

Abstract
Unraveling functional and ancestral relationships between proteins as well as structure-prediction procedures require powerful protein-alignment methods. A structure-alignment method is presented where the problem is mapped onto a cost function containing both fuzzy (Potts) assignment variables and atomic coordinates. The cost function is minimized by using an iterative scheme, where at each step mean field theory methods at finite “temperatures” are used for determining fuzzy assignment variables followed by exact translation and rotation of atomic coordinates weighted by their corresponding fuzzy assignment variables. The approach performs very well when compared with other methods, requires modest central processing unit consumption, and is robust with respect to choice of iteration parameters for a wide range of proteins.
Keywords: algorithm, dynamical programming, fuzzy assignment, mean field annealing
Protein structure alignment is a subject of utmost relevance. It enables the study of functional relationships between proteins and is very important for homology and threading methods in structure prediction. Furthermore, grouping protein structures into fold families and subsequent tree reconstruction may unravel ancestral and evolutionary issues.
Structure alignment amounts to matching two 3D structures such that potential common substructures, e.g., α-helices, have priority (see Fig. 1). The latter is accomplished by allowing for gaps in either of the chains. At first sight, the problem may appear very similar to sequence alignment as manifested in some of the vocabulary (gap costs, etc.). However, from an algorithmic standpoint, there is a major difference. Whereas sequence alignment can be solved within polynomial time by using dynamical programming methods (1), this is not the case for structure alignment, because rigid bodies are to be matched. Hence, for all structure-alignment algorithms the scope is limited to high-quality approximate solutions.
Fig. 1.

An example of pairwise structure alignment. The two proteins 1ECD (Upper Left) and 1MBD (Upper Right) are to be structurally matched to each other. (Lower) The resulting alignment by using our algorithm. The proteins are in the backbone representation.
Existing methods for structure alignment fall into two broad classes depending on whether one directly minimizes the interatomic distances between two structures or minimizes the distance between substructures that are either preselected or supplied by an algorithm involving intraatomic distances. For an extensive list of references to structure-alignment methods see ref. 2.
One approach within the first category is the iterative dynamical programming method (3, 4), where one first computes a distance matrix between all pairs of atoms forming a similarity matrix, which by dynamical programming methods gives rise to an assignment matrix mimicking the sequence-alignment procedure. One of the chains then is moved toward the other by minimizing the distance between assigned pairs. This two-step procedure is repeated until convergence. A different interatomic approach is pursued in ref. 5, where the area rather than the distances between two structures is minimized.
In refs. 6 and 7 the approach is different. Here one compares distance matrices within each of the two structures to be aligned, which provide information about similar substructures. These substructures are subsequently matched. A similar framework is used in refs. 8 and 9.
The iterative dynamical programming method (4) has been extensively assessed for backbone structures (10) from the Structural Classification of Proteins (SCOP) database (11), in which protein structures have been classified by visual inspection. Some comparisons with SCOP have also been performed (12) by using the method in ref. 8.
The key ingredients of our approach are (i) a cost-function formulation of the problem simultaneously in terms of binary (Potts) assignment variables and real-valued atomic coordinates and (ii) a minimization of the cost function by an iterative method, in which each iteration contains two steps. First, fuzzy assignment variables are calculated based on the distances between the amino acids in the two proteins to be aligned. Second, exact rotation and translation of amino acid coordinates weighted with the corresponding fuzzy assignment variables are performed. The approach, which is very general, has some very appealing properties. First, probabilistic interpretation of the results is present without tedious Monte Carlo estimates, because the algorithm is deterministic. Among other things, this implies that the approach is less sensitive to the choice of distance metric, because the distances are weighted with fuzzy numbers. Second, almost arbitrary additional constraints are easily incorporated into the formalism including, for example, different functional forms of gap penalties and sequence-matching preferences.
Our method was tested by using the Cα representation of backbones by comparing the results with the approaches in refs. 4, 6, and 7 as implemented in the Yale Alignment Server, Dali and CE, respectively. In choosing benchmark problems, we chose two sets of problems already used in this context: (i) pairs with marginal sequence overlap but in which each protein in a pair belongs to the same SCOP superfamily and (ii) 10 “difficult” problems that have been used previously to evaluate structure-alignment methods (7). In all but one case our method gave results as good as or better than the other algorithms and was of similar speed computationally.
Alignment Method
Consider two proteins with M and N atoms that are to be structurally aligned by a series of weighted rigid body transformations of one of the chains, keeping the other one fixed. We denote by xi (i = 1,..., M) and yj (j = 1,..., N) the atom coordinates of the first and second chain, respectively. The phrase “atom” is used here in a generic sense: it could represent individual atoms but also groups of atoms. In our applications it will mean Cα atoms along the backbone. We use a square distance metric between the chain atoms,
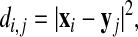 |
[1] |
but the formalism is not confined to this choice.
The alignment of the two proteins is accomplished in an iterative two-step procedure as follows:
Calculation of a fuzzy assignment matrix 𝒲, where element
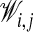 ∈ [0, 1] is the probability that atom i in the first chain is matched to atom j in the second.
∈ [0, 1] is the probability that atom i in the first chain is matched to atom j in the second.Rigid body transformation of one of the chains by using fixed 𝒲.
We will start by describing the latter part.
Weighted Transformations. The objective of the rigid body transformation of one of the chains is to minimize the distance between the matched elements of the two chains. Let x′i be the coordinates of the translated and rotated protein, i.e., 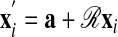 . Based on the fuzzy assignment matrix 𝒲 we determine the translation vector a and the rotation matrix ℛ at each iteration by minimizing the chain error function at fixed yj
. Based on the fuzzy assignment matrix 𝒲 we determine the translation vector a and the rotation matrix ℛ at each iteration by minimizing the chain error function at fixed yj
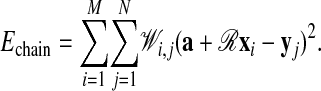 |
[2] |
This minimization problem can be solved exactly with closed-form expressions for ℛ and a that minimizes Echain (13). It should be noted that this solution is rotationally invariant (independent of ℛ) for the special case when the atoms in the two chains match each other with the same weight, i.e., when 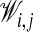 is constant for all i and j.
is constant for all i and j.
Global Sequence Alignment. Structure alignment of two proteins by using a fixed distance measure between the atoms in the two chains is closely related to global sequence alignment of proteins. The latter is conveniently solved by using the Needleman–Wunsch algorithm (1). In fact, our approach to structure alignment is based on the Needleman–Wunsch algorithm but augmented to produce a fuzzy assignment matrix. The fuzziness is controlled by a parameter that is not held constant during the iterative procedure.
We next rephrase the Needleman–Wunsch algorithm in an implementation that will allow for a straightforward introduction of our approach. Let X = (X1, X2,..., XM) and Y = (Y1, Y2,..., YN) denote the two chains containing M and N residues in a dot-matrix representation, respectively. Every possible alignment of the two chains (not including permutations of atoms in a chain) can be represented as a directed path on the (M + 1) × (N + 1) alignment matrix (Fig. 2 Left). Each dot (i, j) has, excluding obvious boundary restrictions, three possible predecessors along the alignment path, which are denoted by k = 1, 2, and 3 (Fig. 2 Right).
Fig. 2.

Aligning two chains. (Left) Alignment matrix for an alignment between the two chains X = (X1, X2,..., XM) and Y = (Y1, Y2,..., YN). (Right) Unit vectors connecting to the three possible predecessors to a dot (i, j).
The alignment cost 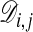 for the optimal alignment of subchains (X1, X2,..., Xi) and (Y1, Y2,..., Yj) is given by
for the optimal alignment of subchains (X1, X2,..., Xi) and (Y1, Y2,..., Yj) is given by
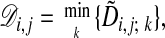 |
[3] |
where 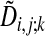 is the alignment cost if the alignment path is forced to pass through the preceding node given by k and is calculated by using the recursive relation
is the alignment cost if the alignment path is forced to pass through the preceding node given by k and is calculated by using the recursive relation
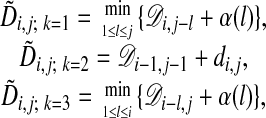 |
[4] |
where α(k) is the gap penalty for a gap of length k. Computing 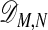 by using Eqs. 3 and 4 yields the cost for the optimal alignment of the two chains. In this formulation we use costs rather than scores, because the objective is to minimize the distance between matched atoms.
by using Eqs. 3 and 4 yields the cost for the optimal alignment of the two chains. In this formulation we use costs rather than scores, because the objective is to minimize the distance between matched atoms.
To obtain the actual optimal alignment path one records, for each node (i, j), which of the three 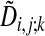 in Eq. 3 achieved the lowest cost. This information is stored in the binary decision variables si,j; k such that
in Eq. 3 achieved the lowest cost. This information is stored in the binary decision variables si,j; k such that
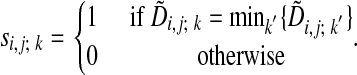 |
[5] |
Using si,j; k, we rewrite Eq. 3 as
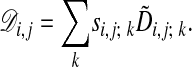 |
[6] |
There are obvious boundary conditions on the first row and column of the alignment matrix that make si,j; k constant (0 or 1) on these boundaries.
Introducing Fuzzy Alignment Paths. We next introduce fuzzy alignment paths that will finally lead to a fuzzy assignment matrix. The above introduction of si,j; k means that we have strictly binary “winner-takes-all” variables; only one alignment path is allowed, the optimal path. We now replace the binary variable si,j; k by the continuous variable vi,j; k, with the property that Σkvi,j; k = 1 (14). This allows for the interpretation that vi,j; k is a probability that an optimal alignment path that passes through (i, j) also passes through the preceding node specified by k. The replacement
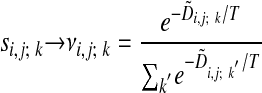 |
[7] |
can be viewed as a soft implementation of the “min” function in Eq. 3 where the parameter T > 0 controls the fuzziness. For large T, almost all paths are equally probable, and in the limit T → 0, the original Needleman–Wunsch algorithm is recovered.
In what follows, we will restrict ourselves to position dependent linear gap penalties of the type
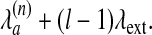 |
[8] |
Here, 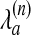 is the penalty for matching atom a in chain n to a gap, λext is the extension penalty, and l is the gap length. Eqs. 3 and 4 now can be expressed in terms of vi,j; k,
is the penalty for matching atom a in chain n to a gap, λext is the extension penalty, and l is the gap length. Eqs. 3 and 4 now can be expressed in terms of vi,j; k, 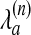 , and λext
, and λext
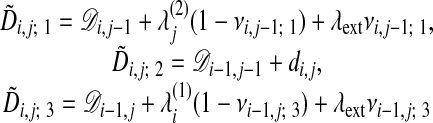 |
[9] |
and finally the optimal alignment cost at node (i, j),
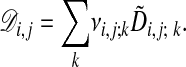 |
[10] |
Obtaining an Assignment Matrix. From the probabilities vi,j; k it is straightforward to calculate Pi,j; the probability that node (i, j) is part of the optimal path. This quantity can be calculated with a similar recursive relation as for 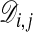 . With the obvious initial value PM,N = 1 one has
. With the obvious initial value PM,N = 1 one has
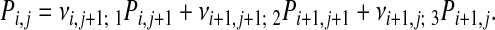 |
[11] |
By construction this leads to the necessary condition P0,0 = 1. Finally, a fuzzy assignment matrix can now be calculated by means of Pi,j and vi,j; 2,
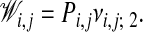 |
[12] |
In other words, the probability for matching atom i in the first chain and atom j in the second chain is the product of the probability that (i, j) is part of the optimal path and the probability that this pair is locally matched.
Summary of the Algorithm. As stated in the beginning, the pairwise structure alignment of two proteins is accomplished by iteratively computing a fuzzy assignment matrix (Eq. 12) and minimizing distances between matched atoms of the two chains (Eq. 2). The T parameter that controls the degree of fuzziness of the assignment matrix is annealed during the iterative procedure from a large value to a small value, which ensures the necessary transition from a fuzzy assignment matrix to a binary one where uniquely matched atoms are identified. The algorithmic steps are shown in Fig. 4, which is published as supporting information on the PNAS web site, www.pnas.org.
Relation to Mean Field Annealing. We recognize the similarity of the vi,j; k variables (Eq. 7) to Potts variables frequently occurring in mean field annealing algorithms. A suitable starting point for the assignment problem is given by Eq. 6, where the binary variable si,j; k encodes one of three possible directions for each lattice point (i, j). The smallest local energy 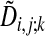 defines the optimal direction k. Now, the mean field version of Eq. 6 is obtained by replacing si,j; k with its thermal average vi,j; k in the mean field approximation given by Eq. 7. By definition, Σkvi,j; k = 1, which allows for a probability interpretation of vi,j; k. This interpretation is exploited in the definition of the fuzzy assignment matrix (Eq. 12), which in turn is used for the rigid body transformations defined by the minimum of Eq. 2. Thus, no mean field approximation of the entire minimization problem is used.
defines the optimal direction k. Now, the mean field version of Eq. 6 is obtained by replacing si,j; k with its thermal average vi,j; k in the mean field approximation given by Eq. 7. By definition, Σkvi,j; k = 1, which allows for a probability interpretation of vi,j; k. This interpretation is exploited in the definition of the fuzzy assignment matrix (Eq. 12), which in turn is used for the rigid body transformations defined by the minimum of Eq. 2. Thus, no mean field approximation of the entire minimization problem is used.
By introducing continuous vi,j; k variables that can evolve in a space not accessible by the original discrete ones, we introduce an (infinitely large) set of possible alignments between the two proteins. When T → 0, the original discrete set of possible alignments is recovered. The annealing procedure is thus a way of producing intermediate (fuzzy) alignments as an efficient pathway to finding the optimal structure alignment of two proteins.
Side Chains. It is difficult to align proteins that consist of strands by using only Cα coordinates. Strands in the two proteins are often matched satisfyingly to one another, whereas the individual atoms are aligned such that one strand is translated with respect to the other. The generality of our method makes it straightforward to address this problem by extending the method to handle more detailed chain representations, e.g., side-chain orientation. The angle between two side chains can be used to construct a suitable distance measure for the two side chains (4). This distance then is added to the distance between the corresponding Cα atoms; e.g.,
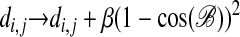 |
[13] |
where β is a constant and ℬ is the angle between the two side chains.
Similar Approaches. Our approach has strong similarities to that in refs. 3 and 4. However, there are two major differences. (i) A scoring procedure is used in refs. 3 and 4, whereas in our approach a cost is minimized, mainly affecting the way gaps are treated. (ii) We compute a fuzzy assignment matrix and minimize distances between atoms based on this matrix, which is different from refs. 3 and 4 where a binary assignment matrix is used.
In the field of 3D point matching in computer vision one can find similarities with the algorithm in ref. 15.
Results and Discussion
To test the quality of our alignment algorithm, we compared alignments of protein pairs with results from other automatic procedures. For many of the tested pairs, each protein belongs to the same SCOP superfamily. Here, the goal was not a full investigation of all families but rather to explore a limited set with representative variation. Pairs were picked both from the SCOP families investigated in ref. 10 and the structures studied in ref. 7. Our choice of pairs was essentially based on two criteria. First, the pairs should include diverse structures. Second, we selected pairs according to how hard the structural similarities have been considered to find. In ref. 10 some families are considered to be very easy, easy, and difficult to align, and we included pairs from all these categories. In addition, we selected 10 of the most difficult pairs from the benchmark developed by Fischer et al. (16) to assess the performance of fold recognition methods that seek to align a protein sequence to a 3D structure. Their benchmark consists of a set of protein sequences matched by superposition to known structures. This set covers a wide range of protein families and includes matching proteins with insignificant sequence similarity. These 10 difficult structures have been used previously to evaluate structure-alignment methods (7).
We compared our results with the Yale Alignment Server (http://bioinfo.mbb.yale.edu/align), Dali (www.ebi.ac.uk/dali), and CE (http://cl.sdsc.edu/ce/ce_align.html). The Yale server applies postprocessing to its alignments by removing aligned atoms with large distances between them in an iterative manner subject to a termination criteria. A similar procedure of course is possible in our approach, but we have chosen at this stage to keep the algorithm simple and not apply any postprocessing.
We denote proteins by their Protein Data Bank (PDB, www.rcsb.org/pdb) (17) identifiers. In the case of multichain or domain proteins, we restricted our alignments to chains or parts of chains, denoted by their SCOP domain labels.
Our algorithm contains parameters of two types: one set of algorithm-specific parameters and one set related to gap penalties. For the results presented here, all algorithm-specific parameters were set by using a training set that consisted of 10 randomly selected pairs from the full data set (see Table 1, which is published as supporting information on the PNAS web site). The parameters used for the gap penalties were chosen to give alignments in which the number of aligned atoms were comparable to the other methods. Once all the parameters were set, we used these parameter settings for all pairs in the data set (see Table 2, which is published as supporting information on the PNAS web site).
A summary of the results for all the protein pairs in terms of the rms distance (rmsd), between aligned atoms, and the number of aligned atoms (N) is shown in Table 1. It is clear that our algorithm performs very well in comparisons with the other methods and generally produced alignments with lower or equal rmsd. An exception is 1CRL-1EDE, for which our method failed to find a good alignment. In general, Dali performed worse than CE and our method in terms of rmsd and N. However, one must keep in mind that it is not straightforward to assess alignment algorithms in terms of rmsd and N, because there are no obvious figure of merits. In particular, the postprocessing applied by the Yale server typically results in alignments with lower N as compared with the other methods. To investigate the dependence between rmsd and N and allow for a better evaluation of our method, we ran our algorithm with varying sizes of the gap penalty parameter for many of the pairs where the alignment methods gave different N. For each of these pairs, we found that our algorithm, for identical N, typically resulted in a lower rmsd than the other algorithms (Fig. 3).
Fig. 3.

Alignment results for a set of protein pairs in terms of rmsd and number of aligned atoms (N). The results from Yale (red circles), Dali (green squares), CE (blue diamonds), and our method (black asterisks) are plotted. For comparison, different rmsd–N pairs for our algorithm, obtained by varying the size of the gap penalty parameter, are shown (solid lines). Our method gives the largest improvements compared to the others for protein pairs from the 10 difficult structures (protein pairs marked with asterisks).
One can also investigate the quality of alignments by comparing them to manual alignments from the literature. This was done in ref. 10, and the conclusion was that an automatic procedure using only Cα atoms works in general, but proteins that contain only strands, such as some immunoglobulins, were problematic. We aligned the domain 7FABl2 with the chain 1REIa and got a competitive alignment in terms of low rmsd and large N (Fig. 3). However, if we look at amino acid pairs considered important in the manual alignments, the results are worse. The manual “gold-standard” alignment of these proteins contains seven core regions consisting of a total of 36 amino acid pairs (18). Looking at our alignment in detail we found that only three of the seven core regions and 19 of the 36 amino acid pairs were aligned correctly. CE and Yale both failed to align any of the core amino acid pairs correctly, whereas Dali aligned five of the seven core regions correctly (27 of 36 amino acid pairs). By modifying the Yale approach to include variable gap costs and take side-chain orientation into account, six core regions could be aligned correctly (10). Therefore, we included side-chain orientation [with β = 0.005 (see Eq. 13) and all other parameters unchanged], which improved our results, and we got five of seven core regions aligned correctly (25 of 36 amino acid pairs).
Some alignment algorithms are designed to be fast, at the potential expense of accurate alignment of core amino acids, to allow for structural comparisons of all pairs of proteins in large protein databases. In these methods, the protein structures are initially reduced to their secondary structures, and these are subsequently aligned (8, 9). Interesting candidate protein pairs then can be evaluated in more detail. Our method was designed to provide accurate alignments at the amino acid level. Nevertheless, our algorithm is relatively fast. It is implemented in c, scales proportional to the chain lengths squared, and on the average requires a few seconds on a Pentium 4 2-GHz personal computer.
In summary, we developed an efficient approach to structure alignment. It was tested by using a wide variety of proteins and protein structures. We used a subset of the protein pairs to tune the parameters of the algorithm and found that, by using these parameter settings, the method produced very robust results for all pairs tested. The results were better than or equal to the other methods tested except in one case for which we failed to get a good alignment. In addition to very good performance, our approach has many appealing properties. It provides a probabilistic interpretation of the result without tedious stochastic simulations. Also, it is readily extended to handle more detailed chain representations (e.g., side-chain orientation) and additional user-provided constraints of almost any kind.
Supplementary Material
Acknowledgments
C.P. thanks the Theory Group at the Stanford Linear Accelerator Center, where this work was initiated, for its hospitality. This work was supported in part by the Swedish Research Council and Swedish Foundation for Strategic Research.
Abbreviations: SCOP, Structural Classification of Proteins; rmsd, rms distance.
References
- 1.Needleman, S. B & Wunsch, C. D. (1970) J. Mol. Biol. 48, 443–453. [DOI] [PubMed] [Google Scholar]
- 2.Szustakowski, J. & Weng, Z. (2000) Proteins 38, 428–440. [DOI] [PubMed] [Google Scholar]
- 3.Laurents, D. V., Subbiah, S. & Levitt, M. (1993) J. Mol. Biol. 3, 141–148. [DOI] [PubMed] [Google Scholar]
- 4.Gerstein, M. & Levitt, M. (1996) in Proceedings of the Fourth International Conference on Intelligent Systems in Molecular Biology, eds. States, D. J., Agarwal, P., Gaasterland, T., Hunter, L. & Smith, R. F. (American Assoc. for Artificial Intelligence Press, Menlo Park, CA), pp. 59–67.
- 5.Falicov, A. & Cohen, F. E. (1996) J. Mol. Biol. 258, 871–892. [DOI] [PubMed] [Google Scholar]
- 6.Holm, L. & Sander, C. (1993) J. Mol. Biol. 233, 123–138. [DOI] [PubMed] [Google Scholar]
- 7.Shindyalov, I. N. & Bourne, P. E. (1998) Protein Eng. 11, 739–747. [DOI] [PubMed] [Google Scholar]
- 8.Gibrat, J.-F., Madej, T. & Bryant, S. H. (1996) Curr. Opin. Struct. Biol. 6, 377–385. [DOI] [PubMed] [Google Scholar]
- 9.Lu, G. (2000) J. Appl. Crystallogr. 33, 176–183. [Google Scholar]
- 10.Gerstein, M. & Levitt, M. (1998) Protein Sci. 7, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hubbard, T. J., Murzin, A. G., Brenner, S. E. & Chothia, C. (1997) Nucleic Acids Res. 25, 236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuo, Y. & Bryant, S. H. (1999) Proteins 35, 70–79. [PubMed] [Google Scholar]
- 13.von Neumann, J. (1937) Tomsk Univ. Rev. 1, 286–300. [Google Scholar]
- 14.Häkkinen, J., Lagerholm, M., Peterson, C. & Söderberg, B. (1998) Neural Comput. 10, 1587–1599. [DOI] [PubMed] [Google Scholar]
- 15.Gold, S., Lu, C. P., Rangarajan, A., Pappu, S. & Mjolsness, E. (1995) in Advances in Neural Information Processing Systems, eds. Tesauro, G., Touretzky, D. & Leen, T. (MIT Press, Cambridge, MA) Vol. 7, pp. 957–964. [Google Scholar]
- 16.Fischer, D., Elofsson, A., Rice, D. & Eisenberg, D. (1996) Proceedings of the 1st Pacific Symposium on Biocomputing, eds. Hunter, L. & Klein, T. (World Scientific, Singapore), pp. 300–318.
- 17.Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000) Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lesk, A. M. & Chothia, C. (1982) J. Mol. Biol. 160, 325–342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.