

Abstract
Standard free energies (ΔGN°) for formation of near attack conformers, those ground state conformers that can convert directly to the transition state, were calculated for the Claisen rearrangement of chorismate to prephenate in six different environments: water, wild-type enzymes from Bacillus subtilis and Escherichia coli, their Arg90Cit and Glu52Ala mutants, and the 1F7 catalytic antibody. Values of the calculated ΔGN°s and the experimentally determined activation energies (ΔG‡) are linearly related with the slope of ≈1. This demonstrates that the relative rate of the chorismate → prephenate reaction is overwhelmingly dependent on the efficiency of formation of near attack conformers in the ground state.
In a chemical reaction of bond formation there is a time in which the two reacting atoms are at a distance of van der Waals contact and at an angle resembling the bond to be formed in the transition state (TS). By our convention this situation is called a near attack conformation (NAC) (1–3). The reaction of interest must proceed to the TS via NAC formation (Eq. 1).
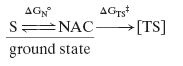 |
[1] |
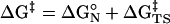 |
[2] |
The free energy for formation of a NAC (ΔGN°) can be determined quantitatively either by calculating the mole fraction of NAC relative to S during molecular dynamics (MD) simulations or by use of free energy calculation methods. Once ΔGN° is obtained, the activation energy on going from NAC to the TS (ΔGTS‡) can be calculated by simply subtracting ΔGN° from ΔG‡ (Eq. 2), where the best determination of ΔG‡ is from the experimental rate constant. The advantage of the enzymatic reaction over the water reaction [ΔΔG‡ = RT·ln(kcat/ko), where kcat and ko are the rate constants for the enzymatic and water reactions, respectively] can be partitioned into ΔΔGN° and ΔΔGTS‡. The value of ΔΔGN° represents the kinetic advantage of the enzyme compared with water in formation of NAC, and the value of ΔΔGTS‡ represents the advantage in the conversion of NAC to the TS.
Chorismate mutase catalyzes the unimolecular Claisen rearrangement of chorismate to prephenate without formation of an enzyme covalent intermediate (Scheme 1). Thus, the reactions in water and the enzyme are both kinetically first order and directly comparable. By long-term MD simulations (1) [and in the present study by thermodynamic integration (TI) methods], we have found that chorismate dianion is present as NAC only at 10–4% of the population of ground state conformers in water, whereas in the enzyme, E·NAC consists of 30% of the Michaelis complex (3). It follows that ΔGN° in the water and the enzymatic reactions is 8.4 and 0.6 kcal/mol, respectively, and accordingly, ΔΔGN° is 7.8 kcal/mol. Because the experimentally determined ΔΔG‡ is 9 kcal/mol, the advantage of the enzymatic reaction was concluded to be primarily in preferential formation of NACs.
Scheme 1.

We now report extensions of our studies using TI methods of free energies for NAC formation in water, Escherichia coli chorismate mutase and Bacillus subtilis chorismate mutase wild-type enzymes (w-EcCM and w-BsCM), the mutant obtained by alanine substitution for Glu-52 of EcCM (E52A), the mutant obtained by citrulline substitution for Arg-90 of BsCM (R90Cit), and the catalytic antibody 1F7 (4).
Methods
Free energy calculations were performed by using TI methods (5) implemented in the program charmm v.27b4 (6). Free energy calculation for chorismate in water was performed on a dianionic chorismate solvated in a 24 × 24 × 24-Å3 periodic box of TIP3P (7) water. The crystal structure of Protein Data Bank ID code 1ECM (8) was used for the w-EcCM and E52A simulations, the structure of ID code 2CHT (9) was used for the w-BsCM and R90Cit simulations, and the structure of ID code 1FIG (4) was used for the 1F7 simulations. The cocrystallized TS analogue [TSA (10); Scheme 1] was replaced by chorismate. The whole structure of each protein was solvated in a periodic box of TIP3P water and equilibrated at 300 K for 500 ps. The final MD structures were used to select residues within a 25-Å sphere from the active site. For the computational efficiency, only these selected residues were included for the TI calculations of protein systems with use of the stochastic boundary condition (6). The free energy derivatives were calculated every 5–10° of dih1 (dihedral angle of C4-C3-O13-C14; see Scheme 1). At each window, the systems were heated for 70 ps for the water simulation and 150 ps for the protein simulations, before the production dynamics of 300 ps for the water simulation and 200 ps for the protein simulations.
An additional free energy calculation was performed for 1F7 by using umbrella sampling methods (11). We also carried out each of 500-ps MD simulations of TSA complexed to R90Cit and 1F7, and an MD simulation of chorismate in water using a combined quantum mechanical and molecular mechanical (QM/MM) potential with a choice of AM1 for QM Hamiltonian. Details of the methods of these calculations, as well as TI calculations, can be found in Supporting Methods, which is published as supporting information on the PNAS web site, www.pnas.org.
Results
Definition of NAC. NACs of chorismate involve the two atoms of the forming bond (C5 and C16) to be within a van der Waals contact distance of 3.7 Å (12) with the π-orbitals of C5 and C16 pointing at each other. Directions of the two π-orbitals can be assessed by measurement of the two angles θ1 and θ2, as shown in Fig. 1. The “b” side orbital of C16 is not conducive for the reaction, because it involves the TS in a boat-like geometry. The boat-like TS is ≈7 kcal/mol more unstable [calculated at the B3LYP/6–311+G** level with use of COSMO solvent continuum methods to represent the energetics in water (unpublished results)] than the chair-like TS, which is formed via an orbital overlap in the “a” side. In the TS, θ1 = 8.2° and θ2 = 18.6°. For NAC, we allow ±20° deviations (Δθ1 and Δθ2) for each of these angles from those of the TS.§ The dependence of our results on the specific values of the distance and the angles will be discussed later.
Fig. 1.

Definition of NAC includes three conditions: (i) the C5—C16 distance ≤3.7 Å, (ii) Δθ1 ≤ 20°, and (iii) Δθ2 ≤ 20°.
Free Energy for NAC Formation. Using the above definition of NAC, we have calculated the free energies (ΔGN°) for NAC formation in six different environments by using TI. The TI methods generate a free energy profile along dih1 (ΔG°dih1; blue line in Fig. 2). NAC is formed in the range of dih1 = 50–80°. Because there is a combination of NACs and non-NACs at each dih1 of this range, the free energy for NAC additionally includes the free energy of NAC formation at a given dih1 (ΔG°dih1→N; the length of the red bars in Fig. 2), which is calculated from the probability of chorismate being NAC when dih1 is fixed at the value. Thus, the free energy content of NAC at a given dih1 is ΔG°dih + ΔG°dih1→N, and the free energy required for NAC formation (ΔGN°) is the lowest value of ΔG°dih1 + ΔG°dih1→N.
Fig. 2.

Free energy profiles (ΔG°dih, blue line) along dih1 of chorismate in water (A), w-EcCM (B), w-BsCM (C), 1F7 (D), E52A (E), and R90Cit (F). The red bars represent the free energies (ΔG°dih1→N) for NAC formation at a given dih1 value.
Water. NAC formation requires the chorismate ring to be in a diaxial rather than diequatorial configuration (see below). Our free energy calculation is carried out using the force fields that generate diaxial conformations. In our previous MD simulations (1) of chorismate in water (with use of the same force fields), we identified seven diaxial conformers, I–VII (Fig. 3). They were distinguished in their occupation of the conformational space along dih1 and dih2. NACs are subspecies of conformer I and are formed from I by a thermal motion of the side chain rotation along dih1.
Fig. 3.

Conformers of chorismate in water, plotted in the conformational space along dih1 (C4-C3-O13-C14) and dih2 (C3-O13-C14-C16). The structures of conformers are presented in ref. 1.
The free energy profile in our present study (Fig. 2 A) identifies two local minima in water. One minimum is at dih1 = –130° and the other is at dih1 = 20–30°. The comparison of structures with the previous results in Fig. 3 shows that the most stable structure at dih1 = –130° in Fig. 2 A corresponds to IV in Fig. 3, and the conformer at dih1 = 20–30° in Fig. 2 A corresponds to I in Fig. 3. The most stable NAC occurs at dih1 = 60° with ΔG°dih1=60°→N = 2.1 kcal/mol. The free energy of 4.2 kcal/mol (ΔG°dih=60°) is required to form dih1 = 60° from IV. Finally, using the free energy content of IV (1.8 kcal/mol) relative to the total ground state in water (13),¶ it follows that NAC is 8.1 (i.e., 2.1 + 4.2 + 1.8) kcal/mol above the most stable ground state in water. This agrees with our previous estimation (8.4 kcal/mol) from a number of long-term MD simulations (1).
Our estimated ΔGN° is in accord with the findings from other computational and experimental groups. The Karplus and Lipscomb groups (14) have found from their high-level QM/MM (self-consistent density-functional tight binding scheme or CCDFTB) as well as full QM (Hartree–Fock/6–31G*) calculations that reactive conformers of chorismate are not stable in water. The Hilvert and Wright groups (15) have also found from their TRNOE studies that NACs are very rarely formed in water (less than a detectable level by a 600-MHz spectrometer). Reasons for the high energy contents of NAC in water could be (i) the electrostatic repulsion between the two carboxylate functions when bringing the two carbons (C5 and C16) together at a TS-like angle, and (ii) an additional steric conflict between the carboxylate water shell and the ring, which needs to be juxtaposed when NAC is formed (see ref. 1 for detailed discussion).
Recently, the Jorgensen group has reported a computational result (16, 17) that apparently contradicts our and others' conclusions described above. They have calculated the free energy cost of bringing C5 and C16 together to 3.7 Å and presumed this to be the energy for NAC formation. The approaching angles of C16 (θ1) and its orbital (θ2) are ignored by this procedure. The reported energy was 0.1 kcal/mol in water, that is 8 kcal/mol smaller than our estimated ΔGN° in water. To examine whether the conformers of C5—C16 ≤ 3.7 Å sampled in their studies (Jorgensen's NAC or J-NAC) satisfy the θ1 and θ2 criteria, we have performed an MD simulation of chorismate in water with the C5—C16 distance weakly constrained at 3.7 Å. Because it was proposed by Jorgensen et al. that the discrepancy of ≈8 kcal/mol originates from the different Hamiltonian used for chorismate, i.e., AM1 for the Jorgensen's study and molecular mechanical force fields calibrated at the B3LYP/6–311+G** level in our study (3), we have used the AM1 Hamiltonian for chorismate for this simulation. The simulation shows that J-NACs include, in a vast majority, unreactive diequatorial conformers (Fig. 4) where each of the two π-orbitals at C5 and C16 are pointing away from the other orbital, having θ1 and θ2 of 44 ± 5° and 129 ± 9°, respectively (Δθ1 = 36 ± 5° and Δθ2 = 110 ± 9°). We observed only one snapshot of NAC among the 50,000 snapshots of J-NAC during the simulation period of 500 ps. The difficulty of conversion from J-NAC to NAC would explain the discrepancy between our and Jorgensen's results.
Fig. 4.

The most stable conformation of J-NAC has a hydrogen of C16, rather than the C16 orbital, pointing to C5.
Wild-Type Enzymes w-BsCM and w-EcCM. In the two wild-type enzymes w-EcCM and w-BsCM, ΔGN° is 0.1 and 0.3 kcal/mol, respectively. The free energy profiles (Fig. 2 B and C) of these two wild-type enzymes have a very narrow and deep potential minimum located exactly at the point where NAC is formed most efficiently (at dih1 = 60–70°). The well optimized potential shape can be explained from observation of the active site structures of w-EcCM and w-BsCM. Despite the vastly different overall architectures, w-EcCM and w-BsCM share many features at the active site. In our previous studies (3), based on the comparison of the crystal structure of w-BsCM and our MD structure of w-EcCM, we have proposed a possibility of different mechanisms used in w-BsCM and w-EcCM. Now, we have MD structures of both enzymes and we found the following. Two arginines (Arg-28 and Arg-11* in w-EcCM and Arg-63* and Arg-7 in w-BsCM; see Fig. 5 A and C) position the two carboxylate functions such that the two reacting moieties (C5 and C16) of chorismate are placed in proximity with θ1 and θ2 close to those of the TS. In addition, in both wild-type enzymes, the side chain vinyl group and the ring of chorismate are confined in a small space by having van der Waals contacts with hydrophobic protein residues (Fig. 5 A and C). In w-EcCM, the Val-35···C16 and Ile-81···C5 distances are 3.7 ± 0.2 Å and 3.8 ± 0.4 Å, respectively. In w-BsCM, the contact distances of Leu-115···C16 and Phe-57···C4 are 3.8 ± 0.3 Å and 3.7 ± 0.3 Å, respectively. These contacts do not involve a direct pushing, as seen from the absence of a correlation between the enzyme·chorismate contact distance and the C5—C16 distance (data presented in ref. 3 and Fig. 9, which is published as supporting information on the PNAS web site). Thus, the chorismate mutases merely provide an environment where NAC is favored by properly arranging both electrostatic and hydrophobic residues, and the TS is formed without further motion involving enzyme strain.
Fig. 5.

Stereoviews of chorismate bound to the active site of w-EcCM (A), E52A (B), w-BsCM (C), R90Cit (D), and 1F7 (E). The structures are the most stable geometries, as taken from the free energy minimum points in Fig. 2. Yellow arrows in A and C designate hydrophobic interactions. Yellow dotted lines represent electrostatic interactions. Residues with * are from the other subunit (light chain for 1F7) of the protein.
E52A Mutant of EcCM. The active site of E52A supports NAC formation at the free energy cost of 1.3 kcal/mol, which can be compared with 0.1 kcal/mol in w-EcCM. The free energy profile (Fig. 2E) shows that the energy minimum is at dih1 = 50°, whereas NAC is most frequently formed at dih1 = 80°. This slight shift of the potential accounts for the small increase in ΔGN° for E52A compared with w-EcCM. Comparison of the active sites of E52A (Fig. 5B) and w-EcCM (Fig. 5A) reveals that overall architectures of the active sites are conserved. However, a few differences are also observed. In E52A, the bottom part of the active site (as pictured in Fig. 5A) is accessible to bulk solvents through a newly formed water channel. This channel is closed in w-EcCM because of the Glu-52·Arg-47 interaction. The loss of this interaction in E52A also makes the backbone near Arg-47 very mobile, resulting in a weakening of several electrostatic interactions between protein and chorismate [i.e., Asp-48(NH)···O10 and Lys-39(NZ)···O13; see Fig. 5B].
R90Cit Mutant of BsCM. In R90Cit, the free energy minimum point (dih1 = 20°) (Fig. 2F) is significantly moved from that of w-BsCM (dih1 = 70°). Because NAC is formed most frequently at dih1 = 70°, ΔGN° increases from 0.3 kcal/mol in w-BsCM to 4.1 kcal/mol in R90Cit. The structure of the R90Cit active site provides an explanation as to the change in the free energy profile. The active site residues of R90Cit are significantly reorganized when compared with the w-BsCM structure. In w-BsCM (Fig. 5C), the guanidine group of Arg-90 is placed close to the ether O13 of chorismate by forming a salt bridge with Glu-78. In R90Cit (Fig. 5D), the amino group of Cit90 is turned away from O13 and interacts with Glu-78. In w-BsCM (Fig. 5C), the side chain carboxylate of chorismate is held by Arg-7, but in R90Cit (Fig. 5D) it interacts with Arg-116 because of a significant movement of Arg-7 toward Glu-78 to make up the charge imbalance created by the substitution of the neutral Cit90 for the positive Arg-90. In this altered interaction mode, the C5—C16 distance of chorismate is ≈4.5 Å, as compared with 3.5 ± 0.3 Å in w-BsCM.
Our computational results on R90Cit are surprising because the binding affinity of TSA (1/Ki), as well as of substrate chorismate (1/Km), is similar in both R90Cit and w-BsCM (18). To understand the origin of similar Kis, we have further performed a 500-ps MD simulation of TSA complexed to R90Cit.
We find that TSA binds to R90Cit in a different mode as compared with w-BsCM (Fig. 6). In R90Cit, the side chain carboxylate of TSA is held by both Arg-116 and Arg-7, and the ether O13 of TSA forms no hydrogen bond with a protein residue. It would appear that the similarity of Kis of R90Cit and w-BsCM is due to two compensating factors in enzyme and this mutant. These factors are (i) the favorable electrostatic interaction in w-BsCM between Arg-90 and O13 of TSA, which is not present in the R90Cit mutant, and (ii) the better fit of the covalently fixed carboxylates of TSA to Arg-116 and Arg-63 of R90Cit compared with the fit of TSA to Arg-7 and Arg-63 of w-BsCM. The latter was inferred from a comparison of the distances between the two carboxylate oxygens (COO···OOC; see blue double arrow in Fig. 6) of the enzyme-bound chorismate and TSA. Because of the bicyclic constraint of TSA, COO···OOC of TSA is 5.0 Å, independent of the environment (Fig. 6). However, COO···OOC of flexible chorismate is dictated by the enzyme preference, which is 6.6 ± 0.3 Å in w-BsCM and 5.0 Å in R90Cit. Thus, the two carboxylate functions of TSA fit better to R90Cit rather than to w-BsCM.
Fig. 6.

Electrostatic interactions between TSA and w-BsCM (A; crystal structure; Protein Data Bank ID code 2CHT) and between TSA and R90Cit (B; MD simulation data). Red residues interact with the carboxylate functions of TSA, and blue double arrows designate the distance between the two carboxylate oxygens.
Catalytic Antibody 1F7. The binding site of 1F7 resides at the surface of the protein and is significantly exposed to bulk solvents. The free energy profile (Fig. 2D) of 1F7-bound chorismate is very rugged and widely spread. This is in marked contrast to other protein environments examined, where the potential well is narrow and surrounded by high barriers. Intriguingly, the energy profile in 1F7 has a pattern similar to the profile in water, having the lowest energy minimum at dih1 = –110° and the next stable local minimum at dih1 = 50°. The barrier at dih1 =–50–0° is ≈2 kcal/mol higher in 1F7 compared with water, presumably because of the additional energy cost for the protein conformational rearrangement. The most stable NAC in 1F7 resides at dih1 = 80° with ΔGN° of 6.0 kcal/mol. Because of the ruggedness of the energy profile (Fig. 2D), it is possible that errors could be introduced in the course of energy integration to generate the whole profile. Thus, we performed another free energy calculation to compare with the results from the TI calculations. We used umbrella sampling methods (UMB) (11), and the C5—C16 distance was chosen as a driving coordinate. The obtained ΔGN° is ≈5 kcal/mol by UMB. Combining the results from the TI and UMB calculations, we take the average of 5.5 kcal/mol for ΔGN° for discussion purposes.
In the most stable chorismate conformation in 1F7 (at dih1 = –110°; Fig. 5E), the chorismate side chain carboxylate is held by Arg-95 and the Tyr-100 backbone amide, and the hydroxyl group of chorismate interacts with the amino functions of the Asn-33 and the His-32 backbone and the Asn-33 side chain. The ring carboxylate is mainly exposed to bulk solvents, but weakly interacts with Tyr-94* and Asn-50. With dih1 at –110°, the side chain vinyl group of chorismate is positioned away from the ring moiety and is buried in the antibody binding site. When chorismate becomes NAC, the side chain is placed above the ring, and the overall volume of chorismate becomes smaller than in the extended conformation at dih1 = –110°. Some interactions, such as ones made at the chorismate side chain carboxylate and the ring hydroxyl group, remain as in the extended conformation because of the adjustment of Arg-95 side chain and the movement of the loop 30–35 backbone (Fig. 7). However, other interactions are lost when NAC is formed and solvent water fills the empty space created between NAC and the protein. These enzyme-bound waters would cause an entropic cost in formation of NAC, which might be responsible for the more unfavorable entropy of activation as measured in the 1F7 reaction compared with the water reaction (15).
Fig. 7.

Superposition of three structures of 1F7 complexed with TSA (black), chorismate NAC (magenta), and the most stable chorismate conformer at dih1 = –110° (colored by the standard atomic definition). The structures are matched by their protein backbones.
From the previous TRNOE experiments (15), it was suggested that the most dominant form of chorismate at the 1F7 active site is the reactive conformation (NAC). This assignment is based on the observation of NOE transfer from the vinyl C16 hydrogens to the ring hydrogens near C5. However, as has been pointed out in the same paper (15), an indirect NOE transfer via surrounding protein residues is also a possibility. In our simulation, the most stable conformer at dih1 = –110° possesses a potential NOE transfer pathway, such as through Asn-33, Asn-35, and Asn-50 (Fig. 5E). It is surprising that the catalytic antibody 1F7, elicited by TSA as a hapten, binds chorismate in the conformation vastly different from the TSA geometry. To address this question, we compared our MD structures of 1F7·TSA with 1F7·chorismate (at dih1 = –110°) and 1F7·NAC.∥ Superposition of the three structures (Fig. 7) reveals that the protein backbone of 1F7·TSA, as well as protein side chains, matches better with 1F7·chorismate|dih1=–110° than 1F7·NAC. The RMSD of the protein backbone of 1F7·TSA (within 25 Å distant from the active site chorismate) is 0.80 Å relative to 1F7·chorismate|dih1=–110°, and 0.90 Å relative to 1F7·NAC. This suggests that, although TSA mimics NAC in geometry, NAC does not fit the protein structure complementary to TSA. Thus, chorismate binds to 1F7 in a conformation that requires a less pronounced structural rearrangement.
Discussion
In our present study on the chorismate → prephenate reaction (Scheme 1), the standard free energies (ΔGN°) for formation of reactive conformers (NAC) have been calculated in water, the two wild-type enzymes w-BsCM and w-EcCM, and their mutants R90Cit and E52A, as well as the catalytic antibody 1F7 (Table 1). Fig. 8 shows that the calculated ΔGN°s are linearly correlated with the experimentally determined (18–20) activation energies (ΔG‡) with the slope of 1.1 and the correlation coefficient of 0.97. It is unlikely that this linear relationship is an artifact of the force fields, because the effect of the chorismate force fields on ΔGN° varies depending on the environment. The use of the NAC approach has been questioned on the basis that the population of NACs depends on the choice of the boundary definition of NAC (21). To investigate such dependencies, we have repeated the calculation by varying, within reason, the limits for the C5—C16 distance (3.3–3.9 Å) and the θ1/θ2 angle deviations (10–30°) for NAC criteria. The individual values of ΔGN° change by as much as 4 kcal/mol when employing a different definition (Table 3, which is published as supporting information on the PNAS web site). However, relative values of ΔGN° are not dependent on the specific choice of the C5—C16 distance and the angle (Δθ1 and Δθ2) criteria. Table 2 shows that, regardless of the distance and the angle definitions used, the slope is between 1.04 and 1.21, and the correlation coefficient is >0.94. This clearly demonstrates that the NAC approach is valid if the same definition is used for a comparison of the different catalysts of the same reaction.
Table 1. Free energies of experimental ΔG‡ and computational ΔGN° (kcal/mol).
| ΔG‡ | - | ΔGN° | = | ΔGTS‡ | |
|---|---|---|---|---|---|
| Water | 24.2 | 8.1 | 16.1 | ||
| 1F7 | 21.3 | 5.5 | 15.8 | ||
| R90Cit | 21.2 | 4.1 | 17.1 | ||
| E52A | 18.2 | 1.3 | 16.9 | ||
| w-BsCM | 15.4 | 0.3 | 15.1 | ||
| w-EcCM | 15.2 | 0.1 | 15.1 |
Fig. 8.

Plot of ΔG‡ vs. ΔGN° in Table 1. Circles are of 1 kcal/mol diameter. The equation at the bottom right is the linear fit equation for the six data points. R is the correlation coefficient for the linear fit.
Table 2. Dependence of the linear fit on the criteria for NAC.
| Limit | 3.3 Å | 3.5 Å | 3.7 Å | 3.9 Å |
|---|---|---|---|---|
| 10°* | 1.04 (0.95) | 1.04 (0.95) | 1.04 (0.94) | 1.04 (0.94) |
| 20° | 1.04 (0.98) | 1.08 (0.97) | 1.08 (0.97) | 1.08 (0.97) |
| 30° | 1.05 (0.98) | 1.13 (0.97) | 1.18 (0.96) | 1.21 (0.96) |
The values are the slopes and the correlation coefficients (in parentheses) for the type of plots shown in Fig. 8, when using different combinations of the distance and the angle deviation (Δθ1 and Δθ2) criteria.
When using the 10° limit, no water conformers satisfy the angle criteria.
The slope of ≈1.0 in Fig. 8 indicates that the differences in the rate constants for these reactions reside in the mole fraction of substrate present as NACs. The catalysis on NAC → TS, which includes the TS stabilization proposed by Hilvert and colleagues (18) and the NAC strain effect proposed by Jorgensen and colleagues (16, 17), can only be minor contributions (see Results for detailed discussion). Table 2 shows that ΔG‡TS is constant regardless of the catalysts within an 1.0 kcal/mol deviation from 16.1 kcal/mol. The predominant contribution of ΔΔGN° to ΔΔG‡ for the chorismate rearrangement was also observed by the Karplus group in their studies on the BsCM mutants (14).
Cleland and his coworkers have found that kinetic isotope effects at O13 are similar in water and the enzyme (W. W. Cleland, personal communication), implying that the C3—O13 bond-breaking is progressed to the similar extent in both environments. This supports our finding that stabilization of the ether O13 of the TS by a positive charge in the enzyme is no more significant than the stabilization by neutral water. This is apparently counterintuitive and needs a rationalization. In water, the ether O13 is more water-accessible in the TS, because of the elongation of the C—O bond and the development of a partial charge at O13 (22). Thus, although neutral, more water molecules stabilize the TS than the ground state, and this preferential stabilization of the TS by water might be comparable to the stabilization by a charged residue. As for proteins, 1F7, R90Cit, and E52A lack a positively charged residue near O13 in the protein·NAC complex. This would presumably be so in the protein·TS complex. The 1F7 active site is extensively exposed to bulk solvent, and similar TS stabilization as in water might occur during the reaction. The ≈1 kcal/mol-less-efficient conversion of NAC → TS in E52A and R90Cit, compared with water and 1F7, may be attributed to the less efficient solvent accessibility. In E52A and R90Cit, the space near O13 is not as significantly exposed to solvents as in water and 1F7, which may not allow an entrance of additional water molecules in the transiently formed TS.
Conclusion
From the supposition that catalysis of chemical reactions (Eq. 1) invariably involves TS stabilization, it has been reasoned that the free energy (ΔGN°) for formation of reactive ground state conformers (NACs) is dictated by the free energy of the TS (23). In our present study of chorismate → prephenate reaction we find just the opposite. We have determined ΔGN° for the chorismate rearrangements in water, two wild-type enzymes, their mutants, and a catalytic antibody. We found that the common mechanism does not involve TS stabilization. The free energy of conversion of NAC → TS (ΔGTS‡) is independent of the overall free energy of reaction (ΔG‡), and the stability of NAC (ΔGN°) determines the differences in ΔG‡. However, this is not an attribute of enzymatic catalysis in general, because we have observed cases where NAC formation is of little significance and catalytic consequences collected as TS stabilization are the most important factors (2). Thus, these observations, along with our present studies, support the proposal that enzymes use relative contribution from stabilization of NACs and TSs that vary widely as required for most efficient catalysis of markedly divergent chemistry.
Supplementary Material
Abbreviations: BsCM, Bacillus subtilis chorismate mutase; EcCM, Escherichia coli chorismate mutase; MD, molecular dynamics; NAC, near attack conformer; TI, thermodynamic integration; TS, transition state; TSA, TS analogue; w-BsCM and w-EcCM, BsCM and EcCM wild-type enzymes.
See commentary on page 11931.
Footnotes
The 20° allowance for Δθ1 matches with the definition (C16 approaching angle <30°) used in refs. 1 and 2. Although θ2 criterion was not explicitly stated in those previous studies, it was taken into account by an inspection of dih1 and dih2.
0.4 kcal/mol (computational) on going from diaxial conformer to IV (2), and 1.4 kcal/mol (experimental) on going from the total ground state to diaxial conformers (13).
We use our MD structure of 1F7·TSA instead of the crystal structure, because of the low resolution (3.0 Å) of the crystal structure.
References
- 1.Hur, S. & Bruice, T. C. (2003) J. Am. Chem. Soc. 125, 5964–5972. [DOI] [PubMed] [Google Scholar]
- 2.Hur, S. & Bruice, T. C. (2003) J. Am. Chem. Soc. 125, 1472–1473. [DOI] [PubMed] [Google Scholar]
- 3.Hur, S. & Bruice, T. C. (2002) Proc. Natl. Acad. Sci. USA 99, 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haynes, M. R., Stura, E. A., Hilvert, D. & Wilson, I. A. (1994) Science 63, 646–652. [DOI] [PubMed] [Google Scholar]
- 5.Kuczera, K. (1996) J. Comput. Chem. 17, 1726–1749. [Google Scholar]
- 6.Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983) J. Comput. Chem. 4, 187–217. [Google Scholar]
- 7.Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. (1983) J. Chem. Phys. 79, 926–935. [Google Scholar]
- 8.Lee, A. Y., Karplus, P. A., Ganem, B. & Clardy, J. (1995) J. Am. Chem. Soc. 117, 3627–3628. [Google Scholar]
- 9.Chook, Y. M., Gray, J. V., Ke, H. & Lipscomb, W. N. (1994) J. Mol. Biol. 240, 476–500. [DOI] [PubMed] [Google Scholar]
- 10.Bartlett, P. A., Nakagawa, Y., Johnson, C. R., Reich, S. H. & Luis, A. (1988) J. Org. Chem. 53, 3195–3210. [Google Scholar]
- 11.Kottalam, J. & Case, D. A. (1988) J. Am. Chem. Soc. 110, 7690–7697. [Google Scholar]
- 12.Nussinov, A.-J. L. R. (1998) Proteins Struct. Funct. Genet. 32, 111–127.9672047 [Google Scholar]
- 13.Copley, S. D. & Knowles, J. R. (1987) J. Am. Chem. Soc. 109, 5008–5013. [Google Scholar]
- 14.Guo, H., Cui, Q., Lipscomb, W. L. & Karplus, M. (2003) Angew. Chem. Int. Ed. Engl. 42, 1508–1511. [DOI] [PubMed] [Google Scholar]
- 15.Campbell, A. P., Tarasow, T. M., Massefski, W., Wright, P. & Hilvert, D. (1993) Proc. Natl. Acad. Sci. USA 90, 8663–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Repasky, M. P., Guimaraes, C. R. W., Chandrasekhar, J., Tirado-Rives, J. & Jorgensen, W. L. (2003) J. Am. Chem. Soc. 125, 6663–6672. [DOI] [PubMed] [Google Scholar]
- 17.Guimaraes, C. R. W., Repasky, M. P., Chandrasekhar, J., Tirado-Rives, J. & Jorgensen, W. L. (2003) J. Am. Chem. Soc. 125, 6892–6899. [DOI] [PubMed] [Google Scholar]
- 18.Kienhoefer, A., Kast, P. & Hilvert, D. (2003) J. Am. Chem. Soc. 125, 3206–3207. [DOI] [PubMed] [Google Scholar]
- 19.Andrews, P. R., Smith, G. D. & Young, I. G. (1973) Biochemistry 12, 3492–3498. [DOI] [PubMed] [Google Scholar]
- 20.Liu, D. R., Cload, S. T., Pastor, R. M. & Schultz, P. G. (1996) J. Am. Chem. Soc. 118, 17891790. [Google Scholar]
- 21.Shurki, A., Strajbi, M., Villa, J. & Warshel, A. (2002) J. Am. Chem. Soc. 124, 4097–4107. [DOI] [PubMed] [Google Scholar]
- 22.Cramer, C. J. & Truhlar, D. G. (1992) J. Am. Chem. Soc. 114, 8794–8799. [Google Scholar]
- 23.Strajbl, M., Shurki, A., Kato, M. & Warshel, A. (2003) J. Am. Chem. Soc. 125, 10228–10237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.