

Abstract
DNA polymerase η (Polη) has the unique ability to replicate through UV-light-induced cyclobutane pyrimidine dimers. Here we use pre-steady-state kinetic analyses to examine the mechanism of nucleotide incorporation opposite a cis-syn thymine-thymine (TT) dimer and an identical nondamaged sequence by yeast Polη. Polη displayed “burst” kinetics for nucleotide incorporation opposite both the damaged and nondamaged templates. Although a slight decrease occurred in the affinity (Kd) of nucleotide binding opposite the TT dimer relative to the nondamaged template, the rate (kpol) of nucleotide incorporation was the same whether the template was damaged or nondamaged. These results strongly support a mechanism in which the nucleotide is directly inserted opposite the TT dimer by using its intrinsic base-pairing ability without any hindrance from the distorted geometry of the lesion.
Cells use several strategies for repairing UV-light-induced DNA lesions, including the enzymatic reversal of the damage by photolyase and the removal of the damage from the DNA duplex by nucleotide excision repair. However, when the damaged nucleotides are not removed, they present a block to replicative DNA polymerases and result in the stalling of the replication fork. As a consequence, cells have evolved strategies for overcoming replication blocks at DNA lesions, and translesion synthesis by specialized DNA polymerases plays an important role in lesion bypass. DNA polymerase η (Polη) is unique among eukaryotic polymerases in its ability to replicate through a cis-syn thymine-thymine (TT) dimer, a major UV photoproduct (1–3). Hence, mutational inactivation of Polη in yeast (4, 5) and in humans (6, 7) results in increased UV mutagenesis, and defects in Polη in humans cause the cancer-prone syndrome, the variant form of xeroderma pigmentosum (8, 9). Thus, Polη acts to prevent skin cancers by reducing the incidence of UV-induced mutations.
Classical DNA polymerases, such as T7 DNA polymerase and Escherichia coli DNA polymerase I, are blocked at DNA lesions presumably because they cannot tolerate the distorted DNA geometry of the lesion. This strict intolerance of geometric distortions in the DNA is believed to be responsible for the high fidelity of classical polymerases (10, 11). Based on the ability of Polη to replicate through a TT dimer, we hypothesized that Polη possesses an active site that is unusually tolerant of geometric distortions in the DNA, and, consistent with this hypothesis, with the use of a steady-state kinetics assay, we found that Polη indeed synthesizes DNA with a low fidelity, misincorporating nucleotides with a frequency of 10–2 to 10–3 (2, 12). Steady-state kinetic studies with yeast and human Polη have also indicated that both enzymes incorporate two As opposite the two Ts of a TT dimer with nearly the same catalytic efficiency (kcat/Km) as opposite nondamaged Ts (2, 3). However, because the dissociation of the polymerase from the DNA substrate is likely to be rate-limiting under steady-state conditions, these studies do not provide any mechanistic information about the similarities in the nucleotide incorporation reaction opposite the TT dimer vs. undamaged nucleotides.
One of the most important questions regarding Polη is how does this enzyme manage to incorporate nucleotides opposite the TT dimer efficiently and accurately. To understand the mechanism of nucleotide incorporation by Polη opposite this DNA-distorting lesion, we have performed a pre-steady-state kinetic analysis of this reaction. Because pre-steady-state kinetic studies allow one to directly measure the incorporation of nucleotides during the first turnover of the polymerase (13, 14), with this approach, it is possible to directly examine the elementary steps of nucleotide binding and incorporation without complications arising from the slow dissociation of the enzyme from the DNA that limits the subsequent steady-state turnovers. Studies with several classical DNA polymerases, such as T7 DNA polymerase and the Klenow fragment of E. coli DNA polymerase I, have shown that the nucleotide incorporation reaction is composed of several distinct elementary steps including the following: (i) DNA binding, (ii) nucleotide binding, (iii) one or more conformational changes to a catalytically competent state, (iv) phosphodiester bond formation, (v) pyrophosphate release, (vi) translocation to the next template residue, or (vii) DNA dissociation. It is believed that the nucleotide-binding step and the subsequent conformational change and/or chemistry steps (steps ii–iv) are principally responsible for the high fidelity of these enzymes (15–18).
We have used pre-steady-state kinetic studies to examine the mechanism of correct and incorrect nucleotide binding and incorporation by yeast Polη opposite a nondamaged DNA template to investigate the mechanistic basis of its low fidelity (19). We found that, at the initial nucleotide-binding step (step ii), the correct nucleotide bound with ≈5-fold higher affinity than the incorrect nucleotide, whereas at the nucleotide incorporation steps (steps iii and iv), the nucleotide was incorporated ≈150-fold faster when the correct nucleotide was bound than when the incorrect nucleotide was bound. The evidence also indicated that the rate of nucleotide incorporation was determined by an induced-fit conformational change step (step iii) rather than the chemical step of phosphodiester bond formation (step iv). Thus, the low fidelity of Polη likely results from a poorly discriminating induced-fit mechanism.
Here, we have carried out a pre-steady-state kinetic analysis of yeast Polη-catalyzed incorporation of an A nucleotide opposite a TT dimer to understand the mechanism of nucleotide incorporation opposite this lesion and to delineate the impact of this DNA-distorting lesion on the elementary steps of nucleotide binding and incorporation. We find that yeast Polη incorporates nucleotides opposite the TT dimer without any hindrance from the distorted geometry of the lesion. These results strongly support a mechanism in which the nucleotide is directly inserted opposite the TT dimer by using the intrinsic base-pairing ability of this lesion.
Materials and Methods
Purification of Polη. Yeast Polη was purified as a GST fusion protein from yeast strain BJ5464 carrying plasmid pR30.175, and the GST portion of the fusion protein was removed by treatment with PreScission protease (Amersham Pharmacia) as described (19). Purified Polη was stored in 10-μl aliquots at –70°C. The concentration of total protein was determined by UV absorbance at 280 nm in the presence of 8 M urea by using a molar extinction coefficient equal to 78,896 M–1·cm–1. The concentration of total protein was also determined by the Bio-Rad Protein Assay with BSA as a standard. Both methods yielded similar values of protein concentration. Comparison of the concentration of total protein with the concentration of active protein determined by active-site titrations (Fig. 1) showed that the preparation of Polη used here was 95% active. The concentration of Polη used in all experiments was corrected for the amount of active enzyme.
Fig. 1.

Pre-steady-state kinetics of nucleotide incorporation opposite a TT dimer (indicated by ν) and the analogous nondamaged sequence (both shown above the graphs) by Polη and active-site titrations. (A) Polη (120 nM) and the damaged 3′ T DNA substrate (300 nM) were mixed with dATP (500 μM) by using a rapid chemical quench flow instrument for various reaction times. The data were fit to the burst equation with an amplitude of 100 ± 3 nM and rate constants equal to 1.0 ± 0.06 s–1 and 0.050 ± 0.003 s–1. (B) Polη (120 nM) and various concentrations of the damaged 3′ T DNA substrate (•, 25 nM; ○, 50 nM; ▪, 100 nM; □, 150 nM; ▴, 200 nM; ▵, 300 nM) were mixed with dATP (500 μM) for various reaction times. The solid lines represent the best fits to the burst equation. (C) The amplitudes of the burst phases in B (•) were graphed as a function of DNA concentration, and the solid line represents the best fit to the quadratic equation with a 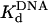 for the Polη–DNA complex equal to 40 ± 5 nM and an active-site concentration equal to 120 ± 4 nM. (D) Polη (120 nM) and the nondamaged 3′ T DNA substrate (300 nM) were mixed with dATP (200μM) for various reactions times. The data were fit to the burst equation with an amplitude of 100 ± 2 nM and rate constants equal to 1.4 ± 0.06 s–1 and 0.054 ± 0.002 s–1. (E) Polη (120 nM) and various concentrations of the nondamaged 3′ T DNA substrate (•, 25nM; ○, 50nM; ▪, 100 nM; □, 150 nM; ▴, 200 nM; ▵, 300 nM,) were mixed with dATP (300 μM) for various reaction times. The solid lines represent the best fits to the burst equation. (F) The amplitudes of the burst phases in E (•) were graphed as a function of DNA concentration, and the solid line represents the best fit to the quadratic equation with a
for the Polη–DNA complex equal to 40 ± 5 nM and an active-site concentration equal to 120 ± 4 nM. (D) Polη (120 nM) and the nondamaged 3′ T DNA substrate (300 nM) were mixed with dATP (200μM) for various reactions times. The data were fit to the burst equation with an amplitude of 100 ± 2 nM and rate constants equal to 1.4 ± 0.06 s–1 and 0.054 ± 0.002 s–1. (E) Polη (120 nM) and various concentrations of the nondamaged 3′ T DNA substrate (•, 25nM; ○, 50nM; ▪, 100 nM; □, 150 nM; ▴, 200 nM; ▵, 300 nM,) were mixed with dATP (300 μM) for various reaction times. The solid lines represent the best fits to the burst equation. (F) The amplitudes of the burst phases in E (•) were graphed as a function of DNA concentration, and the solid line represents the best fit to the quadratic equation with a 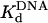 for the Polη–DNA complex equal to 32 ± 2 nM and an active-site concentration equal to 120 ± 2 nM.
for the Polη–DNA complex equal to 32 ± 2 nM and an active-site concentration equal to 120 ± 2 nM.
DNA Substrates. Two oligodeoxynucleotide primers were used to measure the incorporation of nucleotides opposite the 3′ T and the 5′ T of a template TT dimer and the identical nondamaged template residues. The primer for measuring incorporation opposite the 3′ T was an 11-mer with the sequence 5′-CAC CAC GCC GC, and the primer for measuring the incorporation opposite the 5′ T was a 12-mer with the sequence 5′-CAC CAC GCC GCA. Two oligodeoxynucleotide 16-mer templates were used with the sequence 5′-AAA TTG CGG CGT GGT G; in one of which the underlined bases were a TT dimer and, in the other, the underlined bases were nondamaged. A third oligodeoxynucleotide 16-mer template was used with the sequence 5′-AAA AXG CGG CGT GGT G, where X represents an abasic site. Primer strands (10 μM) were 32P-labeled at the 5′ end by incubation with [γ-32P]ATP [6,000 Ci/mmol (1 Ci = 37 GBq); Amersham Pharmacia] and polynucleotide kinase (Boehringer Mannheim) for 1 h at 37°C. Labeled primers were separated from unreacted [γ-32P]ATP by a Sephadex G-10 spin column (Amersham Pharmacia). DNA substrates were annealed by incubating the labeled primer (2 μM) and the template (2.5 μM) in 50 mM Tris·Cl (pH 7.5)/50 mM NaCl at 90°C for 2 min and slowly cooling the samples to room temperature over several hours. Various control experiments indicated that <1% of the molecules in the preparation of the TT dimer oligodeoxynucleotide were nondamaged (20). Solutions of dATP (0.1 M sodium salt, pH 7.0, ultrapure grade) were purchased from United States Biochemical and were stored at –70°C.
Pre-Steady-State Kinetics Assays. All experiments were carried out in 32.5 mM Tris·HCl (pH 7.5)/7.5 mM NaCl/5 mM MgCl2/5 mM DTT/10% glycerol at 22°C by using a Rapid Chemical Quench Flow instrument (Kin Tek Instruments, University Park, PA). Preincubated Polη (120 nM final concentration) and DNA (25–300 nM final concentration) were loaded into one sample loop (15 μl), and dATP (5–500 μM final concentration) was loaded into the other sample loop. Reactions were quenched with 0.3 M EDTA, and the products were analyzed by 20% PAGE in the presence of 8 M urea. The intensities of the gel bands were quantitated by using the PhosphorImager and imagequant 5.0 software (Molecular Dynamics). To ensure reproducibility, time courses for each set of DNA and dATP concentrations were repeated at least two times, and the amplitudes and rate constants obtained were in very close agreement.
Data Analysis. The amount of product formed (P) was graphed as a function of time (t), and the data were fit by nonlinear regression by using sigmaplot 7.0 (SPSS, Chicago) to the burst equation
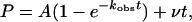 |
[1] |
where A is the amplitude of the burst phase, kobs is the rate constant of the burst phase, and v is the rate of the linear phase. The amplitudes of the burst phases (A) were graphed as a function of total DNA concentration ([DNA]), and the data were fit to the quadratic equation
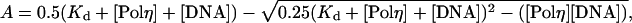 |
[2] |
where Kd is the dissociation constant for the Polη–DNA complex and [Polη] is the concentration of active Polη molecules. The observed rate constants of the burst phases (kobs) were graphed as a function of total dATP concentration ([dATP]), and the data were fit to the hyperbolic equation
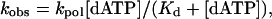 |
[3] |
where kpol is the maximum rate constant of the pre-steady-state burst phase and Kd is the dissociation constant of the Polη–DNA–dATP complex.
Results
The presence of a TT dimer in the DNA substrate was verified by mass spectroscopy and by T4 UV endonuclease V assays. Also, the substrate was a complete block to synthesis by yeast Polδ and human Polκ, and from such observations, we determined the purity of the TT dimer-containing DNA template to be 99% or better.
We performed a pre-steady-state analysis of the mechanism of nucleotide incorporation opposite a template TT dimer by Polη. We measured the Kd, the dissociation constant for the nucleotide-binding step, and the kpol, the maximal first-order rate constant for incorporating the bound nucleotide, for nucleotide binding and subsequent incorporation opposite the 3′ T and the 5′ T of the TT dimer and an identical nondamaged DNA sequence. Before measuring these values, however, we needed to determine whether Polη displayed biphasic or “burst” kinetics on the TT dimer and to determine the Kd for DNA binding and the concentration of active Polη.
Pre-Steady-State Kinetics of Nucleotide Incorporation Opposite the TT Dimer. First, we determined whether yeast Polη displayed biphasic kinetics during the incorporation of dATP opposite the 3′ T of the TT dimer. Preincubated Polη (120 nM final concentration) and DNA (300 nM final concentration) from one syringe were rapidly mixed with dATP (500 μM final concentration) from the other syringe, in a rapid chemical quench flow instrument. The reactions were quenched after 0.2–15 s, and the amount of product formed was graphed as a function of time (Fig. 1 A). A similar experiment was performed for the incorporation of dATP opposite the 3′ T of an identical nondamaged TT sequence (Fig. 1D). With both the damaged and the nondamaged DNA substrates, two phases of nucleotide incorporation were clearly observed, indicating that incorporation during the first enzyme turnover (the pre-steady-state phase) is faster than in subsequent turnovers (the steady-state phase). This difference is because the subsequent turnovers are limited by an elementary step of the nucleotide incorporation reaction that follows the chemical step of phosphodiester bond formation, and this step is likely the dissociation of the enzyme from the DNA substrate.
Active-Site Titration and Determination of the Kd for DNA Binding. The presence of a clear pre-steady-state burst phase provided an opportunity to examine the kinetics of dATP binding and incorporation in the first turnover. However, to better define the conditions necessary to carry out these measurements, it was first necessary to determine the concentration of active Polη molecules and the dissociation constant (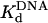 ) for the Polη–DNA complex. Because the amplitude of the pre-steady-state burst phase is equal to the amount of active Polη–DNA complex at the start of the reaction, these values could be determined by examining the variation of the burst amplitude with different concentrations of the DNA substrate. Fig. 1B shows the kinetics of dATP incorporation opposite the 3′ T of the TT dimer at different concentrations of DNA from 25 to 300 nM, and Fig. 1C shows the amplitudes of the pre-steady-state burst phases graphed as a function of DNA concentration. From the best fit of this data, we obtained an active-site concentration of 120 nM and a
) for the Polη–DNA complex. Because the amplitude of the pre-steady-state burst phase is equal to the amount of active Polη–DNA complex at the start of the reaction, these values could be determined by examining the variation of the burst amplitude with different concentrations of the DNA substrate. Fig. 1B shows the kinetics of dATP incorporation opposite the 3′ T of the TT dimer at different concentrations of DNA from 25 to 300 nM, and Fig. 1C shows the amplitudes of the pre-steady-state burst phases graphed as a function of DNA concentration. From the best fit of this data, we obtained an active-site concentration of 120 nM and a 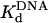 for the Polη–DNA complex of 40 nM. Fig. 1 E and F shows the analogous results for incorporation opposite the 3′ T of the nondamaged TT sequence. From the curve fit, we obtained an active-site concentration of 120 nM and a
for the Polη–DNA complex of 40 nM. Fig. 1 E and F shows the analogous results for incorporation opposite the 3′ T of the nondamaged TT sequence. From the curve fit, we obtained an active-site concentration of 120 nM and a 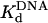 of 32 nM. Thus, whether the DNA template is damaged or nondamaged, the amount of active Polη–DNA complex and the dissociation constant for this complex were nearly the same.
of 32 nM. Thus, whether the DNA template is damaged or nondamaged, the amount of active Polη–DNA complex and the dissociation constant for this complex were nearly the same.
Nucleotide Incorporation Opposite the 3′ T of the TT Dimer. The kinetics of dATP binding and incorporation opposite the 3′ T of the TT dimer and the analogous nondamaged sequence were then examined. The maximal rate constant of nucleotide incorporation in the first turnover (kpol) and the dissociation constant for the Polη–DNA–dATP complex (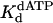 ) were determined by examining the dATP concentration dependence of the observed rate constant of the pre-steady-state burst phase. Fig. 2A shows the kinetics of dATP incorporation opposite the 3′ T of the TT dimer at different concentrations of dATP from 10 to 500 μM, and the observed burst rate constant (kobs) was graphed as a function of dATP concentration (Fig. 2B). From the best fit of these data, a kpol of 1.2 s–1 and a
) were determined by examining the dATP concentration dependence of the observed rate constant of the pre-steady-state burst phase. Fig. 2A shows the kinetics of dATP incorporation opposite the 3′ T of the TT dimer at different concentrations of dATP from 10 to 500 μM, and the observed burst rate constant (kobs) was graphed as a function of dATP concentration (Fig. 2B). From the best fit of these data, a kpol of 1.2 s–1 and a 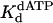 of 130 μM were obtained. Fig. 2 C and D shows the analogous experiments with the identical nondamaged DNA sequence, and a kpol of 1.5 s–1 and a
of 130 μM were obtained. Fig. 2 C and D shows the analogous experiments with the identical nondamaged DNA sequence, and a kpol of 1.5 s–1 and a 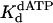 of 28 μM were obtained.
of 28 μM were obtained.
Fig. 2.

Nucleotide incorporation opposite the 3′ T of the TT dimer and the analogous nondamaged template residue. (A) Polη (120 nM) and the damaged 3′ T DNA substrate (300 nM) were mixed with various concentrations of dATP (•, 10 μM; ○, 20 μM; ▪, 50 μM; □, 100 μM; ▴, 200 μM; ▵, 500 μM) for various reaction times. The solid lines represent the best fits to the burst equation. (B) The observed rate constants of the burst phases in A (•) were graphed as a function of dATP concentration, and the solid line represents the best fit to the hyperbolic equation with a kpol equal to 1.2 ± 0.07 s–1 and a 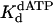 for the Polη–DNA–dATP complex equal to 130 ± 20 μM. (C) Polη (120 nM) and the nondamaged 3′ T DNA substrate (300 nM) were mixed with various concentrations of dATP (•, 5 μM; ○, 10 μM; ▪, 20 μM; □, 50 μM; ▴, 100 μM; ▵, 200 μM) for various reaction times. The solid lines represent the best fits to the burst equation. (D) The observed rate constants of the burst phases in C (•) were graphed as a function of dATP concentration, and the solid line represents the best fit to the hyperbolic equation with a kpol equal to 1.5 ± 0.08 s–1 and a
for the Polη–DNA–dATP complex equal to 130 ± 20 μM. (C) Polη (120 nM) and the nondamaged 3′ T DNA substrate (300 nM) were mixed with various concentrations of dATP (•, 5 μM; ○, 10 μM; ▪, 20 μM; □, 50 μM; ▴, 100 μM; ▵, 200 μM) for various reaction times. The solid lines represent the best fits to the burst equation. (D) The observed rate constants of the burst phases in C (•) were graphed as a function of dATP concentration, and the solid line represents the best fit to the hyperbolic equation with a kpol equal to 1.5 ± 0.08 s–1 and a 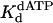 for the Polη–DNA–dATP complex equal to 28 ± 5 μM.
for the Polη–DNA–dATP complex equal to 28 ± 5 μM.
Nucleotide Incorporation Opposite the 5′ T of the TT Dimer. Next, we examined the kinetics of dATP binding and incorporation opposite the 5′ T of the TT dimer and the analogous nondamaged sequence. Fig. 3A shows the kinetics of dATP incorporation opposite the 5′ T of the TT dimer at different concentrations of dATP from 10 to 500 μM, and the kobs was graphed as a function of dATP concentration (Fig. 3B). From the best fit of these data, a kpol of 2.5 s–1 and a 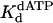 of 72 μM were obtained. Fig. 3 C and D shows the analogous experiments with the identical nondamaged DNA sequence, and a kpol of 1.6 s–1 and a
of 72 μM were obtained. Fig. 3 C and D shows the analogous experiments with the identical nondamaged DNA sequence, and a kpol of 1.6 s–1 and a 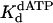 of 39 μM were obtained.
of 39 μM were obtained.
Fig. 3.

Nucleotide incorporation opposite the 5′ T of the TT dimer and the analogous nondamaged template residue. (A) Polη (120 nM) and the damaged 5′ T DNA substrate (300 nM) were mixed with various concentrations of dATP (•, 10 μM; ○, 20 μM; ▪, 50 μM; □, 100 μM; ▴, 200 μM; ▵, 500 μM) for various reaction times. The solid lines represent the best fits to the burst equation. (B) The observed rate constants of the burst phases in A (•) were graphed as a function of dATP concentration, and the solid line represents the best fit to the hyperbolic equation with a kpol equal to 2.5 ± 0.1 s–1 and a 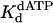 for the Polη–DNA–dATP complex equal to 72 ± 11 μM. (C) Polη (120 nM) and the nondamaged 5′ T DNA substrate (300 nM) were mixed with various concentrations of dATP (•, 5 μM; ○, 10 μM; ▪, 20 μM; □, 50 μM; ▴, 100 μM; ▵, 200 μM) for various reaction times. The solid lines represent the best fits to the burst equation. (D) The observed rate constants of the burst phases in C (•) were graphed as a function of dATP concentration, and the solid line represents the best fit to the hyperbolic equation with a kpol equal to 1.6 ± 0.04 s–1 and a
for the Polη–DNA–dATP complex equal to 72 ± 11 μM. (C) Polη (120 nM) and the nondamaged 5′ T DNA substrate (300 nM) were mixed with various concentrations of dATP (•, 5 μM; ○, 10 μM; ▪, 20 μM; □, 50 μM; ▴, 100 μM; ▵, 200 μM) for various reaction times. The solid lines represent the best fits to the burst equation. (D) The observed rate constants of the burst phases in C (•) were graphed as a function of dATP concentration, and the solid line represents the best fit to the hyperbolic equation with a kpol equal to 1.6 ± 0.04 s–1 and a 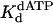 for the Polη–DNA–dATP complex equal to 39 ± 3 μM.
for the Polη–DNA–dATP complex equal to 39 ± 3 μM.
Nucleotide Incorporation Opposite an Abasic Site. One possible model for the of ability Polη to incorporate nucleotides opposite a TT dimer is that it creates an abasic site-like intermediate opposite which it incorporates an A (see Discussion). To compare the mechanism of nucleotide incorporation opposite the TT dimer with that of incorporation opposite a template abasic site, we next examined the kinetics of nucleotide incorporation opposite an abasic site in the same sequence context. The kinetics of dATP incorporation opposite the abasic site were strictly linear, not biphasic, for concentrations of dATP ranging from 20 to 1,000 μM (Fig. 4A). Thus, the steady-state rate of nucleotide incorporation opposite the abasic site is not limited by DNA dissociation or some other step after the chemical step of phosphodiester bond formation; hence, the rate of nucleotide incorporation in the first enzyme turnover is the same as the rate of incorporation in subsequent steady-state turnovers. The linear rate constant, kobs, was graphed as a function of dATP concentration (Fig. 4B), and from the best fit of these data, a kpol of 0.012 s–1 and a 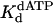 of 70 μM were obtained.
of 70 μM were obtained.
Fig. 4.

Nucleotide incorporation opposite the abasic site (indicated by x in the sequence above the graphs). (A) Polη (120 nM) and the abasic site DNA substrate (300 nM) were mixed with various concentrations of dATP (•, 20 μM; ○, 50 μM; ▪, 100 μM; □, 200 μM; ▴, 500 μM; ▵, 1,000 μM) for various reaction times. The position of the abasic site is designated x in the given sequence. The solid lines represent the best fits to the linear equation. (B) The observed linear rate constants in A (•) were graphed as a function of dATP concentration, and the solid line represents the best fit to the hyperbolic equation with a kpol equal to 0.012 ± 0.001 s–1 and a 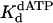 for the Polη–DNA–dATP complex equal to 70 ± 10 μM.
for the Polη–DNA–dATP complex equal to 70 ± 10 μM.
Discussion
Two Possible Mechanisms of Nucleotide Incorporation Opposite the TT Dimer. A TT dimer disrupts the DNA helix, bending it by ≈30° and unwinding it by ≈9°; the ability of the two Ts in the dimer to form Watson–Crick base pairs with As, however, is not affected significantly (21–24). Classical DNA polymerases accommodate only a single unpaired template base in their active site, whereas the 5′ unpaired template base is directed out of the active site at a 90° angle (25–27). However, because of the covalent cis-syn cyclobutane linkage of the 5′ T of a TT dimer to the 3′ T, the 5′ T cannot be flipped out of the active site of the polymerase, preventing classical DNA polymerases from replicating through this lesion. Two possibilities can be envisioned to explain the proficient ability of Polη to replicate through this DNA lesion (Fig. 5). One is that Polη transiently flips the TT dimer out of its active site and then inserts an A opposite the resultant abasic site-like intermediate, acting as an “A rule” polymerase (28). This would then be followed by the insertion of an A opposite the 5′ T, which would occur when the dimer is inside the active site of the enzyme. Such a mechanism has been proposed for the inefficient bypass of a TT dimer by the exo– T7 DNA polymerase (29, 30). The second possibility is that Polη retains both the bases of the TT dimer in the active site and directly inserts an A opposite both Ts of the dimer by using the intrinsic base-pairing ability of the lesion.
Fig. 5.

Two possible models for the insertion of nucleotides opposite a TT dimer by Polη. In the “A rule” insertion model (Left), Polη flips both bases of the TT dimer out of its active site and inserts the first A opposite the resultant transient abasic site-like intermediate. Polη then flips the TT dimer inside its active site and directly inserts the second A opposite the 5′ T of the dimer. In the direct-insertion model (Right), Polη retains both bases of the TT dimer in its active site and directly inserts an A opposite both the 3′ T and 5′ T of the dimer.
Nucleotide Incorporation Opposite a TT Dimer Differs from That Opposite an Abasic Site. Although Polη accurately and efficiently incorporates nucleotides opposite the TT dimer, it is highly inefficient at inserting nucleotides opposite an abasic site (31). Here we directly compared the pre-steady-state kinetics of nucleotide incorporation by yeast Polη opposite a TT dimer and opposite an abasic site in the same sequence context. Incorporation by Polη opposite the TT dimer clearly displayed biphasic or burst kinetics, whereas incorporation opposite the abasic site was strictly linear. The 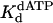 of 70 μM opposite the abasic site was 1.6-fold stronger than for nucleotide binding opposite the 3′ T of the TT dimer and was 2.6-fold weaker than for the binding opposite the corresponding nondamaged T residue (Table 1). These slight variations in
of 70 μM opposite the abasic site was 1.6-fold stronger than for nucleotide binding opposite the 3′ T of the TT dimer and was 2.6-fold weaker than for the binding opposite the corresponding nondamaged T residue (Table 1). These slight variations in 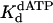 are not surprising given that Polη is not particularly discriminating at the initial nucleotide-binding step (19). In striking contrast, the kpol of 0.012 s–1 for nucleotide incorporation opposite the abasic site was 92-fold less than the kpol value for incorporation opposite the 3′ T of the TT dimer and 130-fold less than the kpol for incorporation opposite the corresponding nondamaged T (Table 1). Because of these clear differences between the kinetics of nucleotide incorporation opposite the abasic site and the 3′ T of the TT dimer, we conclude that it is highly unlikely that Polη incorporates an A opposite the 3′ T of the TT dimer by flipping the TT dimer out of the active site and incorporating the A opposite the resultant abasic site-like intermediate, as has been suggested for exo– T7 DNA polymerase (29, 30).
are not surprising given that Polη is not particularly discriminating at the initial nucleotide-binding step (19). In striking contrast, the kpol of 0.012 s–1 for nucleotide incorporation opposite the abasic site was 92-fold less than the kpol value for incorporation opposite the 3′ T of the TT dimer and 130-fold less than the kpol for incorporation opposite the corresponding nondamaged T (Table 1). Because of these clear differences between the kinetics of nucleotide incorporation opposite the abasic site and the 3′ T of the TT dimer, we conclude that it is highly unlikely that Polη incorporates an A opposite the 3′ T of the TT dimer by flipping the TT dimer out of the active site and incorporating the A opposite the resultant abasic site-like intermediate, as has been suggested for exo– T7 DNA polymerase (29, 30).
Table 1. Kinetic parameters and free-energy differences for yeast Polη-catalyzed nucleotide incorporation opposite a TT dimer.
| 3′ T
|
5′ T
|
|||||
|---|---|---|---|---|---|---|
| Parameter | TT dimer | TT nondamaged | TT dimer | TT nondamaged | ||
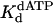 , μM , μM |
110 ± 20 | 27 ± 9 | 76 ± 6 | 38 ± 2 | ||
| Kd/Kd* | 4.1 | 2.0 | ||||
| ΔG, kcal/mol† | 0.06 | -0.76 | -0.16 | -0.57 | ||
| ΔΔG, kcal/mol§ | 0.82 | 0.41 | ||||
| kpol, s-1 | 1.1 ± 0.1 | 1.6 ± 0.1 | 2.4 ± 0.1 | 1.4 ± 0.3 | ||
| kpol/kpol¶ | 0.7 | 1.7 | ||||
| ΔG‡, kcal/mol | 17 | 17 | 17 | 17 | ||
| ΔΔG‡, kcal/mol∥ | 0.2 | -0.3 | ||||
| kpol/Kd, μM-1·s-1 | 0.010 | 0.059 | 0.032 | 0.037 | ||
| frel** | 5.9 | 1.2 | ||||
Values represent the mean and standard errors calculated from at least two complete and independent data sets.
Calculated by 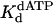 for TT dimer divided by
for TT dimer divided by 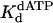 for nondamaged TT sequence.
for nondamaged TT sequence.
Calculation assumes a concentration of 100 μM dATP (1 kcal = 4.18 kJ).
Calculated by ΔG for TT dimer minus ΔG for nondamaged TT sequence.
Calculated by kpol for TT dimer divided by kpol for nondamaged TT sequence.
Calculated by ΔG‡ for TT dimer minus ΔG‡ for nondamaged TT sequence.
Relative efficiency, calculated by kpol/Kd for nondamaged TT sequence divided by kpol/Kd for TT dimer.
Nucleotide Incorporation Opposite a TT Dimer Resembles That Opposite Nondamaged DNA. We directly compared the pre-steady-state kinetics of nucleotide incorporation opposite the 3′ T and 5′ T of the TT dimer and opposite the corresponding nondamaged T residues. In all cases, biphasic or burst kinetics was observed. Overall, we find only slight differences between the 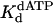 and kpol values for nucleotide incorporation opposite the TT dimer and the nondamaged T residues. The affinity for nucleotide binding opposite the TT dimer was slightly lower than opposite the nondamaged template, being reduced 4.1-fold for incorporation opposite the 3′ T and 2.0-fold for incorporation opposite the 5′ T (Table 1). These correspond to differences in free-energy changes (ΔΔG) for dATP binding opposite the TT dimer vs. the nondamaged TT sequence of ≈0.82 and 0.41 kcal/mol, respectively (Table 1). In the TT dimer, the tilting of the bases from a strictly parallel base-stacking arrangement could preclude optimal base-stacking interactions, and that may account for the slight reduction in affinity for dATP in the presence of the TT dimer.
and kpol values for nucleotide incorporation opposite the TT dimer and the nondamaged T residues. The affinity for nucleotide binding opposite the TT dimer was slightly lower than opposite the nondamaged template, being reduced 4.1-fold for incorporation opposite the 3′ T and 2.0-fold for incorporation opposite the 5′ T (Table 1). These correspond to differences in free-energy changes (ΔΔG) for dATP binding opposite the TT dimer vs. the nondamaged TT sequence of ≈0.82 and 0.41 kcal/mol, respectively (Table 1). In the TT dimer, the tilting of the bases from a strictly parallel base-stacking arrangement could preclude optimal base-stacking interactions, and that may account for the slight reduction in affinity for dATP in the presence of the TT dimer.
Remarkably, we find that the kpol values for nucleotide incorporation opposite the TT dimer and the nondamaged T residues are nearly equivalent (Table 1). The rate-limiting step of nucleotide incorporation in the first turnover could occur at either the chemical step of phosphodiester bond formation or at a conformational change step preceding and limiting the chemical step. The similarities in kpol values indicate that the transition state for this rate-limiting step is entirely insensitive to the geometric distortion introduced by the TT dimer, with differences in the free energy of activation (ΔΔG‡) for this step in the presence of the TT dimer vs. the nondamaged TT sequence of less than half a kilocalorie (<2.092 kJ) per mole (Table 1).
Based on these observations, we conclude that Polη proficiently replicates through a TT dimer by retaining both bases of the dimer within the active site and directly incorporating an A opposite the two Ts of the dimer by using the intrinsic base-pairing ability of the lesion (Fig. 5). Although the structure of the ternary complex of Polη has not been determined, the structure of yeast Polη with modeled DNA and an incoming dNTP indicating that the active site of Polη can accommodate two template nucleotides, the templating residue and the next 5′ unpaired residue (32), supports this inference. Furthermore, the similarities between the 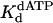 and kpol values with the TT dimer and the corresponding nondamaged sequence show that the active site of Polη is arranged to accommodate the distorted geometry of this lesion in a manner that does not noticeably interfere with the elementary steps of the nucleotide incorporation reaction.
and kpol values with the TT dimer and the corresponding nondamaged sequence show that the active site of Polη is arranged to accommodate the distorted geometry of this lesion in a manner that does not noticeably interfere with the elementary steps of the nucleotide incorporation reaction.
By contrast to its proficient ability to replicate through a TT dimer, lesions such as a (6-4) TT photoproduct (33) or an abasic site (31) present a considerable block to Polη. Whereas a TT dimer has no effect on Watson–Crick base pairing, this potential is greatly impaired for the 3′ T of the (6-4) TT lesion and is absent for an abasic site. As we have pointed out, Watson–Crick hydrogen bonding makes a very significant contribution to DNA synthesis by Polη, and its ability to bypass certain lesions but not others can be explained by the effect the lesion has on normal base pairing (34).
Biological Implications of the Proposed Mechanism of Dimer Bypass. UV is a major risk factor for skin cancers, which include melanomas, basal cell carcinomas, and squamous cell carcinomas. In the United States, the frequency of skin cancers approaches that of all other cancers combined, and is on the rise because of the depletion of the ozone layer (35–37). In addition to the formation of cyclobutane dimers at dipyrimidine sites, UV induces the formation of (6-4) dipyrimidine photoproducts. Although both types of photoproducts are removed by nucleotide excision repair in human cells, the removal of (6-4) photoproducts is much more efficient than that of cyclobutane pyrimidine dimers (38), making it likely that a pyrimidine dimer would persist in DNA and block DNA replication. Our observations that Polη incorporates As opposite the two Ts of the TT dimer with the same kpol values as opposite undamaged Ts strongly support the premise that Polη does not act as an “A rule” polymerase, incorporating an A opposite an abasic site-like intermediate (28); rather, it is able to include the two nucleotides of a TT dimer in its active site and to directly incorporate the nucleotides opposite the dimer, using the intrinsic base-pairing ability of the lesion. An important implication of this mechanism is that it provides a means for the error-free bypass of cyclobutane dimers formed at sites other than TT, as for example, 5′-TC-3′ and 5′-CC-3′ sites. The incorporation of an A opposite the 3′ C of pyrimidine dimers formed at such sites would result in C → T transition mutations. Polη, however, carries out error-free bypass of dimers formed at TC and CC sites, as the frequency of UV-induced mutations at these sites is elevated considerably in a yeast mutant lacking Polη over that in the wild-type strain (39), and recent genetic studies in humans have indicated a similar requirement of Polη in the error-free bypass of these UV lesions (40). Polη thus is a unique polymerase to have evolved a highly efficient and relatively accurate mechanism for replicating through cyclobutane dimers formed at the various dipyrimidine sites, and thereby it makes an important contribution to the prevention of sunlight-induced skin cancers in humans.
Acknowledgments
This work was supported by National Institutes of Health Grant GM19261.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Polη, DNA polymerase η; TT dimer, cis-syn thymine-thymine dimer.
References
- 1.Johnson, R. E., Prakash, S. & Prakash, L. (1999) Science 283, 1001–1004. [DOI] [PubMed] [Google Scholar]
- 2.Johnson, R. E., Washington, M. T., Prakash, S. & Prakash, L. (2000) J. Biol. Chem. 275, 7447–7450. [DOI] [PubMed] [Google Scholar]
- 3.Washington, M. T., Johnson, R. E., Prakash, S. & Prakash, L. (2000) Proc. Natl. Acad. Sci. USA 97, 3094–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald, J. P., Levine, A. S. & Woodgate, R. (1997) Genetics 147, 1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson, R. E., Prakash, S. & Prakash, L. (1999) J. Biol. Chem. 274, 15975–15977. [DOI] [PubMed] [Google Scholar]
- 6.Wang, Y.-C., Maher, V. M., Mitchell, D. L. & McCormick, J. J. (1993) Mol. Cell. Biol. 13, 4276–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waters, H. L., Seetharam, S., Seidman, M. M. & Kraemer, K. H. (1993) J. Invest. Dermatol. 101, 744–748. [DOI] [PubMed] [Google Scholar]
- 8.Johnson, R. E., Kondratick, C. M., Prakash, S. & Prakash, L. (1999) Science 285, 263–265. [DOI] [PubMed] [Google Scholar]
- 9.Masutani, C., Kusumoto, R., Yamada, A., Dohmae, N., Yokoi, M., Yuasa, M., Araki, M., Iwai, S., Takio, K. & Hanaoka, F. (1999) Nature 399, 700–704. [DOI] [PubMed] [Google Scholar]
- 10.Echols, H. & Goodman, M. F. (1991) Annu. Rev. Biochem. 60, 477–511. [DOI] [PubMed] [Google Scholar]
- 11.Goodman, M. F. (1997) Proc. Natl. Acad. Sci. USA 94, 10493–10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Washington, M. T., Johnson, R. E., Prakash, S. & Prakash, L. (1999) J. Biol. Chem. 274, 36835–36838. [DOI] [PubMed] [Google Scholar]
- 13.Benkovic, S. J. & Cameron, C. E. (1995) Methods Enzymol. 262, 257–269. [DOI] [PubMed] [Google Scholar]
- 14.Johnson, K. A. (1995) Methods Enzymol. 249, 38–61. [DOI] [PubMed] [Google Scholar]
- 15.Kuchta, R. D., Mizrahi, V., Benkovic, P. A., Johnson, K. A. & Benkovic, S. J. (1987) Biochemistry 26, 8410–8417. [DOI] [PubMed] [Google Scholar]
- 16.Kuchta, R. D., Benkovic, P. & Benkovic, S. J. (1988) Biochemistry 27, 6716–6725. [DOI] [PubMed] [Google Scholar]
- 17.Patel, S. S., Wong, I. & Johnson, K. A. (1991) Biochemistry 30, 511–525. [DOI] [PubMed] [Google Scholar]
- 18.Wong, I., Patel, S. S. & Johnson, K. A. (1991) Biochemistry 30, 526–537. [DOI] [PubMed] [Google Scholar]
- 19.Washington, M. T., Prakash, L. & Prakash, S. (2001) Cell 107, 917–927. [DOI] [PubMed] [Google Scholar]
- 20.Washington, M. T., Johnson, R. E., Prakash, L. & Prakash, S. (2001) Proc. Natl. Acad. Sci. USA 98, 8355–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciarrocchi, G. & Pedrini, A. M. (1982) J. Mol. Biol. 155, 177–183. [DOI] [PubMed] [Google Scholar]
- 22.Kemmink, J., Boelens, R., Koning, T., van der Marel, G. A., van Boom, J. H. & Kaptein, R. (1987) Nucleic Acids Res. 15, 4645–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Husain, I., Griffith, J. & Sancar, A. (1988) Proc. Natl. Acad. Sci. USA 85, 2558–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park, H., Zhang, K., Ren, Y., Nadji, S., Sinha, N., Taylor, J.-S. & Kang, C. (2002) Proc. Natl. Acad. Sci. USA 99, 15965–15970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doublie, S., Tabor, S., Long, A. M., Richardson, C. C. & Ellenberger, T. (1998) Nature 391, 251–258. [DOI] [PubMed] [Google Scholar]
- 26.Kiefer, J. R., Mao, C., Braman, J. C. & Beese, L. S. (1998) Nature 391, 304–307. [DOI] [PubMed] [Google Scholar]
- 27.Li, Y., Korolev, S. & Waksman, G. (1998) EMBO J. 17, 7514–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strauss, B. S. (1991) BioEssays 13, 79–84. [DOI] [PubMed] [Google Scholar]
- 29.Smith, C. A., Baeten, J. & Taylor, J.-S. (1998) J. Biol. Chem. 273, 21933–21940. [DOI] [PubMed] [Google Scholar]
- 30.Sun, L., Wang, M., Kool, E. T. & Taylor, J.-S. (2000) Biochemistry 39, 14603–14610. [DOI] [PubMed] [Google Scholar]
- 31.Haracska, L., Washington, M. T., Prakash, S. & Prakash, L. (2001) J. Biol. Chem. 276, 6861–6866. [DOI] [PubMed] [Google Scholar]
- 32.Trincao, J., Johnson, R. E., Escalante, C. R., Prakash, S., Prakash, L. & Aggarwal, A. K. (2001) Mol. Cell 8, 417–426. [DOI] [PubMed] [Google Scholar]
- 33.Johnson, R. E., Haracska, L., Prakash, S. & Prakash, L. (2001) Mol. Cell. Biol. 21, 3558–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Washington, M. T., Helquist, S. A., Kool, E. T., Prakash, L. & Prakash, S. (2003) Mol. Cell. Biol. 23, 5107–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glass, A. G. & Hoover, R. N. (1989) J. Am. Med. Assoc. 262, 2097–2100. [PubMed] [Google Scholar]
- 36.Magnus, K. (1991) Int. J. Cancer 47, 12–19. [DOI] [PubMed] [Google Scholar]
- 37.Oikarinen, A. & Raitio, A. (2000) Int. J. Circumpolar Health 59, 52–56. [PubMed] [Google Scholar]
- 38.Mitchell, D. L., Haipek, C. A. & Clarkson, J. M. (1985) Mutat. Res. 143, 109–112. [DOI] [PubMed] [Google Scholar]
- 39.Yu, S.-L., Johnson, R. E., Prakash, S. & Prakash, L. (2001) Mol. Cell. Biol. 21, 185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stary, A., Kannouche, P., Lehmann, A. R. & Sarasin, A. (2003) J. Biol. Chem. 278, 18767–18775. [DOI] [PubMed] [Google Scholar]