

Abstract
Proteins, nucleic acids, and small molecules form a dense network of molecular interactions in a cell. Molecules are nodes of this network, and the interactions between them are edges. The architecture of molecular networks can reveal important principles of cellular organization and function, similarly to the way that protein structure tells us about the function and organization of a protein. Computational analysis of molecular networks has been primarily concerned with node degree [Wagner, A. & Fell, D. A. (2001) Proc. R. Soc. London Ser. B 268, 1803–1810; Jeong, H., Tombor, B., Albert, R., Oltvai, Z. N. & Barabasi, A. L. (2000) Nature 407, 651–654] or degree correlation [Maslov, S. & Sneppen, K. (2002) Science 296, 910–913], and hence focused on single/two-body properties of these networks. Here, by analyzing the multibody structure of the network of protein–protein interactions, we discovered molecular modules that are densely connected within themselves but sparsely connected with the rest of the network. Comparison with experimental data and functional annotation of genes showed two types of modules: (i) protein complexes (splicing machinery, transcription factors, etc.) and (ii) dynamic functional units (signaling cascades, cell-cycle regulation, etc.). Discovered modules are highly statistically significant, as is evident from comparison with random graphs, and are robust to noise in the data. Our results provide strong support for the network modularity principle introduced by Hartwell et al. [Hartwell, L. H., Hopfield, J. J., Leibler, S. & Murray, A. W. (1999) Nature 402, C47–C52], suggesting that found modules constitute the “building blocks” of molecular networks.
Large-scale experiments and integration of published data (1) have provided maps of several biological networks such as metabolic networks (2, 3), protein–protein (4, 5) and protein–DNA interactions (6, 7), etc. Although incomplete and, perhaps, inaccurate (8–11), these maps became a focal point of a search for the general principles that govern the organization of molecular networks (12–16). Important statistical characteristics of such networks include power-law distribution (P(k) ∼ k–γ) (e.g., refs. 16 and 17) or a similar distribution of the node degree k (i.e., the number of edges of a node); the small-world property (11, 13, 16) (i.e., a high clustering coefficient and a small shortest path between every pair of nodes); anticorrelation in the node degree of connected nodes (15) (i.e., highly interacting nodes tend to be connected to low-interacting ones); and other properties.
These properties become evident when hundreds or thousands of molecules and their interactions are studied together. Recently discovered motifs (7, 18) that consist of three to four nodes constitute the other end of the spectrum. Large-scale characteristics are usually attributed to massive evolutionary processes that shape the network (6, 14), whereas many small-scale motifs represent feedback and feed-forward loops in cellular regulation (18, 19). However, most important biological processes such as signal transduction, cell-fate regulation, transcription, and translation involve more than four but much fewer than hundreds of proteins. Most relevant processes in biological networks correspond to the mesoscale (5–25 genes/proteins). Meso-scale properties of biological networks have been mostly elusive because of computational difficulties in enumerating midsize subnetworks (e.g., a network of 1,000 nodes contains 1 × 1023 possible 10-node sets).
Here, we present an in-depth exploration of molecular networks on the meso-scale level. We focused on multibody interactions and searched for sets of proteins having many more interactions among themselves than with the rest of the network (clusters). We have developed several algorithms to find such clusters in an arbitrary network. We analyzed a yeast network of protein–protein interactions (20) and found >50 known and previously uncharacterized protein clusters. We analyzed functional annotation of these clusters and found that most of identified clusters correspond to either of the two types of cellular modules: protein complexes or functional modules (see Discussion). Protein complexes are groups of proteins that interact with each other at the same time and place, forming a single multimolecular machine. Examples of identified protein complexes include several large transcription factor complexes, the anaphase-promoting complex, RNA splicing and polyadenylation machinery, protein export and transport complexes, etc. Functional modules, in contrast, consist of proteins that participate in a particular cellular process while binding each other at a different time and place (different conditions or phases of the cell cycle, in different cellular compartments, etc). Examples of identified functional modules include the CDK/cyclin module responsible for cell-cycle progression, the yeast pheromone response pathway, MAP signaling cascades, etc. The discovered complexes and modules have high statistical significance and consistent functional annotation (when available), and match very well to experimentally obtained protein complexes (21, 22). Importantly, by relying on multibody interactions, our method is robust to false-positive interactions present in the network.
The network of protein interactions (20) was represented as an undirected graph with proteins as nodes and protein interactions as undirected edges. The key idea of our analysis was to identify highly connected subgraphs (clusters) that have more interactions within themselves and fewer with the rest of the graph. A fully connected subgraph, or clique, that is not a part of any other clique is an example of such a cluster. In general, we did not require clusters to be fully connected; instead, the density of connections in the cluster was measured by the parameter Q = 2m/(n(n – 1)), where n is the number of proteins in the cluster and m is the number of interactions between them. We developed algorithms that can identify clusters of sufficiently high Q in an arbitrary graph. Note that, despite some similarity, the problem of dense subgraphs is not identical to the problem of clustering objects in a metric space and cannot be solved by traditional clustering techniques.
Methods
Identification of Highly Connected Sets. Our first approach was to identify all fully connected subgraphs (cliques) by complete enumeration. Because the graph is very sparse, this could be done quickly. In fact, to find cliques of size n one needs to enumerate only the cliques of size n – 1 (for details, see Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org). We started with n = 3 and continued until no more cliques were found in the graph. The largest clique found contains 14 nodes.
The second approach used a clustering technique that works on points not embedded in a metric space. A powerful algorithm of this sort is superparamagnetic clustering (SPC) (23). Briefly, this approach assigns a “spin” to each node in the graph. Each spin can be in several (more than two) states. Spins belonging to connected nodes interact and have the lowest energy when they are in the same state. The system (known as the Potts model) is subject to equilibration at nonzero temperature, making spins fluctuate. The concept behind this method is that spins belonging to a highly connected cluster fluctuate in a correlated fashion. By detecting correlated spins, the algorithm can identify nodes belonging to a highly connected area of the graph. Domany and coworkers introduced such a system for clustering points in an arbitrary space (23) and successfully applied it to a variety of clustering problems (24, 25). Here, we applied SPC to identify clusters on a graph.
In the third approach, we formulated the problem of finding highly connected sets of nodes as an optimization problem: find a set of n nodes that maximizes the function Q(m, n) = 2m/(n(n – 1)), where m is the number of interactions between n nodes. The parameter (0 ≤ Q ≤ 1) characterizes the density of a cluster. For a fully connected set of nodes, Q = 1, and for a set not connected to each other, Q = 0. The optimization Monte Carlo (MC) procedure starts with a connected set of n nodes randomly picked on the graph and proceeds by “moving” selected nodes along the edges of the graph to maximize Q. Moves are accepted according to Metropolis criteria. We also developed an algorithm that minimizes the sum of shortest distances between selected nodes. Both algorithms are very efficient and converge fast to identify a highly connected cluster. Both algorithms require the size of the sought cluster as an input parameter. Although the rate of convergence of MC depends on the effective temperature, the algorithm converges fast at a broad range of temperatures (Fig. 6, which is published as supporting information on the PNAS web site). Comparison of MC and SPC algorithms have shown a better performance of MC for clusters that share common nodes and for high density graphs, whereas SPC has an advantage identifying clusters that have very few connections to the rest of the graph (Fig. 7, which is published as supporting information on the PNAS web site).
Found clusters are then subjected to further cleaning, merging, and selection according to criteria of statistical significance (see Supporting Text for more details).
Statistical Significance. To estimate statistical significance of a cluster that has n proteins and m interactions between them, one would need to calculate the expected number of such clusters E(n, m) in a comparable random graph (i.e., random graph that satisfies certain constrains, i.e., fix node degree). Due to combinatorial explosion of possible subgraphs, direct calculation of E(n, m) in random graphs is computationally unfeasible for n > 4. We developed two statistical procedures that estimate expected value E(n, m) and probability P(n, m) to assess statistical significance of identified clusters. Although Q is a good measure of the density of interactions in a cluster, its statistical significance strongly depends on cluster size, n. A cluster of three proteins with Q = 1 is likely to be found in a random graph, whereas a set of 10 proteins with Q = 0.26 may be very unlikely in the same random graph. We introduced two measures of statistical significance that are based on the probability of finding a cluster in a comparable random graph (15, 18, 26, 27). To compute statistical significance, we first generated 1,000 random graphs in which the number of interactions for each protein is preserved. Next, for each cluster of n proteins and m interactions, we computed the P value as the probability of having more than m connections among the same proteins in the corresponding random graphs. A P value computed this way gives the likelihood of having m (or more) interactions among a particular group of proteins, given the number of interactions that each of these proteins has. Although this probability can be very small, the number Ωn of possible comparable clusters of n proteins is huge. To take this into account we computed E value E = PΩn as the expected number of n protein clusters that have m (or more) interactions. The number of possible comparable clusters is estimated by
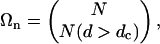 |
where N is the total number of nodes in the graphs and N(d > dc) is the number of nodes with degree greater than dc. We set dc to be the median degree in the cluster of interest. This way, the E value takes into account both the number of proteins in the clusters and the number of interactions each of them has.
Needless to say, all E and P values are approximate and their direct computation is prohibitively computationally expensive. Finally, by applying our search algorithms to the random graphs, we also estimated the Pevd value as the probability of finding any set of n nodes with m or more connections. Because our algorithms seek to maximize m, the Pevd value obeys the Fisher–Tippett extreme value distribution (EVD) Pevd(m) = exp(–exp(–α(m – u))). Parameters α and u of this distribution were obtained by 1,000 MC runs on each of 1,000 random graphs, generated as described above. We observed simple linear scaling of α–1 = a1n + a2 and u = u1n + u2, allowing easy computation of Pevd for clusters of any size. Hence, by establishing Pevd < Pcutoff, one can obtain Q(n)cutoff for clusters of any size n, such that a cluster with Q > Q(n)cutoff is considered to be statistically significant (see Supporting Text and Fig. 8 for details). For our analysis of complexes and modules, we selected highly significant clusters having E < 0.1, P < 1 × 10–4, and Pevd < 1 × 10–4. Fig. 1B shows the comparison of the number of found complexes of a given size and Q versus the number of complexes of this size and Q expected on a random graph.
Fig. 1.

Statistical significance of complexes and modules. (A) Number of complete cliques (Q = 1) as a function of clique size enumerated in the network of protein interactions (red) and in randomly rewired graphs (blue, averaged >1,000 graphs). Inset shows the same plot in log-normal scale. Note the dramatic enrichment in the number of cliques in the protein-interaction graph. Most of these cliques are parts of bigger complexes and modules. (B) Distribution of Q of clusters found by the MC search procedure in the randomly rewired graphs (blue bars). The blue line shows approximation of this distribution by the Fisher–Tippett extreme value distribution (EVD) with two fitted parameters. Red bars show complexes found in the original network of protein interactions. Sizes of the subgraphs are n = 8, 10, and 16. Note that real complexes have many more interactions than the tightest complexes found in randomly rewired graphs.
A similar method used by Milo et al. (18) to identify small network motifs requires exact enumeration of motifs in random graphs. Such enumeration is computationally impossible for larger clusters and modules. Our approach, in contrast, does not involve such enumeration and hence can be expanded to clusters of any size.
We also used importance sampling MC to estimate E(n, m). We sampled a set of n proteins at random and obtained E(m|n) distribution. To make sampling more efficient, we sampled proteins with the probability proportional to their degree, i.e., probability to pick a protein i: Pi = di/Σi=1...N di. E(m|n) estimated by using importance sampling was found to be linear with log(m) and can be accurately extrapolated to higher m. This method was used to estimate the number of cliques in the random graph (Fig. 1 A, blue).
Results
To study the large-scale structure of the protein interaction network, we first enumerated all cliques of size 3 and larger. The relative sparsity of the graph (N = 3,992 nodes and M = 6,500 edges) allowed exact enumeration of the cliques. For comparison, we constructed 1,000 random graphs of the same size and degree distribution (see below) and used them to calculate the expected number of cliques. The graph of protein interactions is known to be a very special graph with a power-law distribution of node degrees. To rule out the possibility that such a cliquish structure is a result of power-law architecture, we constructed our random graphs by using the Maslov–Sneppen procedure (15), which preserves the number of edges of each node. In other words, the obtained protein clusters are controlled for the number of interactions that each protein has. Fig. 1 A presents these results, demonstrating an overwhelming enrichment in cliques of all sizes in the protein graph as compared with the random graphs. Comparison of the observed and expected numbers of cliques also shows that the vast majority of cliques (97% for n = 4, 99.8% for n = 5, etc.) of size 4 and greater are, in fact, statistically significant. High density of interactions in the cliques and their statistical significance show that these cliques did not emerge by chance, pointing at some biological function they carry. The enrichment in the number of cliques reveals an essential modularity in the structure of the network, suggesting that many of these protein interactions are responsible for the formation of protein complexes and functional modules. To further explore this modular structure of the network, we searched the graph for multiprotein clusters that are not cliques (i.e., Q < 1).
The construction of fast algorithms for determining structural properties of graphs is a classic challenge in discrete mathematics and theoretical computer science. Such problems are easy to state and illustrate, but they are often provably difficult in the sense of being NP-hard (NP-hard problems are those for which no known algorithms can find a solution in time polynomial in the problem size, although there are algorithms that can verify a proposed solution in that time). The problem of finding the largest clique, or even of approximating its size, is NP-hard (28). Here, we aimed to find non-fully connected cliques, an even harder problem. Although the stochastic algorithms that we developed cannot be guaranteed to find all solutions, they are efficient when applied to relatively sparse graphs such as a network of protein interactions.
We used two methods to further explore network modularity and find highly connected multiprotein clusters: the MC optimization technique and the SPC algorithm as developed by Domany and coworkers (23, 24). These methods are capable of finding clusters that are highly connected but not necessarily fully connected (Q < 1). Using these techniques, we identified >50 protein clusters of sizes from 4 to 35. Comparable random graphs, in contrast, contained very few, if any, such clusters. Fig. 1B presents distributions of density Q of the same-sized clusters found in the random graphs (blue bars) and in the protein network (red bars). This distribution for random graphs can be fit well by the Fisher–Tippett (29) extreme value distribution (EVD) (also known as the Gumbel distribution) (Fig. 1B, blue line), allowing us to estimate the statistical significance of the protein clusters (see Methods). Strikingly, clusters in the protein network have many more interactions than their counterparts in the random graphs: the probability of finding comparable clusters in random graphs falls below 1 × 10–4 (Pevd < 1 × 10–4). These results demonstrate that, aside from numerous cliques, the protein network contains many significantly dense clusters of interacting proteins. Fig. 2 shows three highly connected clusters and the fragment of network surrounding them, illustrating the difficulty of finding such clusters. These clusters provide additional strong evidence supporting modular architecture in biological networks.
Fig. 2.

Fragment of the protein network. Nodes and interactions in discovered clusters are shown in bold. Nodes are colored by functional categories in MIPS (20): red, transcription regulation; blue, cell-cycle/cell-fate control; green, RNA processing; and yellow, protein transport. Complexes shown are the SAGA/TFIID complex (red), the anaphase-promoting complex (blue), and the TRAPP complex (yellow).
What is the biological role of these highly connected clusters? To answer this question, we analyzed the available functional annotation of Saccharomyces cerevisiae genes (20, 30). We found that genes belonging to the same module or complex have a consistent biological function, obtained from Munich Information Center for Protein Sequences (MIPS) functional annotation tables (www.mips.biochem.mpg.de) (20). Fig. 2 presents examples of discovered complexes, with the proteins colored according to their functional classifications. Fig. 3 gives examples of two functional modules: cell-cycle regulation and the MAP kinase cascade. Gene annotation allowed us to assign function to the identified complexes. The majority of identified complexes and modules belong to the following four functional classes: transcription regulation, cell-cycle/cell-fate control, RNA processing, and protein transport.
Fig. 3.

Examples of discovered functional modules. (A) A module involved in cell-cycle regulation. This module consists of cyclins (CLB1-4 and CLN2) and cyclin-dependent kinases (CKS1 and CDC28) and a nuclear import protein (NIP29). Although they have many interactions, these proteins are not present in the cell at the same time. (B) Pheromone signal transduction pathway in the network of protein–protein interactions. This module includes several MAPK (mitogen-activated protein kinase) and MAPKK (mitogen-activated protein kinase kinase) kinases, as well as other proteins involved in signal transduction. These proteins do not form a single complex; rather, they interact in a specific order.
The largest fully connected complex has 14 proteins, all of which are components of the SAGA/TFIID transcription factor. The 17-member extension of this complex includes some additional transcription factors. Other transcription complexes found in the network include the four-member HAP complex of CCAAT-binding proteins, the seven-member mediator of transcription regulator (MED), and the NOT transcription complex. The next-to-largest fully connected complex consists of 11 proteins: four are the cell-division control proteins CDC16, CDC23, CDC26, and CDC27, and the other seven are subunits of the complex. Together, these 11 proteins constitute the anaphase-promoting complex, an essential component of cell-cycle regulation. Another complex involved in cell-cycle regulation is a six-member ubiquitination complex (CDC34, CDC53, CDC4, MET30, SKP1, and GRR1) known to be scaffolded by Cdc53p and responsible for the transition into S phase. The discovered complexes include several RNA-processing machines: (i) a 12-member complex of several Lsm splicing factors associated with the mRNA-decapping enzyme DCP1, topoII-associated factor, and two 40S small ribosomal subunits; (ii) a 14-member complex of CFI/CFII/PFI factors and poly(A) polymerase; (iii) an rRNA-processing complex (exosome); (iv) a four-member complex of tRNA-splicing endonuclease subunits; and (v) a complex of three pre-mRNA splicing factors, bound to an unknown protein that is homologous to a human breast-tumor associated autoantigen (see Supporting Text).
Modules have more diverse, although still very consistent, functional annotation of their genes. It is important to distinguish between protein complexes and functional modules, because they have different biological meanings. A protein complex is a molecular machine that consists of several proteins (nucleic acids and other molecules) that bind each other at the same place and time (e.g., transcription factors, histones, polymerases, etc.). On the contrary, a functional module (31) consists of a few proteins (and other molecules) that control or perform a particular cellular function through interactions between themselves. These proteins do not necessarily interact at the same time and place, or form a macromolecular complex (e.g., signaling pathway, cell-cycle regulation, etc.). In many cases, it is hard to make this distinction. Because analyzed pairwise protein interactions do not have temporal and spatial information, our method successfully discovers both complexes and modules but does not distinguish between the two.
Fig. 3 presents two identified modules: an eight-member module of cyclin-dependent kinases, cyclins and their inhibitors regulating the cell cycle (32) (Fig. 2 A), and pheromone signal transduction cascade that scaffolds at the STE5 protein (33) (Fig. 2B). Other found modules include a six-member module of proteins involved in bud emergence and polarity establishment (34, 35) (CDC24, CDC42, FAR1, STE20, BEM1, and RSR1); a six-member module of CDCs, septins, and Ser/Thr protein kinases involved in mitotic control; etc. (a complete list of complexes and modules with functional annotation is provided in the Supporting Text).
Comparison of the predicted with the experimentally derived complexes (20–22) showed very good agreement, in terms of both the coverage and specificity of our predictions. We compared identified complexes with the complexes found by (i) tandem-affinity purification (TAP) and mass spectrometry (21), catalogued in Cellzome (http://yeast.cellzome.com); (ii) complexes found by high-throughput mass-spectrometric protein complex identification (HMS-PCI) (22), catalogued in the Biomolecular Interaction Network Database (www.bind.ca); and (iii) other complexes collected from the literature by human experts, catalogued in the MIPS database (20). First, we found experimental complexes that are consistent with the studied network of protein–protein interactions, i.e., that correspond to dense regions of the network. Only 29 experimental complexes satisfied the strict criteria of Q > 0.2 and Pevd < 1 × 10–4, and 69 experimental complexes satisfied the weaker criteria of Q > 0.3 and Pevd < 0.1. This result came as no surprise, because the known protein interactions represent a small fraction of the interactions present in a cell (9, 10).
Next, we compared the experimental with the computationally derived complexes. For each computationally derived complex, we found a best-matching experimental complex by minimizing the probability of a random overlap between the two, using the following equation:
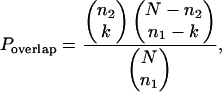 |
where N is the total number of nodes in the network, n1 and n2 are the sizes of two complexes, and k is the number of nodes that they have in common. Fig. 4 presents the overlap k/n1 between found and experimentally derived complexes. In fact, all 29 of the strictly consistent experimental complexes and most of the 69 weakly consistent ones were successfully found with 100% overlap. A few that were missing or only partially recovered are smaller and sparser (Fig. 4 Inset). We also found that of >50 computationally discovered clusters, >80% matched at least one experimental complex. We suggest that the rest constitute previously uncharacterized complexes or modules.
Fig. 4.

Comparison of discovered complexes and modules with complexes derived experimentally (BIND and Cellzome) and complexes catalogued in MIPS. Discovered complexes are sorted by the overlap with the best-matching experimental complex (see Methods and Supporting Text). The overlap is defined as the number of common proteins divided by the number of proteins in the best-matching experimental complex. The first 31 complexes match exactly, and another 11 have overlap above 65%. Inset shows the overlap as a function of the size of the discovered complex. Note that discovered complexes of all sizes match very well with known experimental complexes. Discovered complexes that do not match with experimental ones constitute our predictions (see Discussion for details).
Our study makes four types of predictions: previously uncharacterized protein complexes, previously uncharacterized members in known complexes, membership of uncharacterized proteins in known complexes, and functional modules. We predict 8 possible previously uncharacterized complexes, 7 functional modules, 4 uncharacterized proteins in different complexes, and 13 complexes with possible additional membership. For example, we found a six-member, highly significant complex with Q = 0.73, P = 1 × 10–17, and Pevd = 1 × 10–5 that is not listed among any known protein complexes. Only one protein of the six in that complex has been characterized, as a YIP1 Golgi membrane protein (36); the others have no annotation, although they share some homology with membrane proteins. The best example of previously uncharacterized members in known complexes is a complex of 13 proteins, 11 of which form the Lsm splicing complex, along with two 40S small ribosomal subunits that apparently are new members. These and similar predictions of previously uncharacterized protein complexes (see Supporting Text) can be verified by coimmunoprecipitation or similar techniques.
Discussion
We demonstrated the presence of highly connected clusters of proteins in a network of protein interactions. These results strongly support the suggested modular architecture of biological networks (31). By analyzing the biological function of these clusters, we distinguished two types of clusters: protein complexes and dynamic functional modules. Both complexes and modules have more interactions among their members than with the rest of the network. In a complex, however, these interactions are realized at the same time, bringing participating proteins together (perhaps in a different order, and not simultaneously). In a more generic, dynamic module, interactions are not realized at the same time or place (e.g., interactions between CDKs and cyclins in the module of cell-cycle regulation). Dynamic modules are elusive to experimental purification because they are not assembled as a complex at any single point in time.
An important advantage of computational analysis is that it allows detection of such modules by integrating pairwise molecular interactions that occur at different times and places. Using computational techniques alone, however, we cannot discriminate between complexes and modules or between transient and simultaneous interactions, but instead must target experimental studies toward potential functional modules. For example, the predicted membership of the two ribosomal proteins in the Lsm splicing complex can be transient, conditional, or simultaneous with the rest of the Lsm complex. These ambiguities need to be resolved experimentally.
Computational strategies like ours necessarily rely on experimental data with their limitations and instrumental errors. An important (and unfortunate) aspect of high-throughput experimental data is a high rate of false positives. To investigate the extent to which false positives can derail the search process and affect identified clusters (18), we randomly reconnected, removed, or added 10–50% of interactions in the network. We searched for clusters in the perturbed networks and compared identified clusters with the original ones. Fig. 5 presents the fraction of original clusters that have been recovered in the perturbed networks.
Fig. 5.

The fraction of clusters recovered in the randomly perturbed network, as a function of the fraction of altered links. Black curves correspond to the case when links are randomly rewired; red, randomly removed (true negatives); and green, randomly added (false positives). The original cluster is said to be recovered if the perturbed network has a cluster that shares at least 50% of the nodes with the original one. Each perturbation was repeated 10 times. Also see Fig. 9, which is published as supporting information on the PNAS web site.
Importantly, noise in the form of removal or addition of links has less deteriorating effect than random rewiring. About 75% of clusters can still be found when 10% of links are rewired. More than 80% of clusters sustain addition or removal of 20% of links. The robustness of discovered clusters to noise in the data arises from the use of multiple interactions to identify a cluster. Similar robustness has been demonstrated for smaller motifs (18).
Naturally, our technique fails to identify complexes and modules for which the number of known interactions within the cluster is insufficient. We found several dense complexes and modules that have consistent functional annotation but are not sufficiently dense to be statistically significant clusters. We omitted such clusters from further analysis. Similarly, many cliques of three proteins (total = 1,444) have consistent functional annotation but need to be considered with caution, because a random graph is expected to have ≈500 such cliques (corresponding to the false-positive rate of 38%).
Other techniques to analyze the structure of biological and social networks have been recently developed. Milo et al. (18) looked for small (three- to four-member) motifs that are frequent in a directed network. Shenn-Orr et al. (7) identified three types of structures frequent in the Escherichia coli transcription network. In contrast to these approaches, we were looking for (i) bigger clusters (4–20) and (ii) clusters that have many more interactions within than with the rest of the network. Also, we were concerned not with the frequency of these clusters in the network, but rather with the density of interactions in the clusters. This approach allowed us to uncover large and unique complexes in the protein interaction network (like the anaphase-promoting complex). Another technique, developed by Girvan and Newman (37), attempts to decompose the whole network into weakly connected components. While being a very promising approach, it may not be able to find small, highly connected regions embedded into a highly unstructured network, the apparent situation in the network of protein interactions. The approaches of Milo et al., Girvan and Newman, and this study are highly complementary to each other because they address different questions and study networks at different resolution.
The analyzed network (20) includes interactions obtained by two types of studies: large-scale proteomic experiments (two-hybrid) and traditional, hypothesis-driven studies of protein interactions (i.e., small-scale two-hybrid, coprecipitation, etc.). High-throughput mass-spectrometric protein complex identification (HMS-PCI) and tandem-affinity purification (TAP) derived complexes were not part of the database at the time of downloading. Interestingly, most of the discovered complexes and modules come from traditional studies, rather than from large-scale experiments. This finding indicate significant anthropomorphic bias in the set of known interactions. It also suggests that although large-scale proteomic studies provide a wealth of protein interaction data, the scarcity of the data (and its contamination with false positives) makes such studies less valuable for identification of functional modules. Our results suggest that integration of large-scale two-hybrid data with other types of interactions can help to overcome this limitation. Our computational strategy holds major promise as a tool for integrating various types of data in the search for novel functional modules, in that it can handle different types (“colors”) of interactions, including genetic (e.g., syn-lethality), protein–DNA, and localization data. Integration of networks of physical interactions with graphs of evolutionary relationships (38) can help us to understand the origin of cellular modularity.
We showed that a computational technique can identify complexes and modules of all sizes, including transient complexes and complexes of low stoichiometry, overcoming the limitations of experimental purification of protein complexes (21, 22). Although our technique relies on experimentally derived interactions, the multibody nature of discovered complexes makes our algorithms robust to the high rate of false positives in experimental data. Our results suggest several testable biological hypotheses and reveal an essential meso-scale modularity and multibody structure of molecular networks.
Supplementary Material
Acknowledgments
We are thankful to Shamil Sunyaev, Dennis Vitkup, and Fritz Roth for useful discussions, E. Domany for generously providing the code of the SPC program, and two anonymous referees who suggested MC vs. SPC comparison and the addition of Fig. 5. This work was supported by the John F. and Virginia B. Taplin Award and the Alfred P. Sloan Foundation.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MC, Monte Carlo; MIPS, Munich Information Center for Protein Sequences; SPC, superparamagnetic clustering.
References
- 1.Ideker, T., Thorsson, V., Ranish, J. A., Christmas, R., Buhler, J., Eng, J. K., Bumgarner, R., Goodlett, D. R., Aebersold, R. & Hood, L. (2001) Science 292, 929–934. [DOI] [PubMed] [Google Scholar]
- 2.Kanehisa, M., Goto, S., Kawashima, S. & Nakaya, A. (2002) Nucleic Acids Res. 30, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karp, P. D., Riley, M., Paley, S. M. & Pellegrini-Toole, A. (2002) Nucleic Acids Res. 30, 59–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uetz, P., Giot, L., Cagney, G., Mansfield, T. A., Judson, R. S., Knight, J. R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., et al. (2000) Nature 403, 623–627. [DOI] [PubMed] [Google Scholar]
- 5.Ito, T., Chiba, T., Ozawa, R., Yoshida, M., Hattori, M. & Sakaki, Y. (2001) Proc. Natl. Acad. Sci. USA 98, 4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee, T. I., Rinaldi, N. J., Robert, F., Odom, D. T., Bar-Joseph, Z., Gerber, G. K., Hannett, N. M., Harbison, C. T., Thompson, C. M., Simon, I., et al. (2002) Science 298, 799–804. [DOI] [PubMed] [Google Scholar]
- 7.Shen-Orr, S. S., Milo, R., Mangan, S. & Alon, U. (2002) Nat. Genet. 31, 64–68. [DOI] [PubMed] [Google Scholar]
- 8.Deng, M., Sun, F. & Chen, T. (2003) Pac. Symp. Biocomput., 140–151. [PubMed]
- 9.Gerstein, M., Lan, N. & Jansen, R. (2002) Science 295, 284–287. [DOI] [PubMed] [Google Scholar]
- 10.von Mering, C., Krause, R., Snel, B., Cornell, M., Oliver, S. G., Fields, S. & Bork, P. (2002) Nature 417, 399–403. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg, D. S. & Roth, F. P. (2003) Proc. Natl. Acad. Sci. USA 100, 4372–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Podani, J., Oltvai, Z. N., Jeong, H., Tombor, B., Barabasi, A. L. & Szathmary, E. (2001) Nat. Genet. 29, 54–56. [DOI] [PubMed] [Google Scholar]
- 13.Ravasz, E., Somera, A. L., Mongru, D. A., Oltvai, Z. N. & Barabasi, A. L. (2002) Science 297, 1551–1555. [DOI] [PubMed] [Google Scholar]
- 14.Jeong, H., Tombor, B., Albert, R., Oltvai, Z. N. & Barabasi, A. L. (2000) Nature 407, 651–654. [DOI] [PubMed] [Google Scholar]
- 15.Maslov, S. & Sneppen, K. (2002) Science 296, 910–913. [DOI] [PubMed] [Google Scholar]
- 16.Wagner, A. & Fell, D. A. (2001) Proc. R. Soc. London B 268, 1803–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong, H., Mason, S. P., Barabasi, A. L. & Oltvai, Z. N. (2001) Nature 411, 41–42. [DOI] [PubMed] [Google Scholar]
- 18.Milo, R., Shen-Orr, S., Itzkovitz, S., Kashtan, N., Chklovskii, D. & Alon, U. (2002) Science 298, 824–827. [DOI] [PubMed] [Google Scholar]
- 19.Rosenfeld, N., Elowitz, M. B. & Alon, U. (2002) J. Mol. Biol. 323, 785–793. [DOI] [PubMed] [Google Scholar]
- 20.Mewes, H. W., Frishman, D., Guldener, U., Mannhaupt, G., Mayer, K., Mokrejs, M., Morgenstern, B., Munsterkotter, M., Rudd, S. & Weil, B. (2002) Nucleic Acids Res. 30, 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gavin, A. C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J. M., Michon, A. M., Cruciat, C. M., et al. (2002) Nature 415, 141–147. [DOI] [PubMed] [Google Scholar]
- 22.Ho, Y., Gruhler, A., Heilbut, A., Bader, G. D., Moore, L., Adams, S. L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., et al. (2002) Nature 415, 180–183. [DOI] [PubMed] [Google Scholar]
- 23.Blatt, M., Wiseman, S. & Domany, E. (1996) Phys. Rev. Lett. 76, 3251–3254. [DOI] [PubMed] [Google Scholar]
- 24.Getz, G., Levine, E. & Domany, E. (2000) Proc. Natl. Acad. Sci. USA 97, 12079–12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Getz, G., Vendruscolo, M., Sachs, D. & Domany, E. (2002) Proteins 46, 405–415. [DOI] [PubMed] [Google Scholar]
- 26.Ideker, T., Ozier, O., Schwikowski, B. & Siegel, A. F. (2002) Bioinformatics 18, Suppl. 1, S233–S240. [DOI] [PubMed] [Google Scholar]
- 27.Pilpel, Y., Sudarsanam, P. & Church, G. M. (2001) Nat. Genet. 29, 153–159. [DOI] [PubMed] [Google Scholar]
- 28.Håstad, J. (1996) in Proceedings of the 37th Annual IEEE Symposium on Foundations of Computer Science (IEEE Computer Society, Los Alamitos, CA), pp. 627–636.
- 29.Leadbetter, M. R., Lindgren, G. & Rootzβen, H. (1983) Extremes and Related Properties of Random Sequences and Processes (Springer, New York).
- 30.Weng, S., Dong, Q., Balakrishnan, R., Christie, K., Costanzo, M., Dolinski, K., Dwight, S. S., Engel, S., Fisk, D. G., Hong, E., et al. (2003) Nucleic Acids Res. 31, 216–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartwell, L. H., Hopfield, J. J., Leibler, S. & Murray, A. W. (1999) Nature 402, C47–C52. [DOI] [PubMed] [Google Scholar]
- 32.Spruck, C. H. & Strohmaier, H. M. (2002) Cell Cycle 1, 250–254. [PubMed] [Google Scholar]
- 33.Elion, E. A. (2001) J. Cell Sci. 114, 3967–3978. [DOI] [PubMed] [Google Scholar]
- 34.Krylov, D. M., Nasmyth, K. & Koonin, E. V. (2003) Curr. Biol. 13, 173–177. [DOI] [PubMed] [Google Scholar]
- 35.Segal, M. & Bloom, K. (2001) Trends Cell Biol. 11, 160–166. [DOI] [PubMed] [Google Scholar]
- 36.Yang, X., Matern, H. T. & Gallwitz, D. (1998) EMBO J. 17, 4954–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Girvan, M. & Newman, M. E. (2002) Proc. Natl. Acad. Sci. USA 99, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dokholyan, N. V., Shakhnovich, B. & Shakhnovich, E. I. (2002) Proc. Natl. Acad. Sci. USA 99, 14132–14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.