTEV protease. Line 3, dT34, his-tagged DBP. Reaction in the presence of 10 mM diamide. Line 4, dT34, his-tagged DBP pretreated by 5 mM N-ethylmaleimide. Line 5, ss62-mer, his-tagged DBP. Line 6, ds62-mer, his-tagged DBP. All reactions used to generate these data, except line 3, were carried out in the presence of 5 mM DTT. (C) Competition of DBP and LEF-3 for binding to 63-mer oligonucleotide. The binding assay was carried out in 10-μl reaction mixtures containing 20 mM Tris-HCl, pH 7.5, 50 mM NaCl, 5 mM DTT, 12.5% glycerol, 0.5% Nonidet P-40, and 50 μg/ml BSA. DNA-binding proteins, DBP (lanes 2 and 4), LEF-3 (lanes 3 and 5), and both DBP and LEF-3 (lane 6) were each at concentration of 0.5 μM. After addition of 32P-labeled 63-mer (1 nM), the mixtures were incubated for 15 min at 22ºC. Then LEF-3 and DBP each at concentration 0.5 μM were added to the reactions shown respectively in lanes 4 and 5, and the incubation was continued for 15 min. The reactions were analyzed by EMSA.
The affinity of DBP for ssDNA and dsDNA was compared by using the ss62-mer and ds62-mer oligonucleotides as probes for protein binding. The hisDBP formed one slowly migrating band with the 62-mers similar to the complexes of hisDBP with dT34 shown in Fig. 2A (data not shown). The half saturation of 1 nM 62-mer was observed at hisDBP concentration of 3.6 × 10−8 M (Fig. 2B, line 5). The hisDBP affinity for ds62-mer was much lower than that for ss62-mer, and the half saturation of ds62-mer was achieved at 7.6 × 10−7 M (Fig. 2B, line 6). At the oxidizing conditions, the protein affinity for ss62-mer and ds62-mer was much lower than that at the reducing conditions. In the presence of 10 mM diamide, the half saturation of ss62-mer and ds62-mer with hisDBP was achieved at concentrations of 3.4 × 10−7 M and 1.7 × 10−6 M, respectively (data not shown). These results confirmed the preferential binding of baculoviral DBP to ssDNA and revealed the requirement for reduced cysteines in DBP for efficient interaction with DNA.
The DBP concentrations that saturated dT34 and 62-mers are close to the concentrations required for saturation of these oligonucleotides by LEF-3 (Mikhailov et al., 2005). These data confirmed that similar to LEF-3, DBP has a high affinity for DNA. Because both proteins are abundant in Sf9 cells infected with AcMNPV, they could be competitors for DNA in infected cells. To address this question, we tested the binding of each protein to the ss63-mer in the presence of excessive amounts of the other SSB as competitor (Fig. 2C). DBP and LEF-3 were each added at oversaturating concentrations (0.5 μM) to mixtures containing 1 nM 63-mer. In the absence of competitor, both proteins formed uniform complexes with 63-mers that migrated in polyacrylamide gels at different positions (Fig. 2C, lanes 2–3). When complexes of each protein with 63-mer oligos were formed before addition of the competitor, the protein added last was incapable of replacing the first protein in the complexes under incubation for at least 15 min and was incapable of interacting efficiently with oligos saturated with the other protein (Fig. 2C, lanes 4–5). When 63-mers were added to the equimolar mixture of both proteins, DBP and LEF-3 had approximately the same probability to form a complex with the oligos (Fig. 2C, lane 6). Similar experiments performed with a shorter DNA probe (dT34) and at higher salt concentration (0.2 M instead of 50 mM NaCl) indicated that LEF-3 may be a more efficient competitor than DBP for binding to short DNA fragments or at higher ionic strength (data not shown). These data confirmed that DBP and LEF-3 could compete for ssDNA in infected cells.
Analysis of the thiol groups in DBP
As shown in Fig. 2B, oxidation of cysteine residues in DBP by diamide dramatically reduced protein binding to DNA. The reduced thiol groups of cysteines might be required for structural transitions in DBP that accompany binding to DNA or they might be essential for the structural integrity of this protein. AcMNPV DBP contains six cysteines located at positions 51, 88, 90, 101, 147, and 203. To elucidate possible structural changes in DBP upon the oxidation of cysteines, we analyzed protein treated by diamide by nonreducing SDS-PAGE (Fig. 3A). After incubation with diamide, hisDBP formed oligomeric species with mobility corresponding to protein dimers (31%), tetramers (12%), and higher oligomers (~2%) (Fig. 3A, lane 2). Oxidized hisDBP also formed a mixed species with mobility close to and somewhat higher than the reduced monomers (55%) (Fig. 3A, lane 1). The multimers resulting from the oxidation of the thiol groups in cysteines to produce inter disulfide bonds in DBP, whereas the diffuse monomer band is likely due to intra disulfide bonds that result in a more compact structure. In order to clarify the redox state of cysteine residues in DBP samples isolated from infected cells, we treated the hisDBP and wtDBP with the alkylator AMS and analyzed the samples by standard reducing SDS-PAGE. If all six thiols in DBP are reduced and can react with the alkylator, the treatment with AMS (MW=536 Da) should increase the molecular mass of DBP by 3.2 kDa. After treatment with AMS, we observed an increase in the apparent molecular mass of hisDBP (Fig. 3B, lane 2) and wtDBP (Fig. 3B, lane 5) to a size that would be expected if all thiols in the proteins had reacted with the alkylator. Pretreatment of proteins with the reducing agent TCEP before incubation with AMS to ensure the reduced state of thiols before alkylation did not change the mobility of the alkylated species (Fig. 3B, lanes 3 and 6). This result indicated that the thiol groups of cysteines in the DBP samples purified from infected cells are reduced.
Figure 3.

Crosslinking of DBP by diamide and modification of cysteine residues in DBP by the alkylator AMS. (A) The his-tagged DBP (4.1 μM) was incubated in 12-μl mixture containing 50 mM Tris-HCl, pH 7.5, 0.2 M NaCl, 14% glycerol, and sulfhydryl reagents DTT (50 mM, lane 1) or diamide (20 mM, lane 2). After incubation for 1 h at room temperature, the samples were analyzed by non-reducing SDS-9% PAGE. Positions of putative DBP monomers (x1), dimers (x2) and tetramers (x4) are indicated. Densitometry of lane 2 is shown on the right. (B) Alkylation by AMS was carried out in 12-μl reaction mixtures containing 1.2 μM of his-tagged (lanes 1-3) or wild type (lanes 4–6) DBP, 50 mM Tris-HCl, pH7.5, 0.2 M NaCl, 25% glycerol, and redox reagents, 20 mM DTT (lanes 1 and 4) or 20 mM AMS (lanes 2, 3, 5 and 6). Mixtures shown in lanes 3 and 6 were pretreated with 8 mM TCEP for 20 min at room temperature before the addition of AMS. The reactions were incubated for 1 h at room temperature, then were terminated by addition of 40 mM DTT and subjected to SDS-8% PAGE.
Crosslinking of DBP
The low mobility of the complexes of hisDBP with oligonucleotides in polyacrylamide gels (Fig. 2A) suggested that this protein may be present as oligomers, as was shown previously for BmNPV DBP (Mikhailov et al., 1998). The oligomeric state of AcMNPV DBP also was indirectly confirmed by generation of crosslinked multimers by oxidation by diamide (Fig. 3A), although in the latter case the multimers might be generated after denaturation of DBP in the presence of diamide or upon SDS-PAGE at nonreducing conditions. The presence of reduced cysteines in DBP allows the investigation of oligomerization of the native protein by the use of crosslinkers of sulfhydryl groups. The hisDBP was incubated with the homobifunctional crosslinker BM(PEO)2 that causes conjugation between thiol groups, the reaction was terminated by the addition of excess DTT, and the crosslinked species were analyzed by reducing SDS-PAGE. BM(PEO)2 showed practically the same efficiency of crosslinking in the concentration range of 0.5 to 2 mM. The data obtained at 0.8 and 1.6 mM BM(PEO)2 and hisDBP concentrations of 2.7 and 5.4 μM are shown in Fig. 4A. Treatment of 5.4 μM hisDPB with 0.8 mM BM(PEO)2 resulted in generation of monomers (56%), dimers (30%), tetramers (11%) and a small quantity of higher molecular weight oligomers (~2%) (Fig. 4A, lane 5). Similar data were obtained for the crosslinking at 2.7 μM hisDBP, monomers - 58%, dimers - 27%, tetramers – 13%, and higher oligomers - ~2% (Fig. 4A, lane 2). In another experiment, when the hisDBP concentration was decreased to 1.9 μM, crosslinking with BM(PEO)2 resulted in generation of monomers - 61%, dimers - 24%, tetramers – 13%, and higher oligomers - ~2% (data not shown). These data indicated that generation of the oligomeric species of hisDBP by BM(PEO)2 showed little dependence on protein concentration suggesting that oligomers may be a stable constituent of this sample. However, crosslinking of wtDBP at concentration 1.9 μM resulted in a higher yield of crosslinked monomers (80%) than in the case of hisDBP (see above) and in lower yield of the dimers (17%) and tetramers (3%) (data not shown). It appears that oligomerization in the DBP samples depends on the isolation method and probably on the storage conditions. The wtDBP concentration in the course of purification from infected cells and under storage was much lower than that of the overexpressed hisDBP. The apparent absence of trimers among crosslinked species of DBP suggested that the normal oligomeric form of DBP may be a dimer. Crosslinking of dimers should produce tetramers, hexamers, and etc. This model confirms the data shown in Fig. 3A and 4A. Although, in the case of BM(PEO)2 the intramolecular links in DBP prevailed over intermolecular links thus producing monomers as a major product of crosslinking visible after SDS-PAGE.
Figure 4.

Crosslinking of DBP and LEF-3 by BM(PEO)2. (A) The his-tagged DBP at concentrations of 2.7 μM (lanes 1–3) or 5.4 μM (lanes 4–6) was incubated in 12-μl mixtures containing 50 mM Tris-HCl, pH 7.5, 0.2 M NaCl, 25% glycerol, in the absence of the crosslinker (lanes 1 and 4) or in the presence of BM(PEO)2 at concentrations of 0.8 mM (lanes 2 and 5) and 1.6 mM (lanes 3 and 6). After incubation for 1 h at room temperature, the crosslinking reaction was terminated by addition of 40 mM DTT and the samples were analyzed by SDS-8% PAGE. Densitometry of lane 5 is shown on the right. (B) LEF-3 at concentration of 4.5 μM was incubated in 10-μl mixtures containing 50 mM Tris-HCl, pH 7.5, 0.2 M NaCl, 13% glycerol, in the absence of the crosslinker (lane 1) or in the presence of 1 mM BM(PEO)2 (lane 2) for 1 h at room temperature. Lane 3 shows hisDBP (4.5 μM) crosslinked by BM(PEO)2 at the same conditions. The samples were analyzed by SDS-8% PAGE. Positions of putative protein monomers (x1), dimers (x2) and tetramers (x4) are indicated. Densitometry of lanes 2 and 3 is shown on the right. (C) Yield of protein dimers and tetramers after crosslinking of LEF-3 at concentration 3, 6, and 12 μM in the presence of NaCl varied from 0.1 to 0.5 M. LEF-3 was crosslinked by 1 mM BM(PEO)2 and analyzed as in panel B. Closed and open symbols show respectively the dimers and tetramers at indicated concentrations of LEF-3.
The baculovirus SSB protein LEF-3 also forms oligomers in solution (Evans and Rohrmann, 1997). To compare LEF-3 oligomers with that formed by DBP, we carried out crosslinking experiments with varied concentrations of LEF-3 and BM(PEO)2 as described above for DBP. Treatment of 4.5 μM LEF-3 with 1 mM BM(PEO)2 in the presence of 0.2 M NaCl generated monomers (76%), dimers (19%), and tetramers (~4%) (Fig 4B, lane 2). The yield of the crosslinked oligomers of DBP at the same conditions was higher, dimers - 25%, tetramers - 12%, and higher oligomers - ~3% (Fig 4B, lane 3). Despite the difference in the yield of crosslinked species, the oligomeric forms of both proteins showed similar distributions in the polyacrylamide gel with the dimers and tetramers present as the predominant oligomeric forms. There was no evidence of a major crosslinked trimer species. The yield of LEF-3 dimers was decreased by 9% from 28% to 19% when protein concentration dropped from 12 to 3 μM under crosslinking in the presence of 0.1 M NaCl (Fig. 4C). However, the difference in the yield of dimers was low (~2%) under crosslinking at 0.5 M NaCl, when the dimers comprised respectively 17% and 15% at 12 μM and 3 μM LEF-3. The yield of LEF-3 tetramers showed a low dependence on the protein concentration, but it was decreased, from ~4% to ~2%, when concentration of NaCl under crosslinking was raised from 0.1 to 0.5 M (Fig. 4C). These data suggests that, similar to DBP, dimers may be the predominant oligomeric form of LEF-3.
Hydrolysis of DBP by trypsin
Analysis of DBP interaction with DNA at different conditions revealed that its binding activity could be inhibited by thermal treatment. Incubation of hisDBP at 50ºC caused a gradual decrease in efficiency of its binding to dT34 (Fig. 5A). To clarify possible changes in DBP structure that accompanied the thermal inactivation, we compared domain organization of the native and heated DBP by using limited proteolysis with trypsin (Fig. 5B). Digestion by trypsin revealed specific domains that are resistant to proteolysis in the native DBP (Fig. 5B, lanes 3, 5, and 7). The presence of fragment “e” among proteolytic products appeared to be a marker for the native DBP. The MALDI/TOF mass spectrometry of the partial digest of DBP indicated that the mass of fragment “e” (15,732 Da) corresponded well to a fragment located in the DBP sequence from position 103 to 238 (the calculated mass of 15,731 Da). This suggests that a central portion of DBP is more efficiently protected against trypsin than the N-terminal and C-terminal regions. Heating of DBP at 70ºC for 10 min made the whole protein more resistant to hydrolysis presumably by decreasing its solubility or causing its precipitation (Fig. 5B, lanes 4, 6, and 8). However, heating eliminated most internal domains resistant to proteolysis in the native protein including fragment “e”. This result suggested that the DNA binding activity of DBP requires the domain organization specific for native protein, whereas protein unfolding under thermal treatment disrupts the protein domains and inactivates binding.
Figure 5.

Thermal inactivation of DBP binding to DNA and protein unfolding. (A) The his-tagged DBP (1.8 μM) in the storage buffer (50% glycerol, 10 mM Tris-HCl, pH 7.5, 0.2 M NaCl, 1 mM DTT) supplemented with 1% Nonidet P-40 was incubated at 50ºC as indicated above respective lanes and then was assayed for binding to 32P-labeled dT34 by EMSA. Lanes 1 and 2 show, respectively, controls without DBP and with non-heated DBP. (B) Trypsin digestion of the non-heated DBP and thermally unfolded DBP. Two 60-μl mixtures containing 3.6 μM DBP, 50 mM Tris-HCl, pH 7.5, 0.1 M NaCl, 12.5% glycerol, and 5 mM DTT were assembled on ice. The first mixture was left on ice, while the second was heated for 10 min at 70ºC. 10-μl portions were removed from both mixtures (lanes 1 and 2, respectively), then trypsin (18 μg/ml) was added and digestion was carried out at 30ºC. 10-μl portions were removed from the mixtures at 5 min (lanes 3 and 4), 15 min (lanes 5 and 6), 30 min (lanes 7 and 8), and 60 min (lanes 9 and 10). The reactions were terminated by the addition of 5.1 μl of loading buffer (3 mM PMSF, 190 mM Tris, pH 6.8, 4.3 M 2-mercaptoethanol, 6% SDS) followed by boiling for 3 min, and then analyzed by SDS-13% PAGE. The lanes with the non-heated and heated DBP are marked respectively by symbols “−“ and “+”. The arrow shows position of trypsin (T) in the gel.
Changes in the structure of DNA-binding proteins may be caused by binding to DNA (Villemain and Giedroc, 1993; Gomes et al., 1996; Blackwell et al., 1996; Dekker et al., 1998; Dudas et al., 2001; Uprichard and Knipe, 2003). Therefore, we compared the tryptic fragments of DBP obtained before and after binding to ssDNA and dsDNA (Fig. 6A). The binding to dsDNA decreased the rate of DBP hydrolysis by trypsin more efficiently than the binding to ssDNA (compare lanes 4 to 3, and 7 to 6 in Fig. 6A). However, the dsDNA did not change the internal domain structure typical of the native protein, so the fragment “e” remained a major tryptic fragment in the presence of dsDNA (lanes 4, 7, and 10) as well as in the absence of DNA (lanes 2, 5, and 8). In contrast to dsDNA, the binding to ssDNA caused multiple changes in the pattern of tryptic fragments. Lane 2 (hydrolysis for 15 min in the absence of DNA) and lane 6 (hydrolysis for 30 min in the presence of ssDNA) showed approximately similar levels of DBP digestion. Densitometry of these lanes (Fig. 6A, below the gel) revealed that a new fragment “c” (26 kDa) appeared in the presence of ssDNA, and three fragments “a” (34.0 kDa), “d” (17.2 kDa), and “f” (11.9 kDa) were present in higher amounts, whereas two fragments “b” (31.7 kDa) and “e” (15.7 kDa) were present in lower amounts than in the absence of DNA. The most distinct change is that the fragment “d” appeared to be a major resistant fragment in the presence of ssDNA instead of fragment “e” that was most resistant in the absence of DNA. To identify respective domains, the partial digests of DBP in the absence of DNA and in the presence of ssDNA were subjected to SDS-PAGE, the gel pieces with fragments “e” and “d” were cut from the respective lanes as shown in Fig. 6A and treated by trypsin in situ, and then the released tryptic fragments were analyzed by the mass spectrometry. The tryptic digest of fragment “e” generated seven peptides covering the DBP sequence from position 103 to 227 thus confirming the location of fragment “e” to positions 103 to 238 (Fig. 6B) that was predicted by the mass spectrometry of the partial digest of DBP (see above). The digest of fragment “d” generated seven peptides covering the DBP sequence from position 81 to 227 and corresponded to a fragment of 17,165 Da, a mass similar to that expected for fragment “d” (Fig. 6B). Thus, the binding to ssDNA not only decreased the rate of DBP hydrolysis by trypsin, but also changed the domain that is most resistant to the proteolysis.
Figure 6.

Protection of DBP and LEF-3 against trypsin by ssDNA and dsDNA. (A) Three 35-μl mixtures containing 3.6 μM DBP, 50 mM Tris-HCl, pH 7.5, 0.1 M NaCl, 12.5% glycerol, and 5 mM DTT were assembled on ice. The mixtures #1, #2, and #3 contained no DNA, 100 μg/ml of M13 ssDNA, and 100 μg/ml of M13 dsDNA, respectively. The mixtures were incubated for 15 min at 25ºC to form complexes of DBP with DNA. Then trypsin (12 μg/ml) was added and the incubation was continued. 10-μl portions were removed from the mixtures at 15 min (lanes 2–4), 30 min (lanes 5–7), and 60 min (lanes 8–10). The reactions were terminated by the addition of 6.1 μl of loading buffer (3 mM PMSF, 0.65 M NaCl, 190 mM Tris, pH 6.8, 4.3 M 2-mercaptoethanol, 6% SDS) followed by boiling for 3 min, and then analyzed by SDS-13% PAGE. Trypsin was not added to the time zero reaction (lane 1). Densitometry of lanes 2 and 6 is shown below the panel. Symbols a to f correspond to respective bands in the gel. Brackets show the bands taken for MALDI/TOF analysis. (B) Location of major tryptic fragments in the DBP sequence. The putative bipartite nuclear localization sequence (NLS, residues 5–23), fragments e (residues 103–238) and d (residues 81–227) are shown. The Cys residues are underlined. (C) Three 35-μl mixtures containing 4.5 μM LEF-3 and other ingredients as in the experiment shown in panel A were assembled on ice. The mixtures #1, #2, and #3 contained no DNA, 40 μg/ml of M13 ssDNA, and 40 μg/ml of M13 dsDNA, respectively. After pre-incubation of LEF-3 with DNA, trypsin (10 μg/ml) was added and the portions were removed from the mixtures at 15 min (lanes 2–4), 30 min (lanes 5–7), and 60 min (lanes 8–10) and processed as described above. Trypsin was not added to the time zero reaction (lane 1). The arrows show position of trypsin (T), and fragments L and S of LEF-3.
To compare the effect of DNA binding on proteolysis of DBP and LEF-3, we carried out the experiments on partial digestion by trypsin for LEF-3. The domain structure of LEF-3 probed by trypsin was previously described in detail and three major tryptic fragments (XL, L, and S) were identified (Mikhailov et al., 2006). The time course of LEF-3 hydrolysis by trypsin in the presence of ssDNA and dsDNA is shown in Fig. 6C. The dsDNA provided only limited protection of LEF-3 against trypsin (Fig. 6C, lanes 4, 7, and 10) when compared to the hydrolysis in the absence of DNA (Fig. 6C, compare lanes 4, 7, and 10 to lanes 2, 5, and 8). The tryptic fragments L and S were evident under hydrolysis in the presence of dsDNA (Fig. 6C, lanes 7 and 10). In contrast to the limited protection by dsDNA, LEF-3 bound to ssDNA was practically resistant to the hydrolysis in the presence of 10 μg/ml trypsin (Fig. 6C, lanes 3, 6, and 9). The 5-fold increase in the concentration of trypsin to 50 μg/ml did not increase the yield of the tryptic fragments from LEF-3 bound to ssDNA (data not shown). Thus, the interaction with dsDNA produced some protection of both DBP and LEF-3, but apparently did not change the domain structure of these proteins. The ssDNA appeared to be a much more effective protector of LEF-3 than for DBP, although the binding to ssDNA presumably changes the domain structure of DBP.
Unwinding and renaturation activity of DBP
As with other SSB proteins, baculovirus DBP was capable of destabilizing DNA duplexes. The unwinding activity of DBP was assayed in reaction mixtures containing a Y-shaped structure having a 37-bp duplex region that was formed by 63-mer and 62-mer oligonucleotides. The his-tagged DBP promoted a dose-dependent unwinding of the duplex under oversaturation of the Y-shaped structure (Fig. 7A). The DBP sample lacking the His6-tag showed the same unwinding activity as the his-tagged DBP (data not shown). The unwinding reaction was facilitated by the increase in temperature from 23ºC to 30ºC (Fig. 7B) and was highly sensitive to salts (Fig. 7C). NaCl at concentrations higher than 60 mM completely inhibited the unwinding. The sulfhydryl reagents diamide and NEM blocked the unwinding reaction promoted by hisDBP (Fig. 7D). Therefore, the reduced cysteines in DBP are essential for the unwinding activity of this protein, as well as for its DNA binding activity (Fig. 2).
Figure 7.

Unwinding activity of DBP. (A) The unwinding assay was carried out in 10-μl reaction mixtures containing 0.5 nM 32P-labeled 63-mer annealed to 0.75 nM of 62-mer, 20 mM Tris-HCl, pH 7.5, 40 mM NaCl, 12.5% glycerol, 100 μg/ml BSA, and 2 mM DTT. The his-tagged DBP was added in the following concentrations: control, no DBP (lane 1), 0.55 μM (lane 2), 1.1 μM (lane 3), and 2.2 μM (lane 4). The reactions were incubated for 1 h at 30ºC, then treated with SDS (0.5%) and proteinase K (150 μg/ml) for 15 min at 22ºC, and analyzed by PAGE. Diagrams of the DNA substrates are shown at the right. The asterisk indicates the radioactive label in the 63-mer. (B) Time course of the unwinding reaction promoted by DBP at 23ºC and 30ºC. Reactions containing 1.5 μM his-tagged DBP were assembled and analyzed as in panel A. (C) Inhibition of the unwinding activity of DBP by salt. Reactions containing 0.28 μM his-tagged DBP and different concentrations of NaCl were assembled and analyzed as in panel A. (D) Inhibition of the unwinding activity by alkylation or oxidation of sulfhydryl groups in DBP. Reactions containing the sulfhydryl reagents, DTT, N-ethylmaleimide (NEM), or diamide, each at concentration of 5 mM were assembled and analyzed as in panel A.
The renaturation activity of DBP was assayed with the 2289-bp fragment of the double-stranded replicative form of M13mp9 DNA that was denatured by boiling before incubation with DBP. The incubation with DBP caused a dose-dependent renaturation of complementary single strands of DNA into the duplexes (Fig. 8A). No large DNA networks were detectable in the wells (data not shown). DBP facilitated renaturation of ssDNA for at least two hours during incubation at 37ºC (Fig. 8B). The yield of dsDNA upon reaction in the absence of DBP was about seven times lower than in the presence of 2.2 μM DBP (Fig. 8B, -DBP). The renaturation activity of DBP was moderately stimulated (~30%) by the presence of 3 and 5 mM Ca2+ ions, but 10 mM CaCl2 was inhibitory (data not shown). DBP facilitated renaturation of ssDNA at all NaCl concentrations tested in a range of 35 to 220 mM showing a broad optimum at 75 to 100 mM NaCl (data not shown). The renaturation activity of DBP was efficiently inhibited by diamide but only moderately by N-ethylmaleimide (Fig. 8C,D). The inhibition by diamide was more pronounced at higher DBP concentrations than at lower concentrations (compare lanes 10, 11, and 12 in Fig. 8C). Although alkylator NEM only moderately suppressed the generation of DNA duplexes by DBP, it induced the formation of aberrant DNA species that migrated in the gel slower than the duplexes and were seen as a diffuse signal above the duplexes (Fig. 8C, lanes 6–8). These DNA species are presumably unresolved intermediates formed during renaturation. The partial inhibition of DNA renaturation and generation of slowly migrating species of DNA were observed also in the experiment when DBP was pretreated with NEM before incubation with DNA (data not shown). Thus alkylation of thiol groups in DBP, although not completely abolishing the renaturation activity of this protein, suppressed the resolution of complex hybridization intermediates. An increase in DBP concentration allowed this obstacle to be overcome (compare lanes 6 and 8 in Fig. 8C). The DNA-binding protein LEF-3 was inactive in the renaturation assay described above.
Figure 8.

Renaturation activity of DBP. (A) The renaturation assay was carried out in 10-μl reaction mixtures containing 2 nM 32P-labeled 2289-bp fragment of RF DNA M13mp9, 20 mM Tris-HCl, pH 7.5, 75 mM NaCl, 3 mM CaCl2, 10% glycerol, 100 μg/ml BSA, and 2 mM DTT. DNA was denatured by boiling for 5 min followed by chilling on ice before the addition to the mixtures. DBP was added in the following concentrations: control, no DBP (lane 1), 0.55 μM (lane 2), 1.1 μM (lane 3), 2.2 μM (lane 4), and 4.4 μM (lane 5). The reactions were incubated for 1 h at 37ºC, then EDTA (12 mM) and SDS (0.5%) were added, and the samples were treated with proteinase K (0.5 mg/ml) for 15 min at 22ºC and analyzed by electrophoresis in a 1% agarose gel. The position of double-stranded DNA (ds) and single-stranded DNA (ss) in the gel is shown on the right. (B) Time course of the renaturation reaction promoted by DBP at 37ºC. Reactions containing 2.2 μM DBP were assembled and analyzed as in panel A. The filled symbol shows the amount of dsDNA in the reaction mixture incubated for 120 min at 37ºC in the absence of DBP. (C) and (D) Redox sensitivity of the renaturation reaction promoted by DBP. The renaturation assay was carried out as in panel A. The reactions contained the following redox agents, 5 mM DTT (lanes 1–4), 10 mM N-ethylmaleimide (NEM) (lanes 5–8), and 10 mM diamide (lanes 9–12). DBP was added in the following concentrations: control, no DBP (lanes 1, 5, and 9), 1.1 μM (lanes 2, 6, and 10), 2.2 μM (lanes 3, 7, and 11), and 4.4 μM (lanes 4, 8, and 12). Panel D shows quantification of the data shown in panel C. The amounts of dsDNA generated in the absence of DBP were taken as zero points.
Inhibition of DNA hydrolysis by DBP
An essential function of SSB proteins may be connected to the protection of DNA against hydrolysis by nucleases. Viral DNA may be a target for both host-cell nucleases and nucleases encoded by the viral genome. Baculoviruses can induce apoptosis in infected cells that results in the degradation of host-cell chromatin and viral DNA (Clem et al., 1991). They also express a nuclease that is highly active at alkaline conditions (Li and Rohrmann, 2000). The AcMNPV alkaline nuclease (AN) forms a complex with the SSB-protein LEF-3 (complex AN/L3) and possesses potent 5′→3′ exonuclease activity and weak endonuclease activity both specific for ssDNA (Mikhailov et al., 2003; Mikhailov et al., 2004). Because AN may be a primary source of nuclease activity in infected cells, we analyzed the effect of DBP on hydrolysis of ssDNA by the AN/L3 complex. 3H-labeled E.coli DNA (3 μg/ml) denatured by boiling was used as a substrate for AN/L3 complex (0.13 μg/ml) in the reaction mixture at pH 8.3. In the experiment shown in Fig. 9, AN/L3 hydrolyzed approximately 70% of the DNA after incubation for 30 min at 30ºC in the absence of DBP. The addition of DBP (40 μg/ml) almost completely blocked the DNA hydrolysis after a lag period of about 10 min. The final yield of acid-soluble products in the presence of DBP was approximately 30%, more than two-fold lower than in the absence of DBP. This result showed that DBP is capable of protecting DNA against the nuclease activity of AN/L3.
Figure 9.

Inhibition of ssDNA hydrolysis by DBP. Two 70-μl reaction mixtures containing 3 μg/ml [3H]ssDNA E. coli, 25 mM Tris, pH 8.3, 50 mM KCl, 5 mM MgCl2, 100 μg/ml BSA, 12.5% glycerol, and 2 mM DTT were assembled on ice. DBP was added to one mixture to final concentration of 40 μg/ml, and both mixtures were incubated for 15 min at 30ºC. To initiate hydrolysis, 9 ng of the complex of AcMNPV alkaline nuclease with LEF-3 (complex AN/L3) was added to each reaction and incubation at 30ºC was continued. The 20-μl portions were taken from the mixtures at 10, 20, and 45 min and the release of acid-soluble radioactivity from [3H]ssDNA was measured. The radioactivity of undigested sample (5020 cpm) was taken as 100%.
Discussion
In this report we describe the purification and characterization of DBP from AcMNPV infected cells. This protein was isolated both in its native form from infected Sf9 cells and also as an N-terminal His6-tag fusion expressed in a recombinant baculovirus. The latter product was purified to near homogeneity and obtained with a high yield (2.4 mg from a 100-ml cell culture). Analysis of DBP binding to DNA (Fig. 2) and its unwinding and renaturation activities (Fig. 7 and 8) confirmed the original identification of DBP as an SSB protein. DBP showed a high affinity for ssDNA and was an effective competitor of LEF-3 in binding to oligonucleotides (Fig. 2C). Reduced cysteines were found to be required for efficient binding of DBP to DNA and for DNA unwinding and renaturation. Oxidation or alkylation of the sulfhydryl groups inhibited the binding, unwinding, and renaturation activities of DBP (Fig. 2C, 7, and 8). Treatment of DBP by the thiol-conjugating agent, NEM, generated complex forms of DNA that migrated slowly in agarose gels (Fig. 8) indicating a decreased capacity of the alkylated DBP to resolve intermediates in renaturation. Data from other systems indicate that redox regulation can play a role in the function of DNA-binding factors. This has been observed with transcriptional factors (reviewed in (Bauer et al., 1999; Kim et al., 2002)) and some other SSB proteins including the eukaryotic protein RP-A (You et al., 2000), herpes simplex virus -1 (HSV-1) protein ICP8 ( Knipe et al., 1982; Ruyechan, 1988; Dudas and Ruyechan, 1998), and baculovirus LEF-3 (Mikhailov et al., 2005). Similar to ICP8 and LEF-3, the DBP activities were also inhibited by alkylation with NEM.
Although all cysteines in the DBP samples purified from infected cells appeared to be in a reduced state (Fig. 3B), we could not exclude that some thiol groups of DBP in nuclei were oxidized during infection thus producing crosslinked oligomeric forms that had low solubility and were poorly extractable from nuclei by salt. This could have influenced our ability to efficiently extract DBP from nuclei. It was shown previously that baculovirus replication proceeds at conditions of oxidative stress in infected cells (Wang et al., 2001). Therefore, the crosslinked oligomeric forms may occur naturally under these conditions. It has also been demonstrated that oxidation of the SSB protein ICP8 of HSV-1 may proceed in vivo (Knipe et al., 1982). We were able to produce crosslinked oligomeric species of DBP in vitro by oxidation with diamide (Fig. 3A) or by treatment with the sulfhydryl crosslinker BM(PEO)2 (Fig. 4A). The dimers and tetramers were the predominant crosslinked oligomers formed upon treatment of both DBP and LEF-3 samples with BM(PEO)2 (Fig. 4B). This observation suggested that the regular state of both DBP and LEF-3 may be a dimer. The dimers interact with one another producing tetramers, hexamers, and etc. The presence of oligomeric forms of DBP in infected cells was indirectly confirmed by the co-purification of wtDBP lacking the his-tag with the his-tagged DBP (likely derived from heterodimers of his- and non his-tagged DBP) during fractionation of cells extracts on Ni-NTA columns (Fig. 1C). It was suggested previously that LEF-3 forms trimers in solution (Evans and Rohrmann, 1997), however rigorous proof for trimeric structure of LEF-3 was not provided. Recently EMSA analysis has demonstrated that LEF-3 has two binding sites for a 17-mer oligonucleotide (dT17) (Mikhailov et al., 2005). These data, as well as the result of LEF-3 crosslinking with BM(PEO)2 (Fig. 4B,C), agree more with a dimeric rather than a trimeric structure for LEF-3.
Limited proteolysis by trypsin revealed a major fragment of DBP resistant to hydrolysis (fragment “e”) that was located at position 103 to 238 (Fig. 6B) indicating that the N- and C-terminal portions of DBP were more exposed and accessible to trypsin than the central portion. Binding to ssDNA caused multiple changes in the pattern of proteolytic fragments of DBP (Fig. 6A). The major fragment resistant to trypsin in the presence of ssDNA was shifted towards the N-terminus and is located at positions 81 to 227 (fragment ‘d”, Fig. 6B). This result suggested that the structure of DBP undergoes a major alteration during binding to ssDNA. Such structural transitions during the binding to DNA have been reported for other SSB proteins (Villemain and Giedroc, 1993; Blackwell et al., 1996; Gomes et al., 1996; Dekker et al., 1998; Dudas et al., 2001; Uprichard and Knipe, 2003). Although the trypsin digestion data suggests a structural change, it could also result from direct protection by the binding to ssDNA. However, dsDNA protected DBP more efficiently against proteolysis than ssDNA, but dsDNA did not cause significant changes in pattern of proteolytic fragments and did not shift the position of the resistant fragment from “e” to “d” (Fig. 6A).
Similar to BmN cells infected with BmNPV (Okano et al., 1999), DBP appeared to be about twice as abundant as LEF-3 in Sf9 cells infected with AcMNPV. The Sf9 cells at 20 hpi contained approximately 3 × 107 molecules of DBP and 1.5 × 107 molecules of LEF-3. The reason for the possession of two SSBs, LEF-3 and DBP, by many baculoviruses is not clear. Although both SSBs are required for the production of viable virions, DBP is not required for viral DNA synthesis (Vanarsdall et al., 2007; Quadt et al., 2007). However, the knockout of the dbp gene decreases the yield of nascent viral DNA and appears to prevent the production of mature full-size viral genomes (Vanarsdall et al., 2007).
Although the function of DBP in the baculovirus infection cycle remains unknown, it may be directly related to the activities revealed by our in vitro experiments. It has been shown previously that Sf9 cells transfected with bacmid DNA containing a full set of viral genes but lacking dpb accumulate DNA of apparent high molecular weight (“well DNA”) and fragments that are shorter than full-size viral genome (Vanarsdall et al., 2007). The DNA fragments might represent degradation products of viral genomes or replicative intermediates. Two different activities of DBP might be responsible for these observations. Firstly, DBP may prevent the enzymatic degradation of viral genomes. It has been shown that DBP inhibits hydrolysis of DNA by the proofreading activity of phage T4 DNA polymerase (Mikhailov et al., 1998) or by the nuclease activity of the AN/L3 complex (Fig. 9). Therefore, DBP may protect mature viral genomes against nucleases and stabilize them at stages that precede packaging into virions. On the other hand, the unwinding and renaturation activity of DBP may be required for processing of replicative intermediates by annealing and strand invasion reactions involved in DNA recombination that may be essential for the complete replication and processing of baculovirus genomes (Hajos et al., 2000; Kamita et al., 2003; Okano et al., 2007). LEF-3 likely participates in recombination as a complex with alkaline nuclease AN (Mikhailov et al., 2003; Mikhailov et al., 2004). The AN/L3 complex can generate DNA duplexes with ss tails which can function as recombination intermediates. However, the annealing and invasion of ssDNA may require the renaturation activity of DBP. Although LEF-3 unwinds short DNA duplexes in the same manner as DBP (Mikhailov et al., 2005), its renaturation activity for long ssDNA strands was demonstrated only for the unfolded (thermally denatured) form of LEF-3 (Mikhailov et al., 2006). In this report, we showed that DBP promotes renaturation of complementary strands of DNA at physiological conditions. The bacmid deleted for the dbp gene shows a phenotype similar in some aspects as the bacmid with the an (alkaline nuclease) gene deleted (Okano et al., 2007). The an knockout bacmid results in the accumulation of DNA of high molecular weight that is trapped in wells under field inversion gel electrophoresis and fragments that are shorter than genome size, but it was incapable of producing genome-length DNA (Okano et al., 2007). These data could indicate that DBP and the AN/L3 complex may both participate in the processing of replicative intermediates.
Baculoviruses generate an electron dense nuclear structure called the virogenic stroma where replication of viral genomes and packaging into viral particles occur. Viral DNA and RNA serve presumably as a scaffold for the organization of the stroma (Young et al., 1993). Both proteins, LEF-3 and DBP, colocalize with nascent viral DNA in replication foci in nuclei (Okano et al., 1999) and are found associated with chromatin when nuclei are fractionated (Vanarsdall et al., 2007). However DBP associates more tightly than LEF-3 with subnuclear structures as demonstrated by salt fractionation (Fig. 1A). The salt extraction of DBP and LEF-3 from chromatin resembles the elution pattern of these proteins upon chromatography on ssDNA-cellulose where LEF-3 elutes mostly at 0.6 M NaCl and DBP elutes in a range of 0.8 to 2 M NaCl (data not shown and (Mikhailov et al., 1998) ). These data suggest that DBP forms more stable and probably less soluble complexes with DNA than LEF-3. The tight association with chromatin and its abundance in infected cells suggest that DBP may serve as a component of the virogenic stroma that is essential for integrity and multiple functions of this structure. It was shown previously that the bacmid with the dbp gene knocked out does not produce a normal virogenic stroma (Vanarsdall et al., 2007). As a component of the virogenic stroma, DBP may stabilize viral genomes by protecting them against nucleases, whereas its unwinding and renaturation activity may facilitate structural transitions that accompany maturation of the viral genomes.
Materials and Methods
Source of Chemicals
N-ethylmaleimide (NEM) (Sigma), 1,1′-azobis(N,N-dimethylformamide) (diamide) (TCI America), tris-(2-carboxyethyl)phosphine, hydrochloride (TCEP) and 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid, disodium salt (AMS) (Molecular Probes), 1,8-bismaleimidodiethylene glycol (BM(PEO)2) (Pierce).
Cells and recombinant baculoviruses
Spodoptera frugiperda 9 (Sf9) cells were cultured in Sf900II serum-free media (Invitrogen), penicillin G (50 units/ml), streptomycin (50 μg/ml, Whittaker Bioproducts), and fungizone (amphotericin B, 375 ng/ml, Flow Laboratories) as previously described (Harwood et al., 1998). To construct a bacmid overexpressing a His6-tagged DBP, the DBP gene was amplified with primers 5′-CGGTACCATGGCACCTAAAC-3′ and 5′-TTATTGTTCAATAATAACAA-3′ from an AcMNPV genomic template and subcloned into the pCR 2.1 TOPO cloning vector (Invitrogen). The DBP coding region was sequenced to confirm its fidelity, and then subsequently excised with NcoI and XhoI, ligated in-frame into the pFastBacHTa plasmid, and transposed into the AcMNPV bacmid bMON14272 according to the methods described in the Bac-to-Bac protocol (Invitrogen).
DNA substrates
The following oligonucleotides were used for assay of DBP activities: dT34; 63-mer, CAACGGCATAAAGCTTGACGATTACATTGCTAGGACATGCTGTCTAGAG GATCCGACTATCGA; 62-mer, TGGGTGAACCTGCAGGTGG GCAAAGATGTCCT AGCAATGTAATCGTCAAGCTTTATGCCGTT; 62-mer(c), (5′-P)AACGGCATAAA GCTTGACGATTACATTGCTAGGACATCTTTGCCCACCTGC AGGTTCACCCA. The oligonucleotides were 5′-end-labelled using T4 polynucleotide kinase and [γ-32P]ATP (Perkin-Elmer). The 63:62-mer pseudo-Y fork structure with a 37-bp duplex region and single-stranded (ss) 5′- and 3′-tails was formed by annealing of 32P-labeled 63-mer to a 1.5-fold molar excess of unlabeled 62-mer. The 62-mer duplex was formed by annealing of 32P-labeled 62-mer to 62-mer(c). The replicative form of M13mp9 DNA (7,599 bp) (10 μg) was digested for 3 h at 37º with 20 units of endonuclease XmnI to produce dsDNA fragments of 5,310 and 2,289 bp. The fragments were gel-purified. The 2,289 bp fragment was labeled at the 3′-ends with terminal transferase (Invitrogen) and [α-32P]dCTP (Perkin-Elmer) and purified by using ProbeQuant G-50 Micro Column (Amersham Biosciences). Escherichia coli DNA labeled with [3H]thymidine was denatured by boiling for 10 min followed by chilling on ice and was used as a substrate for AcMNPV alkaline nuclease (AN) associated with LEF-3 as described previously (Mikhailov et al., 2003).
Purification of DBP
To obtain wild type DBP (wtDBP), Sf9 cells (500 ml) at a density of 1.5 x 106/ml in shaker flasks were infected with AcMNPV at a multiplicity of infection (MOI) of about 5 and collected 22 h post infection (hpi). All procedures were performed at 4ºC. The infected cells were pelleted by centrifugation and resuspended in 10 ml of lysis buffer containing 50 mM Tris-HCl, pH 7.5, 0.14 M NaCl, 1.5 mM MgCl2, 1% Nonidet P-40, and a set of protease inhibitors (1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μM pepstatin, 5 μg/ml leupeptin, 5 μg/ml aprotinin, 2 μg/ml E64). After incubation for 10 min with gentle shaking, the nuclei were pelleted by centrifugation for 10 min at 1,000 x g and the cytoplasmic fraction was collected. The nuclear pellet was resuspended in 5 ml of the lysis buffer, incubated for 5 min, and then the nuclei were pelleted again. The supernatant was combined with the cytoplasmic fraction, and the preparation was clarified by centrifugation for 30 min at 105,000 x g. After addition of EDTA (5 mM), the sample was loaded onto a ssDNA-cellulose column (2.1 ml) equilibrated with a buffer containing 0.2 M NaCl, 10 mM Tris-HCl, pH 7.5, 10% glycerol, 1 mM dithiothreitol (DTT), and 1 mM EDTA. The column was washed successively with 10-ml portions of the same buffer containing NaCl in final concentrations of 0.4, 0.6, and 0.8 M. DBP was eluted with the buffer containing 2 M NaCl. Proteins from each fraction were analyzed by SDS-PAGE followed by staining with Coomassie brilliant blue, and fractions enriched in DBP were combined and dialyzed against an excess of a buffer containing 10 mM Tris-HCl, pH 8.5, 10% glycerol, 1 mM DTT, and 1 mM EDTA. The sample was loaded onto a DEAE-Toyopearl 650 (TosoHaas) column (0.6 ml) equilibrated with the dialysis buffer, and DBP was eluted with 70 mM NaCl in the same buffer. Fractions collected were analyzed by SDS-10% PAGE followed by staining with Coomassie brilliant blue, and were combined or dialyzed separately against buffer 0.2 M NaCl, 10 mM Tris-HCl, pH 7.5, 50% glycerol, 1 mM dithiothreitol, and 0.2 mM EDTA, and stored at −20°C.
The his-tagged DBP was purified from a 100-ml culture of Sf9 cells infected with recombinant virus vAcHisDBP at a MOI of about 5 and collected at 72 hpi. The infected cells were pelleted by centrifugation for 5 min at 500 x g and resuspended in 7 ml of lysis buffer containing 50 mM Tris-HCl, pH 8.5, 200 mM KCl, 1% Nonidet P-40, 5 mM 2-mercaptoethanol and the set of protease inhibitors. After extraction for 10 min at 4°C on a rotating shaker, the preparation was clarified by centrifuged at 30,000 x g for 30 min. The supernatant was loaded onto a Ni-NTA-agarose (Qiagen) column (0.8 ml) and processed as recommended by the manufacturer. His-tagged DBP was eluted with 150 mM imidazole in buffer containing 20 mM Tris-HCl, pH 8.5, 75 mM KCl, 10% glycerol, and 5 mM 2-mercaptoethanol. The sample was loaded onto a DEAE-Toyopearl column (0.6 ml) equilibrated with the same buffer containing 1 mM EDTA. The column was processed successively with 2-ml portions of a buffer containing 10 mM Tris-HCl, pH 8.5, 10% glycerol, 1 mM DTT, 1 mM EDTA, and NaCl in final concentrations of 80, 110, 150, 200, and 250 mM. The fractions containing hisDBP were dialyzed against buffer 0.2 M NaÑl, 10 mM Tris-HCl, pH 7.5, 50% glycerol, 1 mM DTT, and 0.2 mM EDTA, and stored at −20°C.
To remove the N-terminal His6-tag from hisDBP, 0.4 mg of his-tagged DBP was treated with 120 units of AcTEV protease (Invitrogen) in a mixture (0.8 ml) containing 25 mM Tris-HCl, pH 8.0, and 5 mM 2-mercaptoethanol for 3 h at room temperature. The digestion was terminated by the addition of 0.25 ml Ni-NTA agarose suspension (0.1 ml of bed volume) in buffer 0.1 M Tris-HCl, pH 8.5, 0.5 M KCl, and 5 mM 2-mercaptoethanol. The mixture was incubated for 1 h at 4ºC with gentle shaking, and then the slurry was removed by centrifugation. The supernatant containing DBP lacking the his-tag was dialyzed against buffer 0.2 M NaÑl, 10 mM Tris-HCl, pH 7.5, 50% glycerol, 1 mM DTT, and 0.2 mM EDTA, and stored at −20°C.
AcMNPV protein LEF-3 and AN (alkaline nuclease) associated with LEF-3 (complex AN/L3) were purified as described previously (Mikhailov et al., 2004).
Chromatin fractionation
Nuclei were purified from Sf9 cells (2.6 x 107) infected with AcMNPV at a MOI of about 5 and collected at 20 hpi. The cells were washed in PBS and then incubated for 10 min on ice in 1.4 ml of buffer 20 mM Tris-HCl, pH 8.0, 140 mM NaCl, 3 mM MgCl2, 1 mM DTT, 1% Nonidet P-40 and the set of protease inhibitors. The nuclei were centrifuged for 10 min at 1,000 x g, resuspended in 1.4 ml of the same buffer, incubated for 10 min and centrifuged again. The purified nuclei were extracted with buffer 20 mM Tris-HCl, pH 8.0, 75 mM NaCl, 25 mM EDTA, 1 mM DTT, 0.5 mM PMSF, and then with buffer 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM DTT, 0.5 mM PMSF. The chromatin was sequentially extracted with buffers containing 20 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, and NaCl in final concentrations of 0.35, 0.6, 0.8, 1.0, 1.4, and 2.0 M. Extractions were performed twice. The volume of each fraction was equal to 1.2 ml. Portions from the fractions obtained were subjected to SDS-11% PAGE and Western blotting using polyclonal antibodies to DBP and LEF-3.
Quantitative Western blotting
Lysates of Sf9 cells infected with AcMNPV at a MOI of 5 and collected 20 hpi were used for quantification of the DBP and LEF-3 content as described previously for BmN cells infected with BmNPV (Okano et al., 1999). The cells lysates corresponding to 0.7 × 104, 1.8 × 104, and 3.6 × 104 cells were analyzed on three lanes by SDS-11% PAGE. The purified DBP and LEF-3 each in amounts of 20, 50, and 100 ng were loaded on the other lanes of the same gel and were used as reference standards. After electrophoresis, the proteins were transferred from the gel to PVDF-Plus membrane (Osmonics Inc) by electroblotting and were probed with polyclonal antibodies to BmNPV DBP (Okano et al., 1999) and with polyclonal antibodies to AcMNPV LEF-3 (Evans and Rohrmann, 1997). The fluorescence signals of the reference proteins were used for generation of the calibration curve.
DNA binding assay
DBP binding to DNA was analyzed by electrophoretic mobility shift assays (EMSA) using 32P-labeled oligonucleotide probes. The binding reaction was carried out in 10-μl mixtures containing 1 nM of 32P-labeled oligonucleotide, 30 mM Tris-HCl, pH 7.5, 50 mM NaCl, 12.5% glycerol, 0.5% Nonidet P-40, and 50 μg/ml BSA. The sulfhydryl reagents (DTT, N-ethylmaleimide (NEM), or diamide) were added as indicated. After addition of DBP (5 nM to 1.3 μM), the mixtures were incubated for 15 min at 22ºC and then subjected to electrophoresis in a 6% polyacrylamide gel as described previously (Mikhailov and Bogenhagen, 1996). The gels were dried on DE-80 paper and analyzed by a PhosphorImager (Molecular Dynamics) and exposed to X-ray film.
DNA unwinding assay
The unwinding assay was carried out in 10-μl mixtures containing the 63:62-mer pseudo-Y fork structure formed by annealing of 0.5 nM 32P-labeled 63-mer to 0.75 nM of 62-mer, 20 mM Tris-HCl, pH 7.5, 40 mM NaCl, 12.5% glycerol, and 100 μg/ml BSA. The sulfhydryl reagents (DTT, NEM, or diamide) were added as indicated. After addition of DBP (0.2 to 2.2 μM), the mixtures were incubated for 1 h at 23ºC or 30ºC, then treated with SDS (0.5%) and proteinase K (150 μg/ml) for 15 min at 22ºC, and analyzed by electrophoresis in a 6% polyacrylamide gel in 20 mM HEPES, pH 8.0, 0.1 mM EDTA. The gels were dried on DE-80 paper and analyzed by PhosphorImager and exposed to X-ray film.
DNA renaturation assay
The renaturation assay was carried out in 10-μl mixtures containing 2 nM 32P-labeled 2289-bp fragment of RF M13mp9 DNA, 20 mM Tris-HCl, pH 7.5, 75 mM NaCl, 3 mM CaCl2, 10% glycerol, and 100 μg/ml BSA. DNA was denatured by boiling for 5 min followed by chilling on ice before the addition to the mixtures. The sulfhydryl reagents (DTT, NEM, or diamide) were added as indicated. After addition of DBP (0.55 to 4.4 μM), the mixtures were incubated for 1 h at 37ºC, then EDTA (12 mM) and SDS (0.5%) were added and the samples were treated with proteinase K (0.5 mg/ml) for 15 min at 22ºC. The samples were subjected to electrophoresis in a 1% agarose gel in TAE buffer (40 mM Tris-acetate, pH 8.0, 1 mM EDTA) at 2.7 V/cm for 3 h. The gels were dried on DE-80 paper and analyzed by PhosphorImager and exposed to X-ray film.
Crosslinking by BM(PEO)2
The standard 12-μl mixture for crosslinking contained 50 mM Tris-HCl, pH 7.5, 0.2 M NaCl, glycerol (13 to 25%) and DBP or LEF-3 (1.9 to 12 μM). BM(PEO)2 (0.5 to 2 mM) were added as indicated. After incubation for 1 h at room temperature, the crosslinking reaction was terminated by addition of 40 mM DTT and the samples were subjected to SDS-8% PAGE followed by Coomassie staining.
Analysis of DBP structure by limited proteolysis with trypsin
The standard 10-μl mixture for hydrolysis by trypsin contained 3.6 μM DBP, 50 mM Tris-HCl, pH 7.5, 0.1 M NaCl, 12.5% glycerol, and 5 mM DTT. The digestion with the sequencing grade modified trypsin (Promega) (12 to 18 μg/ml) was carried out at 25ºC for the indicated times, and the reactions were terminated by the addition of 5.1 μl of loading buffer (190 mM Tris, pH 6.8, 4.3 M 2-mercaptoethanol, 6% SDS) containing 3 mM PMSF and by boiling for 3 min. The samples were analyzed by SDS-13% PAGE.
MALDI/TOF analysis
Two 13-μl reaction mixtures containing 5 μg DBP, 20 mM Tris, pH 7.5, 23% glycerol, 92 mM NaCl, and 2 mM DTT were assembled on ice. Phage M13mp9 ssDNA (0.3 mg/ml) was added to one mixture and the reactions were incubated at 30ºC for 15 min to form a complex of DBP with DNA. Sequencing grade modified trypsin (Promega) (4 μg) was added, and the incubation at 30ºC was continued for 15 min. The hydrolysis was terminated by chilling on ice and by addition of PMSF (2 mM) and the SDS-PAGE loading buffer. The samples were immediately boiled and then subjected to SDS-13% PAGE. The gel pieces with fragments of interest were digested by trypsin in-gel and prepared for MALDI tof-tof analysis (Shevchenko et al., 1996). The samples were mixed in a 1:6 ratio of a-cyano-4-hydroxycinnamic acid in 50% acetonitrile and 0.1% TFA, and 0.5 μl was applied to the sample plate. The crystallized sample material was rinsed with deionized water to remove impurities. Molecular mass analysis was performed by matrix-assisted laser desorption/ionization time of flight mass spectrometry using a Proteomics Analyzer ABI 4700 TOF/TOF mass spectrometer (Applied Biosystems, Inc.) with an accelerating voltage of 20 kV. For analysis of the partial digests, 3-μl aliquots were taken from the reaction mixtures at different time points and were immediately frozen in liquid nitrogen. The MALDI tof-tof analysis was carried out as above.
Inhibition of DNA hydrolysis by DBP
Hydrolysis of DNA was analyzed in reaction mixtures containing 3 μg/ml 3H-labeled ssDNA E. coli (84 × 103 cpm per μg), 25 mM Tris, pH 8.3, 50 mM KCl, 5 mM MgCl2, 100 μg/ml BSA, 12.5% glycerol, and 2 mM DTT. The complex of DBP (40 μg/ml) with DNA was formed upon incubation for 15 min at 30ºC. The DNA hydrolysis by AcMNPV alkaline nuclease (AN) associated with LEF-3 (0.13 μg/ml) was carried out at 30ºC for the indicated times. The reactions were terminated by chilling on ice and by addition of tRNA (2 mg/ml) and trichloroacetic acid (10%). The samples were incubated for 10 min on ice and microcentrifuged for 5 min. The supernatant was mixed with 3.5 ml of liquid scintillation counting cocktail formula 989 (Packard, Inc.) and counted in a liquid scintillation counter.
Other methods
SDS-Polyacrylamide gel electrophoresis (PAGE) was performed as described by Laemmli (Laemmli, 1970). For analysis of LEF-3 samples at non-reducing conditions, a loading buffer without 2-mercaptoethanol was used. Protein concentration in the purified samples was determined by SDS-PAGE followed by optical densitometry of the gels stained with Coomassie brilliant blue. Bovine serum albumin (BSA) loaded in different amounts on separate lanes of the same gel was used for generation of the calibration curve. For quantitative analysis, the stained gels and the fluorescence images were analyzed with ImageQuaNT software (Amersham Biosciences).
Acknowledgments
This research was supported by National Institutes of Health Grant GM060404 to GFR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ayres MD, Howard SC, Kuzio J, Lopez-Ferber M, Possee RD. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology. 1994;202:586–605. doi: 10.1006/viro.1994.1380. [DOI] [PubMed] [Google Scholar]
- Bauer CE, Elsen S, Bird TH. Mechanisms for redox control of gene expression. Annu Rev Microbiol. 1999;53:495–523. doi: 10.1146/annurev.micro.53.1.495. [DOI] [PubMed] [Google Scholar]
- Blackwell LJ, Borowiec JA, Masrangelo IA. Single-stranded-DNA binding alters human replication protein A structure and facilitates interaction with DNA-dependent protein kinase. Mol Cell Biol. 1996;16:4798–4807. doi: 10.1128/mcb.16.9.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Carstens EB. Identification of domains in Autographa californica multiple nucleopolyhedrovirus late expression factor 3 required for nuclear transport of P143. J Virol. 2005;79:10915–10922. doi: 10.1128/JVI.79.17.10915-10922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RJ, Fechheimer M, Miller LK. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science. 1991;254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- Dekker J, Kanellopoulos PN, van Oosterhout JA, Stier G, Tucker PA, van der Vliet PC. ATP-independent DNA unwinding by the adenovirus single-stranded DNA binding protein requires a flexible DNA binding loop. J Mol Biol. 1998;277:825–838. doi: 10.1006/jmbi.1998.1652. [DOI] [PubMed] [Google Scholar]
- Dudas KC, Ruyechan WT. Identification of a region of the herpes simplex virus single-stranded DNA-binding protein involved in cooperative binding. J Virol. 1998;72:257–265. doi: 10.1128/jvi.72.1.257-265.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas KC, Scouten SK, Ruyechan WT. Conformational change in the herpes simplex single-strand binding protein induced by DNA. Biochem Biophys Res Commun. 2001;288:184–190. doi: 10.1006/bbrc.2001.5766. [DOI] [PubMed] [Google Scholar]
- Evans JT, Rohrmann GF. The baculovirus single-stranded DNA binding protein, LEF-3, forms a homotrimer in solution. J Virol. 1997;71:3574–3579. doi: 10.1128/jvi.71.5.3574-3579.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JT, Rosenblatt GS, Leisy DJ, Rohrmann GF. Characterization of the interaction between the baculovirus ssDNA-binding protein (LEF-3) and putative helicase (P143) J Gen Virol. 1999;80:493–500. doi: 10.1099/0022-1317-80-2-493. [DOI] [PubMed] [Google Scholar]
- Gomes XV, Henricksen LA, Wold MS. Proteolytic mapping of human replication protein A: evidence for multiple structural domains and a conformational change upon interaction with single-stranded DNA. Biochemistry. 1996;35:5586–5595. doi: 10.1021/bi9526995. [DOI] [PubMed] [Google Scholar]
- Hajos JP, Pijnenburg J, Usmany M, Zuidema D, Zavodzsky P, Vlak JM. High frequency recombination between homologous baculoviruses in cell culture. Arch Virol. 2000;145:159–164. doi: 10.1007/s007050050012. [DOI] [PubMed] [Google Scholar]
- Hang X, Dong W, Guarino LA. The lef-3 gene of Autographa californica nuclear polyhedrosis virus encodes a single-stranded DNA-binding protein. J Virol. 1995;69:3924–3928. doi: 10.1128/jvi.69.6.3924-3928.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood SH, Li L, Ho PS, Preston AK, Rohrmann GF. AcMNPV late expression factor-5 interacts with itself and contains a zinc ribbon domain that is required for maximal late transcription activity and is homologous to elongation factor TFIIS. Virology. 1998;250:118–134. doi: 10.1006/viro.1998.9334. [DOI] [PubMed] [Google Scholar]
- Ito E, Sahri D, Knippers R, Carstens EB. Baculovirus proteins IE-1, LEF-3, and P143 interact with DNA in vivo: a formaldehyde cross-linking study. Virology. 2004;329:337–347. doi: 10.1016/j.virol.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Kamita SG, Maeda S, Hammock BD. High-frequency homologous recombination between baculoviruses involves DNA replication. J Virol. 2003;77:13053–13061. doi: 10.1128/JVI.77.24.13053-13061.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SO, Merchant K, Nudelman R, Beyer WF, Jr, Keng T, DeAngelo J, Hausladen A, Stamler JS. OxyR: a molecular code for redox-related signaling. Cell. 2002;109:383–96. doi: 10.1016/s0092-8674(02)00723-7. [DOI] [PubMed] [Google Scholar]
- Knipe DM, Quinlan MP, Spang AE. Characterization of two conformational forms of the major DNA-binding protein encoded by herpes simplex virus 1. J Virol. 1982;44:736–741. doi: 10.1128/jvi.44.2.736-741.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M, Ahrens CH, Goldbach RW, Rohrmann GF, Vlak JM. Identification of genes involved in DNA replication of the Autographa californica baculovirus. Proc Natl Acad Sci USA. 1994;91:11212–11216. doi: 10.1073/pnas.91.23.11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzio J, Pearson MN, Harwood SH, Funk CJ, Evans JT, Slavicek JM, Rohrmann GF. Sequence and analysis of the genome of a baculovirus pathogenic for Lymantria dispar. Virology. 1999;253:17–34. doi: 10.1006/viro.1998.9469. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Leisy DJ, Rohrmann GF. Characterization of the replication of plasmids containing hr sequences in baculovirus-infected Spodoptera frugiperda cells. Virology. 1993;196:722–730. doi: 10.1006/viro.1993.1529. [DOI] [PubMed] [Google Scholar]
- Li LL, Rohrmann GF. Characterization of a baculovirus alkaline nuclease. J Virol. 2000;74:6401–6407. doi: 10.1128/jvi.74.14.6401-6407.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Blissard GW. Analysis of an Autographa californica nucleopolyhedrovirus lef-11 knockout: LEF-11 is essential for viral DNA replication. J Virol. 2002;76:2770–2779. doi: 10.1128/JVI.76.6.2770-2779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Miller LK. The roles of eighteen baculovirus late expression factor genes in transcription and DNA replication. J Virol. 1995;69:975–982. doi: 10.1128/jvi.69.2.975-982.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XC, Shang JY, Yang ZN, Bao YY, Xiao Q, Zhang CX. Genome sequence and organization of a nucleopolyhedrovirus that infects the tea looper caterpillar, Ectropis obliqua. Virology. 2007;360:235–246. doi: 10.1016/j.virol.2006.10.024. [DOI] [PubMed] [Google Scholar]
- Mainz D, Quadt I, Knebel-Morsdorf D. Nuclear IE2 structures are related to viral DNA replication sites during baculovirus infection. J Virol. 2002;76:5198–5207. doi: 10.1128/JVI.76.10.5198-5207.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougal VV, Guarino LA. Autographa californica nuclear polyhedrosis virus DNA polymerase: measurements of processivity and strand displacement. J Virol. 1999;73:4908–4918. doi: 10.1128/jvi.73.6.4908-4918.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov VS. Helix-destabilizing properties of the baculovirus single-stranded DNA-binding protein (LEF-3) Virology. 2000;270:180–189. doi: 10.1006/viro.2000.0270. [DOI] [PubMed] [Google Scholar]
- Mikhailov VS, Bogenhagen DF. Effects of Xenopus laevis mitochondrial single-stranded DNA-binding protein on primer-template binding and 3′→5′ exonuclease activity of DNA polymerase γ. J Biol Chem. 1996;271:18939–18946. doi: 10.1074/jbc.271.31.18939. [DOI] [PubMed] [Google Scholar]
- Mikhailov VS, Mikhailova AL, Iwanaga M, Gomi S, Maeda S. Bombyx mori nucleopolyhedrovirus encodes a DNA-binding protein capable of destabilizing duplex DNA. J Virol. 1998;72:3107–3116. doi: 10.1128/jvi.72.4.3107-3116.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov VS, Okano K, Rohrmann GF. Baculovirus alkaline nuclease possesses a 5′→3′ exonuclease activity and associates with the DNA-binding protein LEF-3. J Virol. 2003;77:2436–2444. doi: 10.1128/JVI.77.4.2436-2444.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov VS, Okano K, Rohrmann GF. Specificity of the endonuclease activity of the baculovirus alkaline nuclease for single-stranded DNA. J Biol Chem. 2004;279:14734–14745. doi: 10.1074/jbc.M311658200. [DOI] [PubMed] [Google Scholar]
- Mikhailov VS, Okano K, Rohrmann GF. The redox state of the baculovirus single-stranded DNA-binding protein LEF-3 regulates its DNA binding, unwinding, and annealing activities. J Biol Chem. 2005;280:29444–29453. doi: 10.1074/jbc.M503235200. [DOI] [PubMed] [Google Scholar]
- Mikhailov VS, Okano K, Rohrmann GF. Structural and functional analysis of the baculovirus single-stranded DNA-binding protein LEF-3. Virology. 2006;346:469–478. doi: 10.1016/j.virol.2005.11.027. [DOI] [PubMed] [Google Scholar]
- Nagamine T, Kawasaki Y, Matsumoto S. Induction of a subnuclear structure by the simultaneous expression of baculovirus proteins, IE1, LEF3, and P143 in the presence of hr. Virology. 2006;352:400–407. doi: 10.1016/j.virol.2006.04.034. [DOI] [PubMed] [Google Scholar]
- Okano K, Mikhailov VS, Maeda S. Colocalization of baculovirus IE-1 and two DNA-binding proteins, DBP and LEF-3, to viral replication factories. J Virol. 1999;73:110–119. doi: 10.1128/jvi.73.1.110-119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano K, Vanarsdall AL, Mikhailov VS, Rohrmann GF. Conserved molecular systems of the Baculoviridae. Virology. 2006;344:77–87. doi: 10.1016/j.virol.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Okano K, Vanarsdall AL, Rohrmann GF. A baculovirus alkaline nuclease knockout construct produces fragmented DNA and aberrant capsids. Virology. 2007;359:46–54. doi: 10.1016/j.virol.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer DI, Volkman LE. Evidence for rolling circle replication of Autographa californica M nucleopolyhedrovirus genomic DNA. Arch Virol. 1997;142:2107–2113. doi: 10.1007/s007050050229. [DOI] [PubMed] [Google Scholar]
- Quadt I, van Lent JW, Knebel-Morsdorf D. Studies of the silencing of baculovirus DNA binding protein. J Virol. 2007;81:6122–6127. doi: 10.1128/JVI.02768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruyechan WT. N-ethylmaleimide inhibition of the DNA-binding activity of the herpes simplex virus type 1 major DNA-binding protein. J Virol. 1988;62:810–817. doi: 10.1128/jvi.62.3.810-817.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass-spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Analyt Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Uprichard SL, Knipe DM. Conformational changes in the herpes simplex virus ICP8 DNA-binding protein coincident with assembly in viral replication structures. J Virol. 2003;77:7467–7476. doi: 10.1128/JVI.77.13.7467-7476.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemain JL, Giedroc DP. Energetics of arginine-4 substitution mutants in the N-terminal cooperativity domain of T4 gene 32 protein. Biochemistry. 1993;32:11235–11246. doi: 10.1021/bi00092a038. [DOI] [PubMed] [Google Scholar]
- Vanarsdall AL, Mikhailov VS, Rohrmann GF. Characterization of a baculovirus lacking the DBP (DNA-binding protein) gene. Virology. 2007;364:475–85. doi: 10.1016/j.virol.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Oberley LW, Murhammer DW. Evidence of oxidative stress following the viral infection of two lepidopteran insect cell lines. Free Radic Biol Med. 2001;31:1448–1455. doi: 10.1016/s0891-5849(01)00728-6. [DOI] [PubMed] [Google Scholar]
- Wu Y, Carstens EB. A baculovirus single-stranded DNA binding protein, LEF-3, mediates the nuclear localization of the putative helicase P143. Virology. 1998;247:32–40. doi: 10.1006/viro.1998.9235. [DOI] [PubMed] [Google Scholar]
- You JS, Wang M, Lee SH. Functional characterization of zinc-finger motif in redox regulation of RPA-ssDNA interaction. Biochemistry. 2000;39:12953–12958. doi: 10.1021/bi001206f. [DOI] [PubMed] [Google Scholar]
- Young JC, MacKinnon EA, Faulkner P. The architecture of the virogenic stroma in isolated nuclei of Spodoptera frugiperda cells in vitro infected by Autographa californica nuclear polyhedrosis virus. J Struct Biol. 1993;110:141–153. [Google Scholar]