

Abstract
The proteasome, a multicatalytic protease, is known to degrade unfolded polypeptides with low specificity in substrate selection and cleavage pattern. This lack of well-defined substrate specificities makes the design of peptide-based highly selective inhibitors extremely difficult. However, the x-ray structure of the proteasome from Saccharomyces cerevisiae reveals a unique topography of the six active sites in the inner chamber of the protease, which lends itself to strategies of specific multivalent inhibition. Structure-derived active site separation distances were exploited for the design of homo- and heterobivalent inhibitors based on peptide aldehyde head groups and polyoxyethylene as spacer element. Polyoxyethylene was chosen as a flexible, linear, and proteasome-resistant polymer to mimic unfolded polypeptide chains and thus to allow access to the proteolytic chamber. Spacer lengths were selected that satisfy the inter- and intra-ring distances for occupation of the active sites from the S subsites. X-ray analysis of the proteasome/bivalent inhibitor complexes confirmed independent recognition and binding of the inhibitory head groups. Their inhibitory potencies, which are by 2 orders of magnitude enhanced, compared with pegylated monovalent inhibitors, result from the bivalent binding. The principle of multivalency, ubiquitous in nature, has been successfully applied in the past to enhance affinity and avidity of ligands in molecular recognition processes. The present study confirms its utility also for inhibition of multicatalytic protease complexes.
Keywords: polyoxyethylene, x-ray structures
The proteasome is a multicatalytic protease complex that is involved in intracellular protein turnover in all three kingdoms of life. The proteasome is located in both the cytosol and the nucleus and acts in the degradation of abnormal, misfolded, or improperly assembled proteins, in stress response, cell cycle control, cell differentiation, metabolic adaptation, and cellular immune response. It also is involved in many pathophysiological processes like inflammation and cancer and constitutes a promising target for drug design. In mammals the proteasomes also are responsible for the production of the bulk of antigenic peptides, which are presented via MHC class I molecules on the cell surface to cytotoxic T lymphocytes. The antiviral cytokine INF-γ induces transcription of three additional β subunits (LMP2, MECL-1, and LMP7), which can replace their constitutive homologs (β1, β2, and β5) in newly assembled proteasomes. The resulting immuno-proteasomes show altered cleavage patterns in vitro; these are thought to be essential for the proteasomal antigen processing (1). Most of these functions are linked to an ubiquitin- and ATP-dependent protein degradation pathway involving the 26S proteasome whose core and proteolytic chamber is formed by the 20S proteasome (2–7). The eukaryotic 20S proteasome consists of seven different α-type and seven different β-type subunits, all of which have been cloned and sequenced and can be grouped by sequence homology (8). As defined by the character of the P1 cleavage sites of chromogenic substrates, trypsin-like, chymotrypsin-like, and post-glutamyl-peptide hydrolytic (PGPH) activities are exhibited by the eukaryotic proteasome. These specificities, however, are not reflected in the cleavage pattern of protein substrates, which seem to be cleaved at almost every position (9–12).
The crystal structure of the 20S proteasome from Saccharomyces cerevisiae revealed an overall assembly of the 28 subunits, which are arranged as a stack of four heptameric α1–7,β1–7,β1′-7′,α1′-7′ rings to form a cylindrical particle (13). Only three of the seven different β-subunits of one ring, i.e., β1, β2 and β5, are autoprocessed with generation of the N-terminal nucleophile, the Thr1 residue essential for activity. Mutational studies in yeast have shown that these three β-subunits are responsible for the three major proteolytic activities of the eukaryotic proteasome against small chromogenic substrates and a large protein (14–16), i.e., β1 for the PGPH, β2 for the trypsin-like, and β5 for the chymotrypsin-like activity.
The S1 pockets of these subunits are the major specificity determinants and are appropriately polar and sized to accommodate acidic, basic, and apolar P1 side chains, respectively, but also bind noncomplementary residues (13), consistent with the low specificity of the proteasome (15). Besides tryptase (17), the proteasome is the only oligomeric protease in eukaryotes with a known and well-defined geometry in the display of the active sites. Its unique arrangement lends itself as a target for specific multivalent inhibition. The principle of multivalency as a tool for enhancing affinity and avidity was first established in the chelate chemistry (18, 19) and later discovered in nature as an universal principle that is ubiquitously exploited to enhance selectivity and avidity in molecular recognition processes (20, 21).
We previously have reported the structure-based design of bifunctional inhibitors of the proteasome that led to maleoyl-β-Ala-Val-Arg-H as a highly specific inhibitor of the trypsin-like activity (22). In the present study an approach to proteasome inhibition is presented, where the unique arrangement of six active sites in one enzyme particle is used for the design of bivalent inhibitors (Fig. 1). By linking the N termini of two tripeptide aldehydes as binding heads with a polymeric spacer that is appropriate for simultaneous binding at two different active sites from the nonprimed subsites, bivalent inhibitors of up to 2 orders of magnitude enhanced binding affinities could be obtained. This principle of proteasome inhibition can be applied to a wide range of monovalent proteasome inhibitors to benefit from the effects of multivalency on avidity and selectivity.
Figure 1.
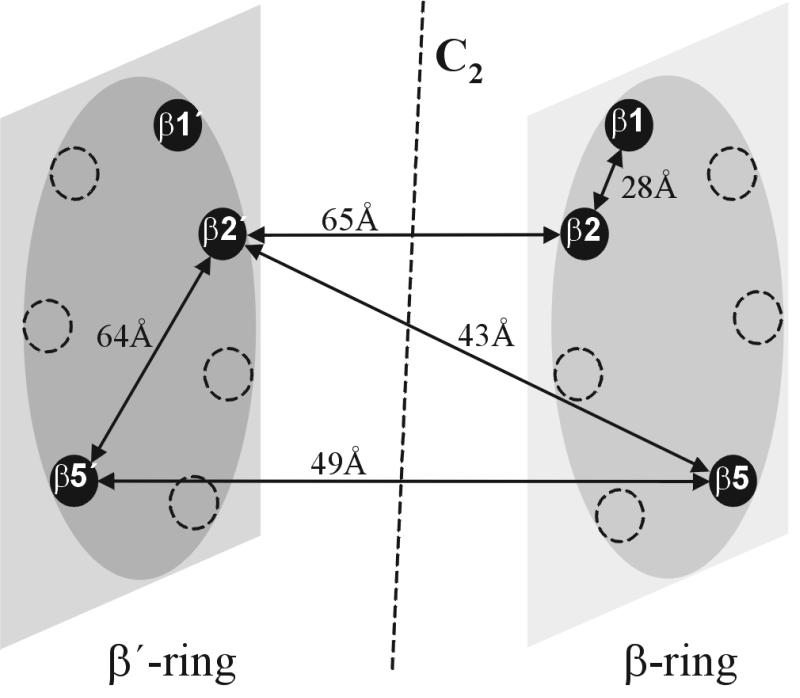
Schematic representation of the central β-rings of the yeast proteasome with selected distances between active sites as derived from the x-ray structure (13).
MATERIALS AND METHODS
The polymeric spacer HOOC-(CH2)2-CO-NH-(PEG)19–25-NH-CO-(CH2)2-COOH was from Rapp Polymere (Tübingen, Germany), the substrates Z-Leu-Leu-Glu-β-naphthylamide, Bz-Phe-Val-Arg-7-amido-4-methyl-coumarin (AMC), and Suc-Leu-Leu-Val-Tyr-AMC were purchased from Bachem, and 20S proteasome from S. cerevisiae was prepared as described (13).
Synthesis of the Inhibitors.
The synthesis of Ac-Leu-Leu-Nle-H (1) and H-Leu-Leu-Nle-semicarbazone (Sc) trifluoroacetate (23) as well as H-Val-Arg(Adoc)2-diethyl acetal (22) have been reported. The tripeptide aldehyde Ac-Arg-Val-Arg-H (2) was prepared as outlined in Scheme S1, and the polyoxyethylene (PEG)/peptide aldehyde conjugates (7-11) were obtained following the synthetic routes of Scheme S2. Stepwise N-terminal elongation of H-Leu-Leu-Nle-Sc with Boc-Gly-Pro-Gly-Gly-OH and (C2H5S)2CH-CO-Leu-OH by the benzotriazol-1-yl-oxytripyrrolidinephosphonium hexafluorophosphate (24) and O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate procedure (25), respectively, and intermediate Nα-Boc cleavage with 25% trifluoroacetic acid (TFA) in CH2Cl2 followed by the final aldehyde deprotection with Tl(NO3)3 in acetonitrile/H2O led to the octapeptide 3. The propeptide-derived inhibitors 4-6 were synthesized on solid support via side-chain attachment of Fmoc-Glu(OH)-Sc (where Fmoc = fluorenylmethoxycarbonyl) to 2-chlorotrityl chloride resin (Alexis, Grünberg, Germany) using standard protocols of the Fmoc/tert-butyl chemistry. The N-terminal succinic acid aldehyde was introduced as Sc derivative. Final cleavage from the resin and deprotection of the aldehydes was achieved with 95% aqueous TFA. Details of the synthesis will be reported elsewhere.
Scheme 1.
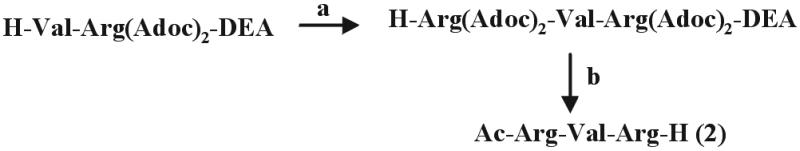
Synthesis of Ac-Arg-Val-Arg-H. (a) i) Z-Arg(Adoc)2-OH, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU)/1-hydroxybenzotriazole/diisopropylethylamine (DIEA), dimethylformamide (DMF); ii) Pd/C (10%), EtOH. (b) i) Ac2O, DIEA, DMF; ii) 95% trifluoroacetic acid.
Scheme 2.
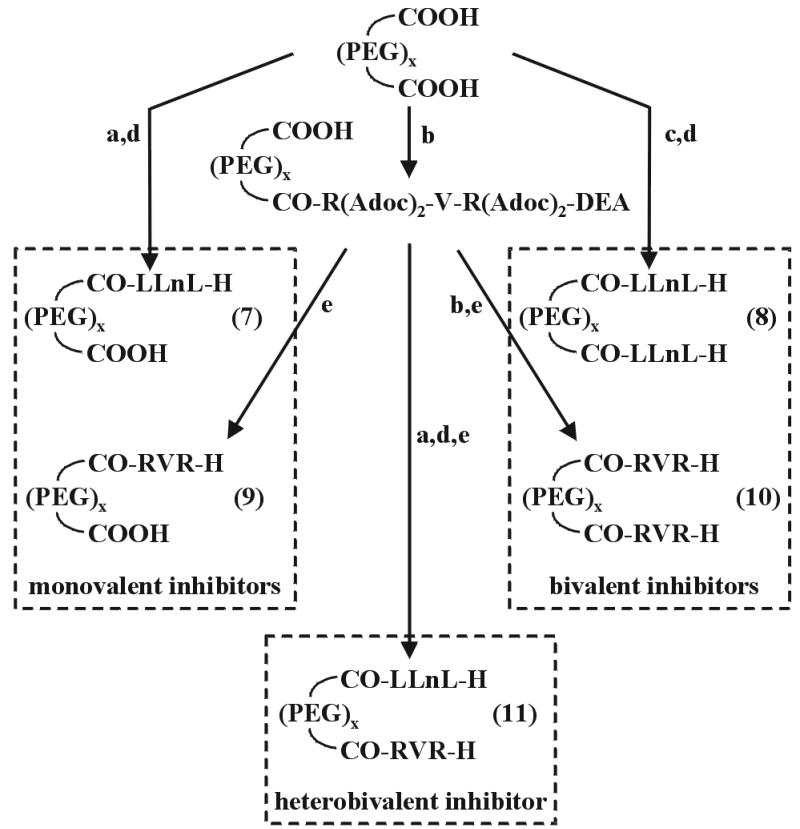
Synthesis of the PEG/peptide aldehyde conjugates. (a) 1 equ. H-Leu-Leu-Nle-Sc, TBTU/1-hydroxybenzotriazole (HOBt)/DIEA, DMF. (b) 1 equ. H-Arg(Adoc)2-Val-Arg(Adoc)2-diethyl acetal, TBTU/HOBt/DIEA, DMF. (c) 2 equ. H-Leu-Leu-Nle-Sc, TBTU/HOBt/DIEA, DMF. (d) AcOH, 37% HCHO, MeOH. (e) 95% trifluoroacetic acid.
Proteasome Assay.
A solution of proteasome from S. cerevisiae in Tris⋅HCl (pH 7.5; 450 μl; 6.67 nM for PGPH, 5.56 nM for trypsin-like, and 1.11 nM for chymotrypsin-like activity) was incubated with the inhibitors at varying concentrations (1 nmol to 100 μmol) for 1 h at 37°C. The fluorogenic substrates for PGPH (Z-Leu-Leu-Glu-β-naphthylamide, 40 μM), trypsin-like (Bz-Phe-Val-Arg-AMC, 8 μM), and chymotrypsin-like assays (Suc-Leu-Leu-Val-Tyr-AMC, 8 μM) were dissolved in the same Tris⋅HCl buffer with a minimum amount of DMSO and added to the enzyme solution at 37°C to reach a final volume of 500 μl. Fluorescence excitation/emission wavelengths were 360 nm/460 nm for AMC and 335 nm/410 nm for β-naphthylamide. The rates of hydrolysis were monitored by the fluorescence increase, and the initial linear portions of the curves (100–300 sec) were used to calculate the IC50 values.
X-Ray Structure Analysis.
Crystals of 20S proteasomes from S. cereisiae were grown in hanging drops at 24°C as described (13). The crystals were soaked at final inhibitor concentrations of 5 mM for 12 h in a cryoprotecting buffer and frozen in a stream of cold nitrogen gas (Oxford Cryo Systems, Oxford, U.K.). Data were collected by using synchrotron radiation with λ = 1.1 Å on the BW6-beamline at the Deutsches Elektronen-Synchroton, Hamburg, Germany (Table 1). The anisotropy of diffraction was corrected by an overall anisotropic temperature factor by comparing observed and calculated structure amplitudes by using x-plor (26). Electron density was averaged 10 times over the 2-fold noncrystallographic symmetry axis by using main (27). Model building was carried out with frodo (28). Modeling experiments were performed by using the coordinates of yeast 20S proteasome with main (27).
Table 1.
Data collection and refinement statistics
| 20S proteasome complex with | 3 | 8 | 10 |
|---|---|---|---|
| Space group | P21 | P21 | P21 |
| Crystal data, Å/° | a = 135.6 | a = 135.4 | a = 134.3 |
| b = 300.3 | b = 298.9 | b = 300.7 | |
| c = 144.0 | c = 144.6 | c = 143.9 | |
| β = 113.0 | β = 112.9 | β = 113.0 | |
| Resolution, Å | 2.7 | 2.3 | 3.0 |
| Observation, >2σ | 834,543 | 1,224,818 | 389,546 |
| Unique reflections | 340,553 | 437,435 | 175,637 |
| Completeness, % | 93.3 | 94.4 | 86.4 |
| Rmerge, % | 11.0 | 9.7 | 17.3 |
| R/Rfree, % | 29.7/36.0 | 25.3/30.0 | 20.7/28.5 |
| rms from bonds, Å | 0.013 | 0.011 | 0.012 |
| rms from angles, ° | 2.043 | 1.875 | 1.966 |
RESULTS
Design of Intra-Ring Bivalent Inhibitors.
Despite the differentiated specificities of the β1, β2, and β5 active sites, the crystal structure of the yeast proteasome when inhibited by the tripeptide aldehyde Ac-Leu-Leu-Nle-H (1) revealed binding of this inhibitor to all active sites via hemiacetal formation with the Thr1 hydroxyl function (13). This observation suggested a simple construct to crosslink the Thr-Oγ of adjacent β1 and β2 subunits, which are 28 Å apart (Fig. 1). For this purpose we have extended the tripeptide aldehyde H-Leu-Leu-Nle-H to the octapeptide 3 containing an N-terminal glyoxylic acid residue. The inhibitory potency of 3 was significantly increased for the chymotrypsin-like activity, i.e., at the β5 active site, but no inhibition was observed for the PGPH (β1) and trypsin-like (β2) activities (Table 2), although its design was directed to a bivalent interaction with the active site pairs β1-β2 and β1′-β2′, respectively. These data suggest an improved monovalent inhibition of the β5 active site. In fact, x-ray analysis of the proteasome/3 complex clearly reveals binding of the C terminus of compound 3 to all six active sites in absence of competing substrates in a mode similar to that of the tripeptide aldehyde 1, but with defined interactions of the peptide in extended conformation from the S1 to the S6 subsites (Fig. 2). The difference electron density map, however, did not allow identification of the N terminus.
Table 2.
Inhibition of 20S proteasome by intra-ring bivalent inhibitors, IC50, μM
| Inhibitor | PGPH | Trypsin-like | Chymotrypsinlike |
|---|---|---|---|
| Ac-LLnL-H (1) | >100 | >100 | 2.1 |
| OHC-CO-LGPGGLLnL-H (3) | >100 | >100 | 0.08 |
| Lev-KKGEVSLE-H (4) | 102 | >100 | >100 |
| Saa-KKGEVSLE-H (5) | 82 | >100 | >100 |
| Saa-AAEVSLE-H (6) | 146 | >100 | >100 |
Lev, levulinic acid; Saa, 4-oxobutyric acid.
Figure 2.

Part of the x-ray structure of the yeast 20S proteasome/OHC-CO-LGPGGLLnL-H (3) adduct. Protein subunits are marked with different colors: blue for β1, red for β2, yellow for β7, and the inhibitor is shown in white (drawn with main; ref. 27).
The x-ray structure of a β1 Thr1-Ala mutant revealed partial processing of the propeptide at Arg−10/Leu−9 by the adjacent β2 active site (29). This propeptide then was used as a possible lead structure to design bivalent inhibitors carrying C terminally a glutamic acid aldehyde for binding from the S subsites to β1 and N terminally the levulinic or 4-oxobutyric acid as anchor for the β2 active site, if an approach to this β2 active site from the S′ subsites would allow for hemiacetal formation with the N-terminal Thr (compounds 4-6). A comparison of the IC50 values of these double-headed inhibitors (Table 2) for the PGPH (β1 or β1′) and trypsin-like activity of the proteasome (β2 or β2′) shows a weak μmolar inhibition of the PGPH activity, but no inhibition of the trypsin-like activity. These results exclude bivalent binding, possibly as a consequence of the difficult access to the β2 Thr residue from the S′ subsites. In the crystal structure of the mutant, the Lys-Lys portion of the propeptide is bent into a turn conformation by a salt bridge interaction with the Glu−5 residue. Therefore compound 6 was prepared in which the lysine residues were replaced by alanine and the peptide itself was shortened by one residue as suggested from modeling experiments. However, even with this bis-aldehyde peptide no improved inhibition of the proteasome could be achieved (Table 2).
Design of Inter-Ring Homobivalent Inhibitors.
To allow for an access of two inhibitory head groups from the S subsites to two active sites of the proteasome, the crystal structure of Ac-Leu-Leu-Nle-H bound to β5 and β5′ was used for the design of a spacer that has to span distances of 50 Å or more. As the spacer should mimic as much as possible the unstructured polypeptide chain of an unfolded protein, peptides of appropriate length should be best suited for such a purpose. However, in full agreement with previous studies (8–10, 30) peptides of this size like gastrin (17 mer) or secretin (27 mer) were found to be rapidly degraded by the yeast proteasome (unpublished data) and are therefore unsuitable. The spacer also should be hydrophilic to avoid formation of compact hydrophobic cores that would prevent the molecule from entering the proteasomal chamber through narrow entrance parts. Consequently, PEG was chosen to mimic random coiled polypeptide chains, as this linear polymer is known to be highly solvated and unstructured (31).
To satisfy the required size of about 50 Å, a PEG with a statistical distribution of 19–25 monomers and with terminal amino groups capped with succinic acid was selected. To analyze the effects of the large PEG moiety on the more or less impeded entrance of inhibitors into the inner cavity of the proteasome (32), it was linked to the N terminus of H-Leu-Leu-Nle-H as shown in Scheme S2, to produce the monovalent inhibitor 7. A comparison of the IC50 values of the pegylated and acetylated tripeptide aldehyde shows little difference that might be caused by the PEG moiety.
This observation led us to synthesize the homo-bivalent inhibitors 8 and 10 containing the tripeptide aldehydes Leu-Leu-Nle-H and Arg-Val-Arg-H as head groups for the β5-β5′ and β2-β2′ active site pairs, respectively (Scheme S2). For comparative purposes Arg-Val-Arg-H was pegylated at its N terminus to produce the monovalent inhibitor 9. The inhibitory potencies of the monovalent and bivalent inhibitors are reported in Table 3 as IC50 values. With both bivalent inhibitors highly selective inhibition was achieved with an increase in potency by 2 orders of magnitude.
Table 3.
Inhibition of 20S proteasome by mono- and bivalent inhibitors (IC50, μM)
| Inhibitor | PGPH | Trypsin-like | Chymotrypsinlike |
|---|---|---|---|
| Ac-LLnL-H (1) | >100 | >100 | 2.1 |
| CO-LLnL-H | |||
| (PEG)x(7) | >100 | >100 | 1.8 |
| COOH | |||
| CO-LLnL-H | |||
| (PEG)x(8) | >100 | >100 | 0.017 |
| CO-LLnL-H | |||
| Ac-RVR-H (2) | >100 | 6.4 | >100 |
| CO-RVR-H | |||
| (PEG)x(9) | >100 | 8.2 | >100 |
| COOH | |||
| CO-RVR-H | |||
| (PEG)x(10) | >100 | 0.071 | >100 |
| CO-RVR-H | |||
| CO-LLnL-H | |||
| (PEG)x(11) | >100 | 0.097 | 0.031 |
| CO-RVR-H |
Design of Heterobivalent Inhibitors.
The geometric arrangement of the three different β1, β2, and β5 active sites in the two inner β-rings allows with homobivalent inhibitors to knock out, with one molecule, a specific activity. In the case of heterobivalent inhibitors, one molecule can neutralize only one active site of the existing pair. Two molecules thus are required for complete inhibition, however, with the advantage of inhibiting two activities concomitantly. To examine this possibility, the heterobivalent inhibitor 11, containing as head groups the tripeptide aldehydes Leu-Leu-Nle-H and Arg-Val-Arg-H, was synthesized following the route outlined in Scheme S2. As expected, both the trypsin- and chymotrypsin-like activities are inhibited (Table 3). The inhibitory potencies against the trypsin- and chymotrypsin-like activities are similar to those of the homobivalent inhibitors if the stoichiometry of this type of inhibitor is taken into account.
Crystal Structures of Proteasome/Inhibitor Complexes.
The mode of binding of the tripeptide aldehyde Ac-Leu-Leu-Nle-H to all six active sites of the S. cerevisiae proteasome has been reported (13). Although inhibitor 3 was designed to crosslink β1 and β2, at the mmolar concentrations used in the soaking experiments and in the absence of competing substrates, it binds as the tripeptide aldehyde to all six active sites by its C terminus via hemiacetal formation with the Thr1 Oγ (Fig. 2). Besides the crystal structure of the propeptide/β1 Thr1-Ala mutant, this complex with the inhibitor 3 shows the binding mode of a peptide aldehyde from the S1 to the S6 subsites. Residues P1-P3 are arranged as in the Ac-Leu-Leu-Nle-H complex (13). The P4 CO is hydrogen-bonded to the imidazole NH of His-116 (β2), whereas for the P5 and P6 residues a defined network of hydrogen bonds is not seen. The contribution of the P4 residue to the binding affinity agrees with previous studies on proteasome inhibitors (33). These additional interactions are most probably responsible for the significantly enhanced binding of the peptide aldehyde 3 compared with the tripeptide aldehyde 1. The electron density map does not allow assignments beyond P6.
As expected from the results obtained with the proteasome/Ac-Leu-Leu-Nle-H complex (13), even the homobivalent inhibitor 8 occupies all six active sites with the inhibitory head groups binding identically to the parent tripeptide aldehyde, as is clearly seen at a resolution of the crystal structure analysis at 2.3 Å (Fig. 3). Because the inhibition data support bivalent binding, occupation of all six active sites in absence of competing substrates is expected to occur both in the intra- and inter-ring mode, because the spacer length is sufficient for all possible bridgings within maximal distances of approximately 65 Å. Moreover, the size of the spacer allows for binding of the inhibitory head groups from the S subsites in all six subunits. The electron density map does not reveal a conformationally restricted PEG moiety in any part of the cavity.
Figure 3.

Part of the electron density map with 2Fo-Fc coefficients after 2-fold averaging of the yeast 20S proteasome/(PEG)19–25-[NH-CO-(CH2)2-CO-Leu-Leu-Nle-H]2 (8) complex. The electron density map was calculated with phases of the free enzyme structure and contoured around the inhibitor molecule at 2ó cutoff. The carbon atoms are marked with different colors: green for β5, red for β6, and white for the inhibitor.
In the case of the homobivalent inhibitor 10 of the trypsin-like activity, the x-ray analysis of the complex revealed the presence of the inhibitory head group Arg-Val-Arg-H only in the two β2 and β2′ active sites, despite the absence of competing substrate (data not shown). This result indicates that affinity of a basic P1 residue is highly restricted to the S1 subsite of the trypsin-like activity. Again the PEG spacer is not detectable and probably fluctuates freely in the cavity of the proteolytic chamber.
DISCUSSION
For the difficult problem of selective inhibition of proteasomes, nature teaches the use of nonpeptidic compounds such as the Streptomyces metabolite lactacystin (34). This natural product inhibits the chymotrypsin-like activity of the multicatalytic protease irreversibly by selective modification the active-site Thr1 of β5 via its active clasto-lactacystin-β-lactone intermediate. This β-lactone was found to inhibit only partially the PGPH and trypsin-like activities (35, 36), but surprisingly shows activity against cathepsin A (37). More selective irreversible inhibitors of the chymotrypsin-like activity were obtained with peptidyl-vinylsulfones (33, 38), although their proteasome specificity is not yet sufficiently documented. In this context the reversible boronic acid-based inhibitors seem to be promising (39).
Another approach to developing highly potent and selective inhibitors of eukaryotic proteasomes is to exploit the unique topography of the six active sites of this protease for the design of bi- or multivalent inhibitors. The results of the present study demonstrate that a polymeric spacer of appropriate length can be used to link two monovalent binding head groups to yield homo- or heterobivalent inhibitors of remarkably enhanced binding affinity. Interestingly, this improved inhibition already was achieved by using a heterogeneous polymeric spacer with a statistical length distribution from 19 to 25 monomers to bridge various distances between different active sites (Fig. 1). Even better results may be expected with homogeneous spacers of defined optimal length for the various interactive site distances. Furthermore, this general principle of bivalency is not at all restricted to the use of peptide aldehydes as binding head groups, but could, in combination with more potent and selective monovalent inhibitors, result in a new generation of highly specific proteasome inhibitors.
The question whether such PEG-linked bivalent inhibitors retain membrane permeability to extents useful for intracellular tools has not yet been specifically addressed. However, PEG is known to be nontoxic, to lower immunogenicity, to lower clearance rates, to increase water solubility, and more importantly, to move molecules across membranes (31).
Acknowledgments
The help of G.B. Bourenkow and H. Bartunik (Max-Planck Arbeitsgruppen, Hamburg) with data collection is gratefully acknowledged. The study was supported by the Deutsche Forschungsgemeinschaft (SFB 469; Grants A1 and A2).
ABBREVIATIONS
- PEG
polyoxyethylene
- AMC
7-amido-4-methyl-coumarin
- PGPH
post-glutamyl-peptide hydrolytic
- DIEA
diisopropylethylamine
- DMF
dimethylformamide
- TBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate
- Sc
semicarbazone
References
- 1.Groettrup M, Schmidtke G. DDT. 1999;4:63–71. doi: 10.1016/s1359-6446(98)01292-6. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg A L, Stein R, Adams J. Chem Biol. 1995;2:503–508. doi: 10.1016/1074-5521(95)90182-5. [DOI] [PubMed] [Google Scholar]
- 3.Hochstrasser M. Curr Opin Cell Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 4.Coux O, Tanaka K, Goldberg A L. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 5.Stock D, Nederlof P M, Seemüller E, Baumeister W, Huber R, Löwe J. Curr Opin Biotechnol. 1996;7:376–385. doi: 10.1016/s0958-1669(96)80111-x. [DOI] [PubMed] [Google Scholar]
- 6.Chen P, Hochstrasser M. EMBO J. 1995;14:2620–2630. doi: 10.1002/j.1460-2075.1995.tb07260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groettrup M, Soza A, Kuckelkorn U, Kloetzel P M. Immunol Today. 1996;17:429–435. doi: 10.1016/0167-5699(96)10051-7. [DOI] [PubMed] [Google Scholar]
- 8.Heinemeyer W, Tröndle N, Albrecht G, Wolf D H. Biochemistry. 1994;33:12229–12237. doi: 10.1021/bi00206a028. [DOI] [PubMed] [Google Scholar]
- 9.Ehring B, Meyer T H, Eckerskorn C, Lottspeich F, Tampé R. Eur J Biochem. 1996;235:404–415. doi: 10.1111/j.1432-1033.1996.00404.x. [DOI] [PubMed] [Google Scholar]
- 10.Dick L R, Aldrich C, Jameson S C, Moomaw C R, Pramanik B C, Doyle C K, DeMartino G N, Bevan M J, Forman J M, Slaughter C A. Immunology. 1994;152:3884–3894. [PMC free article] [PubMed] [Google Scholar]
- 11.Wenzel T, Eckerskorn C, Lottspeich F, Baumeister W. FEBS Lett. 1994;349:205–209. doi: 10.1016/0014-5793(94)00665-2. [DOI] [PubMed] [Google Scholar]
- 12.Kuckelkorn U, Frentzel S, Kraft R, Kostka S, Groettrup M, Kloetzel P-M. Eur J Immunol. 1995;25:2605–2611. doi: 10.1002/eji.1830250930. [DOI] [PubMed] [Google Scholar]
- 13.Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik H D, Huber R. Nature (London) 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 14.Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf D H. J Biol Chem. 1997;272:25200–25209. doi: 10.1074/jbc.272.40.25200. [DOI] [PubMed] [Google Scholar]
- 15.Nussbaum A K, Dick T P, Keilholz W, Schirle M, Stefanovic S, Dietz K, Heinemeyer W, Groll M, Wolf D H, Huber R, et al. Proc Natl Acad Sci USA. 1998;95:12504–12509. doi: 10.1073/pnas.95.21.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arendt C S, Hochstrasser M. Proc Natl Acad Sci USA. 1997;94:7156–7161. doi: 10.1073/pnas.94.14.7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbosa Pereira P J, Bergner A, Macedo-Ribeiro S, Huber R, Matschiner G, Fritz H, Sommerhoff C P, Bode W. Nature (London) 1998;392:306–311. doi: 10.1038/32703. [DOI] [PubMed] [Google Scholar]
- 18.Page M I, Jencks W P. Proc Natl Acad Sci USA. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spike C G, Parry R W. J Am Chem Soc. 1953;75:2726–2729. [Google Scholar]
- 20.Crothers D M, Metzger H. Immunochemistry. 1972;9:341–357. doi: 10.1016/0019-2791(72)90097-3. [DOI] [PubMed] [Google Scholar]
- 21.Mammen M, Choi S-K, Whitesides G M. Angew Chem Int Ed Engl. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 22.Loidl G, Groll M, Musiol H-J, Ditzel L, Huber R, Moroder L. Chem Biol. 1999;6:197–204. doi: 10.1016/S1074-5521(99)80036-2. [DOI] [PubMed] [Google Scholar]
- 23.Schaschke N, Musiol H-J, Assfalg-Machleidt I, Machleidt W, Rudolph-Böhner S, Moroder L. FEBS Lett. 1996;391:297–301. doi: 10.1016/0014-5793(96)00752-1. [DOI] [PubMed] [Google Scholar]
- 24.Coste J, Le-Nguyen D, Castro B. Tetrahedron Lett. 1990;31:205–208. [Google Scholar]
- 25.Carpino L A. J Am Chem Soc. 1993;115:4397–4398. [Google Scholar]
- 26.Brunger A. x-plor: A System for X-Ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1992. , Version 3.1. [Google Scholar]
- 27.Turk D. Ph.D. thesis. Munich: Technical University; 1992. [Google Scholar]
- 28.Jones T A. J Appl Crystallogr. 1978;11:268–272. [Google Scholar]
- 29.Ditzel L, Huber R, Mann K, Heinemeyer W, Wolf D H, Groll M. J Mol Biol. 1998;279:1187–1191. doi: 10.1006/jmbi.1998.1818. [DOI] [PubMed] [Google Scholar]
- 30.Kisselev A F, Akopian T N, Woo K M, Goldberg A L. J Biol Chem. 1999;274:3363–3371. doi: 10.1074/jbc.274.6.3363. [DOI] [PubMed] [Google Scholar]
- 31.Harris J M, editor. Poly(ethylene glycol) Chemistry. New York: Plenum; 1992. [Google Scholar]
- 32.Escherich A, Ditzel L, Musiol H-J, Groll M, Huber R, Moroder L. Biol Chem. 1997;378:893–898. doi: 10.1515/bchm.1997.378.8.893. [DOI] [PubMed] [Google Scholar]
- 33.Bogyo M, Shin S, McMaster J S, Ploegh H. Chem Biol. 1998;5:307–320. doi: 10.1016/s1074-5521(98)90169-7. [DOI] [PubMed] [Google Scholar]
- 34.Fenteany G, Standaert R F, Lane W S, Choi S, Corey E J, Schreiber S L. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 35.Dick L R, Cruikshank A A, Destree A A, Grenier L, McCormack T A, Melandri F D, Nunes S L, Palombella V J, Parent L A, Plamondon L, et al. J Biol Chem. 1997;272:182–188. doi: 10.1074/jbc.272.1.182. [DOI] [PubMed] [Google Scholar]
- 36.Corey E J, Reichard G A, Kania R. Tetrahedron Lett. 1993;34:6977–6980. [Google Scholar]
- 37.Ostrowska H, Omura S, Wojcik C, Worowski K. Biochem Biophys Res Commun. 1997;234:729–732. doi: 10.1006/bbrc.1997.6434. [DOI] [PubMed] [Google Scholar]
- 38.Bogyo M, McMaster J S, Gaczynska M, Tortorella D, Goldberg A L, Ploegh H. Proc Natl Acad Sci USA. 1997;90:6629–6634. doi: 10.1073/pnas.94.13.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams J, Behnke M, Chen S, Cruickshank A A, Dick L R, Grenier L, Klunder J M, Ma Y-T, Plamondon L, Stein R L. Bioorg Med Chem Lett. 1998;8:333–338. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]