

Abstract
This report demonstrates that a single set of identical synthetic multifunctional pores can detect the activity of many different enzymes. Enzymes catalyzing either synthesis or degradation of DNA (exonuclease III or polymerase I), RNA (RNase A), polysaccharides (heparinase I, hyaluronidase, and galactosyltransferase), and proteins (papain, ficin, elastase, subtilisin, and pronase) are selected to exemplify this key characteristic of synthetic multifunctional pore sensors. Because anionic, cationic, and neutral substrates can gain access to the interior of complementarily functionalized pores, such pores can be the basis for very user-friendly screening of a broad range of enzymes.
There are compelling reasons to believe that the “universal enzyme sensor,” a user-friendly, noninvasive device that can detect all existing enzyme activities, belongs to the world of fiction and, despite functional proteomics, will never become reality (1, 2, ‡). However, it would be erroneous to conclude that efforts to maximize adaptability of noninvasive enzyme sensors to as many enzymes as possible are a simple waste of time. In this study, selected examples from biopolymer enzymology are used to demonstrate that a set of identical synthetic multifunctional pores (SMPs) can be used for noninvasive detection of the activity of many different enzymes (Fig. 1).
Fig. 1.

Enzymes detected with a single set of identical SMPs. a, Reported in ref. 1.
In brief, if substrates bind and block the pore better than products, enzyme activity gradually removes the blocking agent and the pore can open. On the other hand, if products bind and block the pores better than the substrates, enzyme activity gradually produces blocking agents that can block the pores (Fig. 2). Thus, if there is substantial molecular recognition of either substrate or product by the same pore, the only prerequisite for detecting enzyme activities with SMP sensors is a simple method to distinguish between blocked and unblocked pores.
Fig. 2.

Enzyme sensing with SMPs shown as schematic oversimplification to outline the concept. (A) If substrates block the pore better than products, enzyme action produces reaction mixtures with gradually reduced ability to block the pore. (B) Enzyme-gated pore closing occurs if products block the pore better than substrates. (A and B) Rigid-rod β-barrel SMPs are depicted in axial view with β-strands as solid (backbone) and dotted (hydrogen bonds) lines. (C) Rigid-rod β-barrel SMPs 1 and 2 depicted as schematic cutaway suprastructures with peptide sequences B–F depicted dark on white for external and white on dark for internal amino acid residues (single-letter abbreviations). We reiterate that all reported suprastructures may be viewed as, at worst, simplifying but productive working hypotheses compatible with experimental data.
As SMPs we introduced rigid-rod β-barrels 1 and 2 (Fig. 2C) (2). Whereas various other rigid-rod β-barrel SMPs are available to cope with particular sensing requirements, SMPs other than rigid-rod β-barrels remain underexplored (3–9), particularly when compared with the remarkable progress made with bio-engineered multifunctional pores as stochastic sensors of single analytes (10–13).
The syntheses of barrel-stave supramolecules 1 (14) and 2 (15) from commercial biphenyl and amino acid derivatives in 19 steps overall each have been described. The characteristics of pores formed by rigid-rod β-barrels 1 and 2 in spherical and planar lipid bilayer membranes have also been reported (2, 14–16). Even without optimization, the extraordinary permeabilizing activity of these p-octiphenyl β-barrels makes it possible to perform >300,000 enzyme assays per mg of polypeptide (1).
l-histidine (H) and l-arginine (R) residues at the inner barrel surface account for the multifunctionality of pore 1. Lining the ion-conducting pathway of SMP 1, these cationic residues recognize anionic substrates and products >10,000 times better than biological pores such as melittin (N.S. and S.M., unpublished work). In SMP 2, the ion-conducting pathway is functionalized with anionic l-aspartates (Ds) instead.
Both barrel-stave supramolecules comprise hydrophobic amino acid residues at the outer barrel surface to interact with the hydrophobic core of bilayer membranes, i.e., to form pores. The external LWV triads in SMP 1 further serve to destabilize external surfaces formed by l-leucines (Ls) only as in SMP 2 (W, l-tryptophan; V, l-valine). The advantageous characteristics of unstable (rather than stable) pores for many practical applications of SMPs hint to the poorly defined importance of supramolecular dynamics for function.
Among many methods available to detect “enzyme-gated” pore opening and closing, we used the emission of 5(6)carboxyfluorescein (CF) as a convenient “naked-eye” read-out (1). This method is based on the encapsulation of CF within vesicles at self-quenching concentrations. This self-quenching vanishes as soon as CF can move across an open pore and dilute into the external medium, reporting unblocked pores by an increase in emission intensity. CF self-quenching therefore remains unchanged as long as the pore is blocked by a substrate. If substrates bind better than enzyme products, enzyme activity is reported by a gradual increase in CF emission (Figs. 2 A and 3); conversely, if products bind better than substrates, enzyme activity is reported by a gradual decrease in CF emission (Fig. 2B).
Fig. 3.

Fluorometric detection of enzyme activity with SMPs for 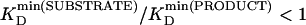 . The addition of an aliquot taken from the reaction mixture at the beginning of the reaction to EYPC-LUVs ⊃ CF followed by SMP addition does not result in an increase in CF emission because CF efflux is blocked by substrates bound to the SMP. Performance of the same experiment with an aliquot taken at the end of the reaction gives an increase in CF emission because the good substrate blocker is now converted into a poor product blocker.
. The addition of an aliquot taken from the reaction mixture at the beginning of the reaction to EYPC-LUVs ⊃ CF followed by SMP addition does not result in an increase in CF emission because CF efflux is blocked by substrates bound to the SMP. Performance of the same experiment with an aliquot taken at the end of the reaction gives an increase in CF emission because the good substrate blocker is now converted into a poor product blocker.
In the following, we first use the example of DNA exonuclease III to describe the methodology of noninvasive, fluorometric enzyme sensing with SMPs in detail. Then, we demonstrate adaptability of the same enzyme sensor (i.e., SMP 1) to watch enzymes catalyzing either synthesis or degradation of DNA, RNA, polysaccharides, and proteins work. Proteases are finally used to demonstrate access to cationic, anionic, and neutral substrates by using SMPs with complementary internal functionalization.
Materials and Methods
Materials. Egg yolk phosphatidylcholine (EYPC) was from Avanti Polar Lipids, and CF, buffers, salts, substrates, products, and enzymes were from Sigma or Fluka–Aldrich. Average molecular weights of polymers were calculated from the number n, of monomers per polymers in Table 1 {n = KD(MONO)/KD; (deoxyadenylic acid, thymidylic acid) copolymer duplex [poly(dA,dT)]2, n = KD(MONO) /2KD}. 13,23,32,43,52,6372,83-Octakis(-OCH2CO-Leu-Arg-Trp-His-Val-NH2)-p-octiphenyl (1m) was synthesized as reported (14). Concentration of monomer stock solutions in DMSO were corroborated by UV-visible in diluted MeOH [ε(p-octiphenyl) = 46.1 mM–1·cm–1 (320 nm); Varian Cary 1 Bio spectrophotometer]. Concentrations of pore 1 refer to tetrameric self-assemblies of 1m. Stock solutions of large unilamellar vesicles (LUVs) composed of EYPC and loaded with CF (i.e., EYPC-LUVs ⊃ CF) were prepared by extrusion (see Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org). The characteristics are: 1.8 mM EYPC [phosphate analysis, ≥22 nM LUVs (assuming diameter ≤ 100 nm)]; inside, 50 mM CF/10 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (Hepes)/10 mM NaCl, pH 7.4; outside, 10 mM Hepes/107 mM NaCl, pH 7.1. For the general procedure below, 0.1 ml of EYPC-LUVs ⊃ CF stock solution was added to 1.9 ml of buffer (10 mM Hepes/107 mM NaCl, pH 6.5).
Table 1. Enzyme screening with SMPs.
| Enzyme | Pore* | Substrates (S)† | KD, M‡§ | KD(MONO), M¶ | Products (P)†∥ | KD, M‡§ | S, M** | SR†† | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | DNA exonuclease III | 1 | [poly(dA,dT)]2 | 1.9 × 10-10 | 2.3 × 10-6 | 5′-dAMP | 1.2 × 10-3 | 1.9 × 10-10 | 2.0 × 10-3 |
| 5′-dTMP | 2.5 × 10-3 | ||||||||
| 2 | DNA polymerase I | 1 | dATP | 2.9 × 10-5 | [poly(dA,dT)]2 | 1.9 × 10-10 | 1.9 × 10-10 | 1.3 × 101 | |
| Klenow fragment | dTTP | n.d. | Pyrophosphate | 1.0 × 10-3 | |||||
| 3 | RNase A | 1 | poly(C) | 1.9 × 10-9 | 9.3 × 10-7 | 3′-CMP | 4.5 × 10-3 | 1.9 × 10-9 | 2.1 × 10-4 |
| 4 | RNase A | 1 | Polyuridylic acid | 2.1 × 10-10 | 5.8 × 10-7 | 3′-UMP | n.d. | 2.1 × 10-10 | n.d. |
| 5 | RNase A | 1 | Polyaderylic acid | 7.2 × 10-10 | 9.7 × 10-7 | 3′-AMP | n.d. | 7.2 × 10-10 | n.d. |
| 6 | Heparinase I | 1 | Heparin | 4.7 × 10-8 | n.d. | ΔDiHs | n.d. | 4.7 × 10-8 | n.d. |
| 7 | Hyaluronidase | 1 | Hyaluronan | 1.6 × 10-10 | 1.0 × 10-6 | ΔDiHAs | n.d. | 1.9 × 10-10 | n.d. |
| 8‡‡ | Galactosyl-transferase | 1 | UDPGal | >7 × 10-2 | 5′-UDP | 1.2 × 10-3 | 1.2 × 10-3 | >4 × 101 | |
| GlcNAc | >5 × 10-2 | Galβ1 → GlcNAc | >5 × 10-2 | ||||||
| 9 | Pronase | 1 | PLE | 1.4 × 10-8 | 1.3 × 10-6 | l-glutamate (E) | >5 × 10-4 | 1.4 × 10-8 | <3 × 10-3 |
| 10 | Papain | 1 | PLE | 1.4 × 10-8 | 1.3 × 10-6 | l-glutamate (E) | >5 × 10-4 | 1.4 × 10-8 | <3 × 10-3 |
| 11 | Ficin | 1 | PLE | 1.4 × 10-8 | 1.3 × 10-6 | l-glutamate (E) | >5 × 10-4 | 1.4 × 10-8 | <3 × 10-3 |
| 12 | Elastase | 1 | PLE | 1.4 × 10-8 | 1.3 × 10-6 | l-glutamate (E) | >5 × 10-4 | 1.4 × 10-8 | <3 × 10-3 |
| 13 | Subtilisin | 1 | PLE | 1.4 × 10-8 | 1.3 × 10-6 | l-glutamate (E) | >5 × 10-4 | 1.4 × 10-8 | <3 × 10-8 |
| 14 | Subtilisin | 1 | PDE | 1.9 × 10-8 | 1.6 × 10-6 | d-glutamate | >5 × 10-4 | 1.9 × 10-8 | <3 × 10-3 |
| 15 | Papain | 2 | PLR | 1.9 × 10-8 | 1.3 × 10-6 | l-arginine (R) | >5 × 10-4 | 1.9 × 10-8 | <3 × 10-3 |
| 16 | Papain | 2 | PLK | 1.1 × 10-8 | 1.5 × 10-6 | l-lysine (K) | >5 × 10-4 | 1.1 × 10-8 | <3 × 10-3 |
| 17 | n.d. | 1 | PLN | 3.7 × 10-8 | 3.5 × 10-6 | l-asparagine (N) | >1 × 10-2 | 3.7 × 10-8 | <4 × 10-4 |
n.d., not determined.
See Fig. 2.
ΔDiH, heparin disaccharide product; ΔDiHA, hyaluronan disaccharides; PLK, poly-l-lysine; PLN, poly-l-asparagine.
KD = global dissociation constant of [(supra)macro] molecular pore blockers.
All reported KD values are conditional to the employed method of detection (see text) and can vary substantially with experimental conditions.
KD(MONO) = average KD per monomer in (supra)macromolecular guests.
Eventual oligomer products are not indicated for clarity only.
S = sensitivity of pore sensor.
SR = selectivity requirement for pore sensor.
Data are from ref. 1. [Reproduced with permission from ref. 1 (Copyright 2002, AAAS, www.sciencemag.org).]
Enzyme Sensing with SMP 1 (General Procedure). Heparin (20 μM) was incubated with heparinase I (50 units/ml; EC 4.2.2.7, Flavobacterium heparinum) in buffer (10 mM Hepes/107 mM NaCl, pH 7.5) at room temperature. After 30 min, 20 μl of the reaction mixture was taken and added to 2 ml of gently stirred EYPC-LUVs ⊃ CF suspension in a thermostated fluorescence cuvette [90 μM EYPC/50 mM CFINSIDE/≤200 nM heparin (KD = 47 nM) [see Dissociation Constants (KD Values) of Substrate/Product Blockers]/10 mM Hepes/107 mM NaCl, pH 6.5]. Then, pore 1 was added (20 μl of 25 μM monomer in DMSO, 63 nM final; lipid/pore = 1.4 × 103) (see Calibration of SMP 1), and the change in CF emission intensity It with time was monitored (λem = 510 nm, λex = 495 nm; FluoroMax-2, JobinYvon, Longjumeau, France). Approximately 250 sec after pore addition, 40 μl of 1.2% aqueous Triton X-100 was added for lysis. Fluorescence kinetics were normalized to fractional emission intensity I by using
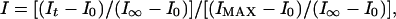 |
[1] |
where I0 = It at pore addition, I0 = It at saturation after lysis, and IMAX = It at maximal emission intensity with blocker-free pore before lysis (see Calibration of SMP 1). From the normalized curves, I(t=0.5) was recorded [I(t=0.5) = maximal I before lysis for pores in presence of an aliquot reaction mixture taken 0.5 h after the beginning of the reaction]. This procedure was repeated with 20 μl of reaction mixture taken after 2, 4, 6, 8, and 11 h of reaction time to obtain I(t=2), I(t=4), I(t=6), I(t=8), and I(t=11). All I(t) values were converted into fractional pore blockage YE (YE = 1 – {[I(t=0) – I(t)]/[I(t=0) – I(t=0)]}) and plotted as a function of reaction time t (Fig. 4D, filled circles). Control experiments included repetition of the above-described set of experiments with less (25 units/ml) and no (Fig. 4D, open circles and X) heparinase (see Special Cases).
Fig. 4.

Real-time enzyme screening monitored as change in fractional blockage YE of pores 1 (A–K) and 2 (L) with reaction time t exemplified for DNA polymerase I Klenow fragment (A), DNA exonuclease III (B), RNase A (C), heparinase I (D), hyaluronidase (E), galactosyltransferase (F) [reproduced with permission from ref. 1 (Copyright 2002, AAAS, www.sciencemag.org)], pronase (substrate: PLE) (G), papain (substrate: PLE) (H), ficin (substrate: PLE) (I), elastase (substrate: PLE) (J), subtilisin {substrate candidates: x = [PLE]/([PLE] + [PDE]) = 1.00 (filled circles), 0.75 (open circles), 0.50 (open squares), and 0.00 (filled squares) at [PLE] + [PDE] = constant} (K), and papain (substrate: PLR) (L) at low (open circles) and high (filled circles) enzyme concentrations against controls (X) except for K. Substrates (S) are as in Table 1.
Dissociation Constants (KD Values) of Substrate/Product Blockers. Stock solutions of blockers (substrates and products) were prepared in buffer (10 mM Hepes/107 mM NaCl) with pH adjusted to 6.5 following the previously described fluorometric method without change (16). Then, x μl of blocker stock solution, (1,900 – x) μl of buffer (10 mM Hepes/107 mM NaCl, pH 6.5), and 100 μl of EYPC-LUVs ⊃ CF were mixed in a fluorescence cuvette, pore 1 was added, and pore blockage was determined following the above-described general procedure (e.g., Fig. 5A, a–g). I(c) values were taken [I(c) = I at maximal emission intensity before lysis for pores in presence of blockers at concentration c], converted into fractional pore blockage Y {Y = [I(c=0) – I(c)]/[I(c=0) – I(c=0)]}, and plotted as a function of blocker concentration c to determine dissociation constants KD by using the Hill equation,
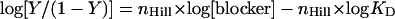 |
[2] |
(nHill = Hill coefficient; e.g., Fig. 5B). For macro- (n > 1, m = 1), supra- (n = 1, m > 1), and supramacromolecules (n > 1, m > 1), the KD per monomer was calculated by using KD(MONO) = KD × n × m (e.g., Fig. 5B, open circles). The sensitivity S of SMP sensors was approximated as 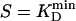 , where
, where 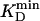 is the lowest KD among all involved compounds for a given enzyme. Initial substrate concentrations, ≈100 to 1,000 ×
is the lowest KD among all involved compounds for a given enzyme. Initial substrate concentrations, ≈100 to 1,000 × 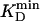 , were ideal to reach
, were ideal to reach 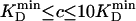 after the 100-fold dilution before detection. The selectivity requirement SR was approximated as
after the 100-fold dilution before detection. The selectivity requirement SR was approximated as
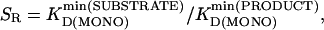 |
[3] |
where 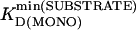 is the lowest KD(MONO) among all substrates, and
is the lowest KD(MONO) among all substrates, and  is the lowest KD(MONO) among all products of a specific enzyme.
is the lowest KD(MONO) among all products of a specific enzyme.
Fig. 5.

Original data for detection of the hydrolysis of [poly(dA,dT)]2 by DNA exonuclease III with SMP 1.(A) Characterization of substrate [poly(dA,dT)]2 as pore blocker. Fractional CF emission I (λem, 510 nm, λex, 495 nm) as a function of time after addition of 0 (a), 0.09 (b), 0.17 (c), 0.29 (d), 0.73 (e), 1.46 (f), and 2.92 (g) nM [poly(dA,dT)]2 and pore 1 (63 nM, arrow) to EYPC-LUVs ⊃ CF. (B) Dependence of fractional pore blockage Y on concentration c of substrate and product blockers {[poly(dA,dT)]2: filled circles, per duplex; open circles, per nucleotide; data are from A; filled squares, dAMP; open squares, dTMP}. (C) Detection of exonuclease activity. Twenty microliters of reaction mixture [0.83 units/ml exonuclease/37 nM [poly(dA,dT)]2 were taken after a reaction time of 0 min and added to 2 ml of EYPC-LUVs ⊃ CF {≈0.37 nM [poly(dA,dT)]2 final}; then, the change in CF emission after pore addition was measured as described for A (a). The same experiment was performed with 20-μl reaction mixtures taken after reaction times of 5 (b), 10 (c), 20 (d), 30 (e), and 130 (f) min.
Calibration of SMP 1. The above-described general procedure was performed in the absence of reaction mixtures [or individual blockers as described in Dissociation Constants (KD Values) of Substrate/Product Blockers]. The concentration of blocker-free SMP 1 was adjusted to cause nearly complete CF efflux (compared with lysis by Triton X-100) within ≈150 sec (Fig. 5A, a). Calibration using Eq. 1 gave the IMAX required for data analysis.
Special Cases. All enzymes listed in Table 1 and Fig. 4 were studied following this general procedure by using initial substrate concentrations c of ≈100 to 1,000 × 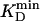 and the following exceptions: (i) YE = [I(t=0) – I(t)]/[I(t=0) – I t=∞)] for product blockage (Fig. 4 A and F) and (ii) SMP 2 and EYPC-LUVs ⊃ [8-amino-1,3,6-trisulfonate (ANTS)/p-xylenebis(pyrimidinium)bromide (DPX)] for cationic substrates (Fig. 4L). Sensing with SMP 2 and specific conditions for each enzyme detected with SMP 1 are reported in Supporting Text.
and the following exceptions: (i) YE = [I(t=0) – I(t)]/[I(t=0) – I t=∞)] for product blockage (Fig. 4 A and F) and (ii) SMP 2 and EYPC-LUVs ⊃ [8-amino-1,3,6-trisulfonate (ANTS)/p-xylenebis(pyrimidinium)bromide (DPX)] for cationic substrates (Fig. 4L). Sensing with SMP 2 and specific conditions for each enzyme detected with SMP 1 are reported in Supporting Text.
Results and Discussion
To detect enzyme activity with SMPs by using fluorometric real-time detection, enzymes and substrates are incubated under appropriate conditions. After a given period, an aliquot of the reaction mixture is removed and added to vesicles containing entrapped CF. Then, the pore sensor is added, and CF release is determined and compared with CF release at the beginning of the reaction (e.g., Fig. 5C, b versus a). This procedure (i.e., removal of an aliquot of reaction mixture, addition to CF-loaded vesicles followed by addition of the SMP, and determination of CF release) is repeated in meaningful intervals as the reaction proceeds (e.g., Fig. 5C,b–f). The individual experiments are then summarized and plotted as fractional pore blockage YE as a function of reaction time t. The obtained YE – t profile reveals enzyme activity as increasing (for product blockers) or decreasing (for substrate blockers) pore blockage with time (e.g., Fig. 4B). To apply this method to a given enzyme, the ability of all involved compounds to block the pore sensor must be determined first. Then, concentrations must be adjusted to assure detectability of consumption/production of the best substrate/product blocker involved. In the following, this calibration process is described in detail by using DNA exonuclease III as an example.
DNA exonuclease III catalyzes the sequential hydrolysis from the 3′ terminus of duplex DNA into nucleotide 5′-monophosphates (Table 1) (17–19). The [poly(dA,dT)]2 is widely used as a model substrate of exonuclease III. Because of its alternating, repeating sequence, duplex [poly(dA,dT)]2 remains double-stranded even after extensive degradation and hydrolysis by exonuclease III continues almost to completion (17).
To detect DNA exonuclease III with SMP 1, the same pore must recognize either substrate [poly(dA,dT)]2 or the products 2′-deoxyadenosine 5′-monophosphate (5′-dAMP) and 2′-deoxythymidine 5′-monophosphate (5′-dTMP). To characterize this supramolecular host–guest chemistry quantitatively, LUVs composed of EYPC were loaded with CF (Fig. 3). CF efflux from EYPC-LUVs ⊃ CF through SMP 1 was easily detectable as an increase in CF emission after pore addition (Fig. 5A, a). Negligible CF efflux was observed without pore. The presence of increasing concentrations of [poly(dA,dT)]2 reduced CF efflux mediated by pore 1 (Fig. 5A, b–g). A plot of blocker concentration as a function of the fractional blockage Y of tetramer 1 gave the dose–response isotherm for [poly(dA,dT)]2 (Fig. 5B, filled circles). Hill analysis of this isotherm yielded a global dissociation constant KD = 185 pM (Table 1, entry 1). Recognition of picomolar concentrations of substrate [poly(dA,dT)]2 implied superb sensitivity S for the detection of DNA exonuclease III with sensor 1 (Table 1). In an unoptimized multiwell end-point assay with a sample volume of 100 μl (1), for example, the detection limit was calculated to ≈19 fmol of [poly(dA,dT)]2.
Application of the same protocol to exonuclease products revealed 107 times poorer affinity of dAMP and dTMP to pore 1 (Fig. 5B, squares). To determine the selectivity requirement SR for SMP 1 to detect [poly(dA,dT)]2 hydrolysis, a KD(MONO) = 2.3 μM per monomer was calculated for supramacromolecule [poly(dA,dT)]2 (Fig. 5B, open circles). Comparison of this value with the KD values of the products revealed SR ≈ 0.002. This selectivity requirement SR ≪ 1 corroborated that differentiation of substrate and products of DNA exonuclease III by SMP 1 would be unproblematic and that enzymatic activity would be detectable as enzyme-gated pore opening (Figs. 2 A and 3).
Fluorometric real-time detection by SMPs was used to monitor DNA exonuclease III. Alternative modes of detection have been described elsewhere [e.g., multiwell screening and end-point detection (1)] or remain to be explored (e.g., continuous and electric detection in black or supported bilayers). For real-time detection of exonuclease III, 37 nM [poly(dA,dT)]2 duplex and 0.833 units/ml of the enzyme were incubated at pH 8.0 and 30°C. At different points in time, 20 μl of the reaction mixture were taken, added to 2 ml of EYPC-LUVs ⊃ CF, and tested for blockage of the subsequently added pore 1 (Fig. 5C). Consistent with conversion of the good blocker [poly(dA,dT)]2 (Fig. 5B, circles) into the poor blockers dAMP and dTMP (Fig. 5B, squares), the ability of the reaction mixture to block SMP 1 decreased with increasing reaction time (Fig. 4B).
Polynucleotides. DNA exonuclease III was selected as an example for fluorometric enzyme sensing with SMPs because of its broad scientific significance (Table 1, entry 1) (17–19). Among diverse applications in biotechnology, the use of exonuclease III in gene sequencing (18, 19) may be particularly noteworthy because combination with exonuclease III may ultimately be necessary to overcome the intrinsic resolution limits of single-gene sequencing with pores (10, 12, 13). Experimental evidence for detectability of exonuclease III with SMP 1 is detailed above (Figs. 4B and 5).
DNA polymerases catalyze the template-directed incorporation of deoxyribonucleotide monophosphates from 5′-deoxyribonucleotide triphosphate substrates to the 3′-hydroxyl terminus of a growing DNA strand (19–23). Because of their central role in biology, biochemistry, and biotechnology (19, 20), polymerases have been the subject of extensive structural and mechanistic studies. DNA polymerase I enzymes, characterized by multidomain architecture including 5′ → 3′ exonucleases, are involved in the repair of DNA lesions. The Klenow fragment of polymerase I, shaped like a right hand that can “hold” the growing DNA duplex with palm, fingers, and thumb, contains the polymerase domain.
Similar to that for the complementary DNA nucleases, duplex [poly(dA,dT)]2 provides a convenient model for studying DNA polymerases because poly(dA,dT) strands can serve both as templates and primers (23). Different than DNA nucleases, however, SMP sensors have to differentiate between nucleotide triphosphate substrates rather than monophosphate products to detect DNA polymerases. A KD = 29 μM was determined for 2′-deoxyadenosine 5′-triphosphate (5′-dATP) as a blocker of SMP 1 (Table 1, entry 2). The ability of substrate 5′-dATP to block pore 1 was ≈5 orders of magnitude weaker than that of the supramacromolecular product; that of 2′-deoxythymidine 5′-triphosphate (5′-dTTP) was assumed to be in the range of 5′-dATP. Contributions from coproduct pyrophosphate to pore blockage were irrelevant. The resulting selectivity requirement SR ≈ 13 together with a superb sensitivity S ≈ 185 pM implied unproblematic detectability of polymerase I (Klenow fragment) as enzyme-gated pore closing. This was corroborated experimentally (Fig. 4A).
RNase A, a pyrimidine specific RNase from bovine pancreas, catalyzes cleavage of the P—O5′ bond and the P—O2′ bond in cyclic intermediates (24). Although cleavage of RNA homopolymers by wild-type RNase A is distributive rather than processive as with the above-mentioned DNA exonuclease III, nucleotide 3′-monophosphates can be assumed as ultimate products of complete hydrolysis. Not surprisingly, substrate polycytidylic acid [poly(C)] turned out to block SMP 1 >5 orders of magnitude better than cytidine 3′-monophosphate (3′-CMP) (Table 1, entry 3). Predictable from SR ≈ 0.0002 and S ≈ 2 nM, unproblematic and sensitive detection of poly(C) hydrolysis by RNase A as enzyme-gated pore opening was confirmed experimentally (Fig. 4C). The ability of homopolymers poly(C), polyuridylic acid, and polyadenylic acid to block SMP 1 was quite similar (Table 1, entries 3–5). Consistent with the literature (24), RNase A sensing with SMP 1 indicated that hydrolysis of polyuridylic acid is almost as fast as poly(C), whereas that of polyadenylic acid was much slower (data not shown).
Polysaccharides. Heparinase I catalyzes the eliminative depolymerization of heparin, a biologically important and chemically unique polysaccharide that is widely used as an anticoagulant drug (25). Spectroscopic detection of either heparin or heparinase is not straightforward, because the substrate contains no chromophore (26, 27). The invention of assays for noninvasive, high-throughput detection of heparinase, however, is desirable to contribute to the search for competitive inhibitors as potential heparin mimics for use in cancer therapy (28, 29). Sensitive detection of heparinase I activity as enzyme-gated pore opening (Fig. 4D) was as unproblematic as expected from superb molecular recognition of the polyanionic heparin substrate by polycation 1 (Table 1, entry 6). Pore blockage by the ultimate mixture of heparin disaccharide products (ΔDiHs) was not determined.
Hyaluronan, discovered 70 years ago by Meyer and Palmer (38), is a viscous, acidic glycosaminoglycan with structural and signaling functions mainly in the extracellular matrix (30). Hyaluronidase converts this polysaccharide into hyaluronan disaccharides (ΔDiHAs). The ability of hyaluronan to block SMP 1 was substantial (Table 1, entry 7). Unproblematic detectability of hyaluronidase activity as enzyme-gated pore opening therefore was not surprising (Fig. 4E).
Detectability of enzymes involved in polysaccharide synthesis with SMP 1 has been reported (Table 1, entry 8, and Fig. 4F) (1). The invention of adaptable assays compatible with glycosyltransferase screening are of high interest in fields of application as diverse as glycobiology, drug discovery, and the invention of automated polysaccharide syntheses (31–33).
Polypeptides. Initial use of poly-l-glutamate (PLE) as the model polypeptide to explore adaptability of pore 1 to detect the activity of proteases was an obvious choice for several reasons. PLE has been used to explore the usefulness of dipole–potential interaction for α-helix recognition by SMPs in polarized bilayers (34). PLE has been used further to demonstrate that molecular recognition by SMP 1 exceeds that by the biological pore melittin >10,000 times (N.S. and S.M., unpublished work).
Unproblematic detectability of monitor hydrolysis of the excellent blocker PLE by proteases was indicated by the fact that l-glutamate did not block SMP 1 under meaningful experimental conditions (Table 1, entry 9). Decreasing ability of reaction mixtures to block pore 1 with increasing reaction time was as expected for PLE hydrolysis by protases (35–37) such as pronase, papain, ficin, elastase, and subtilisin (Fig. 4 G–K).
The binding of poly-d-glutamate (PDE) and PLE to SMP 1 was nearly identical (Table 1, entries 9 and 14). In clear contrast to PLE, however, treatment of PDE with subtilisin did not result in the opening of pore 1 (Fig. 4K, filled squares). This clear difference between PLE and PDE suggested that the latter was not converted by subtilisin. This finding was as expected from the literature (36) and the use of this endoprotease in kinetic resolutions (35). However, treatment of a racemic mixture of PLE and PDE with subtilisin for up to 4 days did not produce reaction mixtures with reduced ability to block pore 1 (Fig. 4K, open squares). Fractional pore blockage YE > 0.6 was observed for a 3:1 mixture of PLE and PDE after incubation with subtilisin for hours, a value clearly above the YE ≤ 0.25 expected for full PLE conversion (Fig. 4K, open circles). One interpretation of this apparently poor degradation of PLE in PLE/PDE mixtures is that the conversion of polyglutamates by subtilisin is stereo-selective, whereas their low-affinity binding (37) is not. Current studies on the use of SMP sensors to screen for enzyme inhibitors focus on examples of medicinal relevance.
Conversion of cationic polypeptides such as poly-l-lysine or poly-l-arginine (PLR) by proteases was not detectable with pore 1 because polycationic substrates are poorly recognized by polycationic pores. The cation-selective (2, 16) polyanionic pores formed by rigid-rod β-barrel 2 with internal aspartates, therefore, were evaluated as sensors for enzymes with cationic substrates. Because SMP 2 is impermeable for anion CF (16), the less ion-selective but also less sensitive ANTS/DPX assay was applied (15). Except for these differences, the fluorometric detection of pore activity with CF and ANTS/DPX assay, however, was identical. According to decreasing efflux of either intravesicular fluorophore ANTS or quencher DPX with increasing polypeptide concentration, polycations poly-l-lysine and PLR blocked anionic SMP 2 about as efficiently as polyanion PLE blocked cationic SMP 1 (Table 1, entries 15 and 16). The opening of pore 2 during PLR hydrolysis by papain confirmed detectability of enzymes with cationic substrates by using anionic pore sensors (Fig. 4L).
Experimental evidence for detectability of enzymes with cationic substrates by using anionic SMP 2 and of those with anionic substrates by using cationic SMP 1 did not answer the question about substrates without charge. We investigated poly-l-asparagine for this purpose. This water-soluble polypeptide was found to hinder CF efflux through SMP 1 almost as efficiently as the anionic polypeptide PLE (Table 1, entry 17).
Conclusion
The reported examples confirm that a single set of identical SMPs can be used to detect the activity of many different enzymes in a straightforward manner. Such broad adaptability of noninvasive and user-friendly enzyme sensors is ideal for practical applications.
Supplementary Material
Acknowledgments
We thank P. Talukdar and D. Gerard for assistance in organic synthesis; N. Sakai, D. Branton, and two anonymous referees for very helpful advice; and the financial support of Swiss National Science Foundation Grant 2000-064818.01 and National Research Program “Supramolecular Functional Materials” 4047-057496.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: SMP, synthetic multifunctional pore; [poly(dA,dT)]2, (deoxyadenylic acid, thymidylic acid)2 copolymer duplex; CF, 5(6)-carboxyfluorescein; EYPC, egg yolk phosphatidylcholine; LUV, large unilamellar vesicle; ANTS, 8-amino-1,3,6-trisulfonate; DPX, p-xylenebis(pyrimidinium)bromide; poly(C), polycytidylic acid; PLE, poly-l-glutamate; PDE, poly-d-glutamate; PLR, poly-l-arginine.
Footnotes
The terms “sensor” and “detector” are poorly distinguishable with enzymes because substrates serve as “cosensors” to sense enzymes in mixed analytes, whereas enzymes serve as cosensors to sense substrates in mixed analytes. For example, ATP can be sensed in mixed analytes containing other nucleotides by using SMP sensors with hexokinase and glucose as cosensors; glucose can be sensed in mixed analytes containing other carbohydrates by using SMP sensors with hexokinase and ATP as cosensors; and hexokinase can be sensed in mixed analytes containing other enzymes by using SMP sensors with glucose and ATP as cosensors (N.S. and S.M., unpublished work).
References
- 1.Das, G., Talukdar, P. & Matile, S. (2002) Science 298, 1600–1602. [DOI] [PubMed] [Google Scholar]
- 2.Sakai, N. & Matile, S. (2003) Chem. Commun. (Advance Article), DOI: 10.1039/b303649a.
- 3.Gokel, G. W. & Mukhopadhyay, A. (2001) Chem. Soc. Rev. 30, 274–286. [Google Scholar]
- 4.Scrimin, P. & Tecilla, P. (1999) Curr. Opin. Chem. Biol. 3, 730–735. [DOI] [PubMed] [Google Scholar]
- 5.Kirkovits, G. J. & Hall, C. D. (2000) Adv. Supramol. Chem. 7, 1–47. [Google Scholar]
- 6.Terrettaz, S., Ulrich, W.-P., Guerrini, R., Verdini, A. & Vogel, H. (2001) Angew. Chem. Int. Ed. Engl. 40, 1740–1743. [PubMed] [Google Scholar]
- 7.Wyman, T. B., Nicol, F., Zelphati, O., Scaria, P. V., Planck, C. & Szoka, F. C., Jr. (1997) Biochemistry 36, 3008–3017. [DOI] [PubMed] [Google Scholar]
- 8.Wang, D., Guo, L., Zhang, J., Jones, L. R., Chen, Z., Pritchard, C. & Roeske, R. W. C. (2001) J. Pept. Res. 57, 301–306. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez-Quesada, J., Isler, M. P. & Ghadiri, M. R. (2002) J. Am. Chem. Soc. 124, 10004–10005. [DOI] [PubMed] [Google Scholar]
- 10.Bayley, H. & Cremer, P. S. (2001) Nature 413, 226–230. [DOI] [PubMed] [Google Scholar]
- 11.Nestorovich, E. M., Danelon, C., Winterhalder, M. & Bezrukov, S. M. (2002) Proc. Natl. Acad. Sci. USA 99, 9789–9794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deamer, D. W. & Branton, D. (2002) Acc. Chem. Res. 35, 817–825. [DOI] [PubMed] [Google Scholar]
- 13.Vercoutere, W. & Akeson, M. (2002) Curr. Opin. Chem. Biol. 6, 816–822. [DOI] [PubMed] [Google Scholar]
- 14.Talukdar, P., Sakai, N., Sordé, N., Gerard, D., Cardona, V. M. F. & Matile, S. (2003) Bioorg. Med. Chem., in press. [DOI] [PubMed]
- 15.Das, G. & Matile, S. (2002) Proc. Natl. Acad. Sci. USA 99, 5183–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das, G., Onouchi, H., Yashima, E., Sakai, N. & Matile, S. (2002) Chembiochem 3, 1089–1096. [DOI] [PubMed] [Google Scholar]
- 17.Richardson, C. C., Lehman, I. R. & Kornberg, A. (1964) J. Biol. Chem. 239, 251–258. [PubMed] [Google Scholar]
- 18.Guo, L.-H. & Wu, R. (1983) Methods Enzymol. 100, 60–97. [DOI] [PubMed] [Google Scholar]
- 19.Stephan, J., Dorre, K., Brakmann, S., Winkler, T., Wetzel, T., Lapczyna, M., Stuke, M., Angerer, B., Ankenbauer, W., Foldes-Papp, Z., et al. (2001) J. Biotechnol. 86, 255–267. [DOI] [PubMed] [Google Scholar]
- 20.Levene, M. J., Korlach, J., Turner, S. W., Foquet, M., Craighead, H. G. & Webb, W. W. (2003) Science 299, 682–686. [DOI] [PubMed] [Google Scholar]
- 21.Steitz, T. A. (1999) J. Biol. Chem. 274, 17395–17398. [DOI] [PubMed] [Google Scholar]
- 22.Carroll, S. S. & Benkovic, S. J. (1990) Chem. Rev. (Washington, D.C.) 90, 1291–1307. [Google Scholar]
- 23.Klenow, H. & Henningsen, I. (1970) Proc. Natl. Acad. Sci. USA 65, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raines, R. T. (1998) Chem. Rev. (Washington, D.C.) 98, 1045–1066. [DOI] [PubMed] [Google Scholar]
- 25.Capila, I. & Linhardt, R. J. (2002) Angew. Chem. Int. Ed. Engl. 41, 390–412. [DOI] [PubMed] [Google Scholar]
- 26.Czarnik, A. W. (1994) Acc. Chem. Res. 27, 302–308. [Google Scholar]
- 27.Zhong, Z. & Anslyn, E. V. (2002) J. Am. Chem. Soc. 124, 9014–9015. [DOI] [PubMed] [Google Scholar]
- 28.Petitou, M., Hérault, J.-P., Bernat, A., Driguez, P.-A., Duchaussoy, P., Lormeau, J.-C. & Herbert, J. M. (1999) Nature 398, 417–422. [DOI] [PubMed] [Google Scholar]
- 29.Neuger, L., Ruge, T., Makoveichuk, E., Vlodavsky, I. & Olivecrona, G. (2001) Atherosclerosis (Shannon, Irel.) 157, 13–21. [DOI] [PubMed] [Google Scholar]
- 30.McDonald, J. & Hascall, V. C. (2002) J. Biol. Chem. 277, 4575–4596. [DOI] [PubMed] [Google Scholar]
- 31.Patenaude, S. I., Seto, N. O. N., Borisova, S. N., Szpacenko, A., Marcus, S. L., Palcic, M. M. & Evans, S. V. (2002) Nat. Struct. Biol. 9, 685–690. [DOI] [PubMed] [Google Scholar]
- 32.Wymer, N. & Toone, E. J. (2000) Curr. Opin. Chem. Biol. 4, 110–119. [DOI] [PubMed] [Google Scholar]
- 33.Hurtley, S., Service, R. & Szuromi, P. (2001) Science 291, 2337–2378. [Google Scholar]
- 34.Sordé, N. & Matile, S. (2002) J. Supramol. Chem. 2, 191–199. [Google Scholar]
- 35.Hedstrom, L. (2002) Chem. Rev. (Washington, D.C.) 102, 4429–4906. [Google Scholar]
- 36.Neumann, H., Sharon, N. & Katchalski, E. (1962) Nature 195, 1002–1003. [DOI] [PubMed] [Google Scholar]
- 37.Miller, W. G. (1964) J. Am. Chem. Soc. 86, 3913–3918. [Google Scholar]
- 38.Meyer, K. & Palmer, J. (1934) J. Biol. Chem. 107, 629–634. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.