

Abstract
Engineered Cys2His2 zinc finger proteins (ZFPs) can mediate regulation of endogenous gene expression in mammalian cells. Ideally, all zinc fingers in an engineered multifinger protein should be optimized concurrently because cooperative and context-dependent contacts can affect DNA recognition. However, the simultaneous selection of key contacts in even three fingers from fully randomized libraries would require the consideration of >1024 possible combinations. To address this challenge, we have developed a novel strategy that utilizes directed domain shuffling and rapid cell-based selections. Unlike previously described methods, our strategy is amenable to scale-up and does not sacrifice combinatorial diversity. Using this approach, we have successfully isolated multifinger proteins with improved in vitro and in vivo function. Our results demonstrate that both DNA binding affinity and specificity are important for cellular function and also provide a general approach for optimizing multidomain proteins.
The Cys2His2 zinc finger domain has emerged as the preferred scaffold for creating customized DNA-binding domains (1–4). A single zinc finger domain, composed of a ββα motif, typically interacts with three to four base pairs of DNA by using key residues in its α-helix (or “recognition helix”). Selection methodologies (e.g., phage display) have been used to identify fingers with altered DNA binding specificities from libraries in which six potential contact residues (within or near the recognition helix) have been randomized (1, 2, 5–8). Synthetic multifinger proteins capable of binding to more extended DNA sequences have been created by a “modular assembly” approach in which preoptimized (9–13), predesigned (14, 15), or naturally occurring finger units (16) are linked into tandem arrays. Artificial transcription factors based on these synthetic zinc finger proteins (ZFPs) can be used to achieve targeted regulation of biologically significant endogenous genes [e.g., vascular endothelial growth factor (VEGF)-A, erbB2] both in tissue culture (9, 14, 16–18) and animal model systems (19).
In both natural and designed multifinger ZFPs, however, individual fingers do not always function as completely modular units (20–23). This interdependence implies that individual fingers optimized in one context may not exhibit precisely the same binding specificity when used in another context (i.e., with different neighboring fingers or different neighboring DNA sites). Thus, to create a multifinger protein that binds optimally to its target DNA sequence, it would be best if all fingers in a protein could be randomized and selected simultaneously. However, randomizing a single finger creates a library composed of ≈2 × 108 potential candidates (using 24 codons at six amino acid positions = 246) (24), and therefore the simultaneous selection of all fingers in a three-finger protein would require a starting library consisting of (2 × 108)3 or ≈8 × 1024 randomized proteins. Some of the previously described strategies have recognized this issue and have tried to balance considerations of interfinger cooperativity with the need to keep combinatorial issues manageable. The sequential optimization strategy of Griesman and Pabo (25) used fingers that were serially selected; the “bi-partite selection” method of Choo and colleagues (26) used preselected “halves” of a three-finger protein that were then assembled together.
In this report we identify optimized multifinger ZFPs by using a novel strategy that accounts for potential interfinger context effects on DNA binding without sacrificing combinatorial diversity. Our method combines a directed domain shuffling strategy with the use of a cell-based selection methodology to rapidly produce ZFPs with superior in vitro and in vivo DNA binding properties. In addition to providing a general approach for engineering a multidomain protein, our results demonstrate that both high affinity and high specificity are required for optimal DNA binding function in a cellular environment.
Materials and Methods
Media. The histidine-deficient medium used for selections has been described (24). Where required, the following antibiotics were added: carbenicillin (50 μg/ml in liquid medium, 100 μg/ml in solid medium), chloramphenicol (30 μg/ml), and kanamycin (30 μg/ml).
Bacterial Two-Hybrid Plasmids and Strains. The αGal4 protein expression plasmid has been described (24). ZFPs were expressed from vectors based on the pBR-GP-Z123 plasmid (24). In these plasmids, the inducible lacUV5 promoter directs the expression of a three-finger ZFP fused to a fragment of the yeast Gal11p protein. The amino acid sequences of the “original” BCR-ABL, erbB2, and HIV ZFPs were obtained from published reports (10, 26, 27). Reporter and selection strains were constructed as described (24). These strains contain a single copy F′-episome with the target DNA-binding site or subsite positioned immediately upstream of a weak lac promoter that controls the transcription of the selectable HIS3 and aadA genes (in selection strains) or the lacZ reporter gene (in reporter strains).
Construction of Master Randomized Libraries. We constructed three master libraries, each based on a synthetic “framework” ZFP (the original BCR-ABL three-finger protein) (27). In each library, recognition helix residues –1, 1, 2, 3, 5, and 6 from a single finger were randomized by cassette mutagenesis. Randomization used a previously described strategy that utilizes 24 codons encoding 16 possible amino acids (excluding the aromatics and cysteine) (24, 25). Each of our libraries consisted of at least 5 × 108 independently derived members.
Low-Stringency Bacterial Two-Hybrid Selections. A master randomized finger library was introduced into an appropriately engineered selection strain bearing the target subsite of interest and transformed cells were plated on histidine-deficient medium containing 50 μM isopropyl β-d-thiogalactoside (IPTG), 10 mM 3-aminotriazole (3-AT), and 20 μg/ml streptomycin and/or 50 μM IPTG, 20 mM 3-AT and 20 μg/ml streptomycin. ZFP-encoding plasmids from surviving colonies were harvested and used to construct the shuffled three-finger libraries.
Construction of Shuffled Three-Finger Libraries. In vitro recombination of finger pools identified in the low-stringency selections was performed by using PCR-mediated random fusion of DNA fragments encoding individual finger units in a way that preserved finger position. For each library, pools of finger sequences isolated in the low-stringency selections were amplified to create partially overlapping cassettes encoding fingers at each position. These cassettes were then randomly recombined together and reamplified by using end primers to create fragments encoding shuffled three-finger proteins (see Fig. 1). Each library we created by using this method contained >108 independently derived members and is likely composed of at least 10,000 distinct ZFP sequences (based on estimates of the likely number of unique sequences in the finger pools used to construct these “shuffled” libraries).
Fig. 1.

Overview of multifinger ZFP optimization strategy. Each master randomized library is derived from a synthetic framework ZFP with known DNA binding specificity (light blue ovals). Randomized fingers are indicated by rainbow-colored ovals. Each target DNA site is divided into its constituent “subsites” followed by substitution within the full binding site of the framework ZFP. Note that members of a randomized library, when matched with the appropriate binding site, can use their constant “anchor” fingers (from the framework ZFP) to position the randomized finger over the target subsite of interest. In stage 1, parallel low-stringency selections are performed by using master randomized libraries to find good fingers for each desired subsite. Ovals of various shades of the same color represent pools of fingers whose members can occupy their associated target DNA subsite (colored rectangles) to varying degrees. In stage 2, finger pools isolated in stage 1 are randomly recombined together to create “shuffled” libraries, and a high-stringency selection is then performed to identify optimal combinations of fingers that bind to the full intact target DNA site.
High-Stringency Bacterial Two-Hybrid Selections. A shuffled ZFP library was introduced into the appropriate bacterial two-hybrid selection strain bearing the full target sequence of interest and these transformants were plated on a series of histidine-deficient plates containing various concentrations of IPTG, 3-AT, and streptomycin. Candidates chosen for sequencing and subsequent analysis were picked from the most stringent selection conditions that permitted colony formation after 2–5 days of growth at 37°C. These conditions were: 0 mM IPTG, 40 mM 3-AT, and 60 μg/ml streptomycin and 0 mM IPTG, 50 mM 3-AT, and 80 μg/ml streptomycin for both the BCR-ABL and HIV selections (six candidates from each set of conditions), and 50 mM IPTG, 25 mM 3-AT, 40 μg/ml streptomycin and 50 mM IPTG, 40 mM 3-AT, 60 μg/ml streptomycin for the erbB2 selections (six candidates from each set of conditions).
Purification of Zinc Finger Peptides. Maltose binding protein–zinc finger protein fusions (MBP-ZFP) were expressed from a T7 promoter (plasmid pEXP1-DEST, Invitrogen) in the Expressway-coupled in vitro transcription/translation system (Invitrogen). Proteins were expressed according to the manufacturer's instructions at 37°C for 3.5 h with the addition of 500 μM ZnCl2 and the omission of the postsynthesis RNase A treatment. Two to three synthesis reactions for each protein were pooled, and the MBP-ZFPs were affinity purified by using amylose resin (New England Biolabs). Purified peptides were aliquoted and frozen for storage at –80°C. Additional details of purification are available upon request.
Determination of Kd and 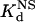 Values. Pairs of DNA oligonucleotides 25 bp in length were designed to contain 5′ TTTT overhangs and a 10-bp BCR-ABL, erbB2, HIV, or Zif268 target binding site. Oligonucleotides were annealed and radiolabeled with [α-32P]dATP. The primary strands of these oligonucleotide pairs were as follows (all sequences written 5′ to 3′ with target sites shown in bold): TTTTCGACACGCAGAAGCCCATTAC (BCR-A BL), T T T TCGACA AGCCGCAGTGGAT TAC (erbB2), TTTTCGACACGATGCTGCATATTAC (HIV), and TTTTGACGGTGCGTGGGCGGTTCAC (Zif268). Electrophoretic mobility-shift assays were performed as described (25) except that (i) binding buffer contained nonacetylated BSA (100 μg/ml), (ii) 0.5 pM (for Zif268 and HIV) or 1 pM (for all other proteins) of the labeled DNA site was used for each binding reaction, and (iii) protein–DNA mixtures were incubated for 1 or 4 h at room temperature. Results for both incubation times were comparable, indicating that the binding reactions had reached equilibrium after 1 h, and, thus, we averaged the results of all of these experiments. Reactions were subjected to gel electrophoresis on Criterion 4–20% native TBE polyacrylamide gels (Bio-Rad). Signal quantitation was performed by using a Bio-Rad Molecular Imager Fx system and quantity one imaging software (Bio-Rad). The percent of DNA bound (θ) was plotted against the concentration of active protein [P] in each binding reaction, and sigmaplot8 (Sigma) nonlinear regression software was used to fit the curve according to equation 1 of Elrod-Erickson and Pabo (28) and to calculate values for the Kd of each protein. Binding site competition experiments to determine
Values. Pairs of DNA oligonucleotides 25 bp in length were designed to contain 5′ TTTT overhangs and a 10-bp BCR-ABL, erbB2, HIV, or Zif268 target binding site. Oligonucleotides were annealed and radiolabeled with [α-32P]dATP. The primary strands of these oligonucleotide pairs were as follows (all sequences written 5′ to 3′ with target sites shown in bold): TTTTCGACACGCAGAAGCCCATTAC (BCR-A BL), T T T TCGACA AGCCGCAGTGGAT TAC (erbB2), TTTTCGACACGATGCTGCATATTAC (HIV), and TTTTGACGGTGCGTGGGCGGTTCAC (Zif268). Electrophoretic mobility-shift assays were performed as described (25) except that (i) binding buffer contained nonacetylated BSA (100 μg/ml), (ii) 0.5 pM (for Zif268 and HIV) or 1 pM (for all other proteins) of the labeled DNA site was used for each binding reaction, and (iii) protein–DNA mixtures were incubated for 1 or 4 h at room temperature. Results for both incubation times were comparable, indicating that the binding reactions had reached equilibrium after 1 h, and, thus, we averaged the results of all of these experiments. Reactions were subjected to gel electrophoresis on Criterion 4–20% native TBE polyacrylamide gels (Bio-Rad). Signal quantitation was performed by using a Bio-Rad Molecular Imager Fx system and quantity one imaging software (Bio-Rad). The percent of DNA bound (θ) was plotted against the concentration of active protein [P] in each binding reaction, and sigmaplot8 (Sigma) nonlinear regression software was used to fit the curve according to equation 1 of Elrod-Erickson and Pabo (28) and to calculate values for the Kd of each protein. Binding site competition experiments to determine 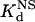 values were performed as described (25) except that 0.5 or 1 pM of radiolabeled target site was used. For all Kd and
values were performed as described (25) except that 0.5 or 1 pM of radiolabeled target site was used. For all Kd and 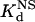 determinations, the concentration of active protein [P] was determined for each experiment by titrating dilutions of fusion ZFP against a fixed excess amount of unlabeled target site (12.5 nM) and a small amount of labeled target site (1 pM). Reactions were incubated and subjected to gel electrophoresis. Active protein concentrations ([P]stock) were determined by plotting θ vs. 1/dilution factor according to the equation:
determinations, the concentration of active protein [P] was determined for each experiment by titrating dilutions of fusion ZFP against a fixed excess amount of unlabeled target site (12.5 nM) and a small amount of labeled target site (1 pM). Reactions were incubated and subjected to gel electrophoresis. Active protein concentrations ([P]stock) were determined by plotting θ vs. 1/dilution factor according to the equation:
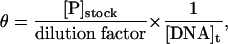 |
[1] |
where [DNA]t is the total concentration of DNA.
Bacterial Cell-Based Reporter Assays. β-Galactosidase assays were performed as described (24).
Mammalian Cell-Based Assays. The original and optimized erbB2 zinc finger proteins were expressed from a tetracycline-inducible cytomegalovirus promoter in plasmid pcDNA5 (Invitrogen). All ZFP proteins were expressed as fusion proteins containing a nuclear localization signal from simian virus 40 (SV40) large T antigen, the human NF-κB p65 subunit activation domain, and a FLAG tag peptide as described (14). Each plasmid encoding a zinc finger fusion protein was transfected into Flp-In TREx HEK 293 cells (Invitrogen) that express tetracycline repressor and stable site-specific integrants were selected and identified according to the manufacturer's instructions (Invitrogen Flp-In TREx system). To perform quantitative RT-PCR, total RNA was isolated by using the RNeasy method with in-column DNase treatment (Qiagen, Valencia, CA) from stable integrants grown for 30 h in the absence and presence of 1 μg/ml doxycycline. Reverse transcription was performed by using Omniscript RT (Qiagen) per the manufacturer's protocol. Quantitative PCR was conducted by using TaqMan universal master mix on an ABI 7700HT machine (Applied Biosystems). Analysis of results was performed by using sds 2.0 software (Applied Biosystems).
Results
Our strategy for optimizing multifinger proteins consists of two stages (Fig. 1). In stage 1, we start with parallel low-stringency selections from premade, master libraries to isolate pools of candidates for each finger position in the desired protein. In stage 2, we then proceed with position-sensitive assembly of finger pools (from stage 1) to create randomly recombined or shuffled libraries of multifinger proteins and then use final high-stringency selections to isolate the optimized ZFPs.
We performed all selection steps by using a previously described bacterial cell-based two-hybrid system adapted for selecting zinc fingers with altered DNA binding capabilities (24). In this system, engineered Escherichia coli cells harbor a specific target DNA site or subsite that is placed adjacent to a test promoter on a single copy episome (Fig. 2). Occupancy of the specific target DNA site by a ZFP (expressed from a separate plasmid in the same cell) triggers transcriptional activation of two selectable marker genes (the yeast HIS3 and the bacterial aadA genes) or a readily quantified reporter gene (the E. coli lacZ gene) expressed from the test promoter. This activation results from recruitment of RNA polymerase (RNAP) mediated by pieces of the interacting yeast Gal11P and Gal4 proteins that are fused to the ZFP and the RNAP α-subunit, respectively (Fig. 2). With this system, E. coli cells expressing a ZFP that efficiently occupies a target DNA sequence of interest can be identified by their ability to grow on selective medium. This system has been used to identify individual zinc finger variants from large randomized libraries >108 in size (24).
Fig. 2.

Bacterial two-hybrid system for selecting ZFPs. Binding of a multifinger ZFP to its cognate DNA binding site triggers transcriptional activation of adjacent reporter gene(s) via recruitment of RNA polymerase (RNAP) to a weak test promoter. Note that this promoter does not function effectively if the ZFP fusion protein fails to occupy the target DNA site.
To test our optimization strategy, we chose three different 10-bp target DNA sequences: one from a BCR-ABL translocation (27), one from the erbB2 gene (10), and one from the HIV promoter (26) (shown in Fig. 3A and hereafter referred to as the BCR-ABL, erbB2, and HIV target sequences). These sequences were attractive targets because multifinger proteins that bind to each of these DNA sequences (Fig. 3B) had been previously constructed by using other strategies (10, 26, 27).
Fig. 3.

(A) BCR-ABL, erbB2, and HIV target DNA sites used for selecting optimized multifinger proteins. (B) Recognition helix sequences of original BCR-ABL, erbB2, and HIV three-finger proteins. Numbering of residues is with respect to the start of the recognition helix. (C) Recognition helix sequences from randomly chosen members of the shuffled BCR-ABL, erbB2, and HIV ZFP libraries. “f.s.” indicates a frameshift mutation introduced during PCR amplification. (D) Recognition helix sequences of optimized ZFPs selected from the shuffled BCR-ABL, erbB2, and HIV libraries. Amino acids highlighted in color appear in at least 75% of the sequenced candidates at that recognition helix position. Boxed sequences indicate candidates chosen for additional characterization.
Optimization Stage 1: Low-Stringency Parallel Selections of Individual Fingers. In this stage, three parallel selections (one for each of the three fingers in the final protein) were performed for each of the target DNA sites. Each of the fingers in a three-finger protein binds a 3- to 4-bp subsite, and these together comprise the 10-bp target DNA sequence (see Fig. 1). The N-terminal finger (finger 1) binds the 3′ subsite; the middle finger (finger 2) binds the middle subsite; and the C-terminal finger (finger 3) binds the 5′ subsite. (This direction along the DNA is established with respect to the strand to which the majority of contacts are made.) We therefore constructed three master libraries, all based on the same synthetic three-finger framework ZFP (see Materials and Methods), in which potential DNA contacting residues in one of the fingers have been randomized (we designate these master libraries LibF1, LibF2, and LibF3 according to the finger randomized; see Fig. 1 and Materials and Methods). To identify fingers that bind to a given subsite, members of a randomized zinc finger library were introduced into a bacterial two-hybrid “selection strain” under conditions in which binding of the randomized finger to the target subsite of interest triggers transcriptional activation of the two selectable marker genes. These low-stringency selections were performed in parallel for each of the three subsites (Fig. 1) in each of the different nine base pair target sequences. [Each of the master libraries contains two fingers that have not been randomized, and these fingers serve to anchor the remaining randomized finger over the target subsite (Fig. 1).]
The recognition helix sequences of the fingers selected in these experiments typically showed only weak or partial consensus sequences. In some cases, multiple consensus sequences were obtained (data not shown and see below). These observations are consistent with the idea that these initial selections yield a large number of different fingers that bind to the target subsite with various affinities. Our intention in these initial selections was to identify all possible fingers that bind to a target subsite and to carry as many of these as possible forward to the next stage.
Optimization Stage 2: High-Stringency Selections of Optimized ZFPs from Shuffled Multifinger Protein Libraries. In the second stage of optimization, we assembled libraries of three-finger proteins consisting of shuffled combinations of the finger pools identified in the initial selections. These libraries were constructed by using an in vitro recombination protocol based on PCR. Our protocol ensures that fingers selected at a given position remain in the same position in the reassembled protein (e.g., fingers selected at the finger 1 position all occupy the finger 1 position in proteins in the recombined library). Each library was constructed from >200 fingers selected at each of the three finger positions (see Materials and Methods for additional details).
We determined the DNA sequence of 12 random members from each of our shuffled libraries to verify the proper assembly and “random” distribution of finger sequences. The results (shown in Fig. 3C) illustrate the weak, partial, and/or multiple consensus sequences obtained for each finger position in the low-stringency selections from stage 1.
To perform the final stringent selections, we introduced each shuffled library into a matched bacterial two-hybrid selection strain in which binding of a ZFP to the final full-length target DNA sequence triggers transcriptional activation of the selectable marker genes. Because nearly every member of the library is predicted to have at least some capability to bind the full-length target sequence, we performed selections under the most stringent conditions possible to isolate proteins that best occupy the target binding sequence (and therefore best activate transcription) in the selection strain (see Materials and Methods for details).
For each target DNA sequence, the recognition helix sequences of 12 candidates that survived stringent selection were determined (shown in Fig. 3D). For nearly all finger positions, the stringent selections appear to have identified only a small number of different recognition helix sequences that closely resemble one another. In addition, we note that none of the finger recognition helix sequences in our optimized proteins exactly matches their corresponding counterpart in the original proteins (compare with the recognition helix sequences of the previously described original BCR-ABL, erbB2, and HIV proteins shown in Fig. 3B). In some cases there appear to be few or no similarities.
Characterization of Optimized Multifinger Proteins. To explore whether sequence differences between our optimized and the original ZFPs correspond with functional differences either in vitro or in vivo, we chose two candidates from each optimization (boxed in Fig. 3D) and compared them with the original BCR-ABL, erbB2, and HIV ZFPs. Initially we tested the ability of each of these proteins to activate transcription of a reporter gene in our bacterial cell-based two-hybrid system. To do this, we replaced the selectable marker genes in each bacterial two-hybrid selection strain with the lacZ gene encoding β-galactosidase. The results of quantitative β-galactosidase assays, shown in Table 1, demonstrate that for all three target DNA sequences, the optimized multifinger proteins activate transcription more efficiently in bacterial cells than the “original” proteins.
Table 1. In vivo and in vitro characterization of the original and optimized BCR-ABL, erbB2, and HIV ZFPs.
| ZFP | Finger sequences* | Fold-activation (bacteria)† | Kd, pM‡ | KdNS, nM§ | Specificity ratio, KdNS/Kd | Fold-activation (mammalian)¶ |
|---|---|---|---|---|---|---|
| Original BCR-ABL | DRSSTR QGGNVR QAATQR | 4 | 28 (±3.9) | 55 (±12) | 1,980 | ND |
| Optimized BCR-ABL #1 | DSPTRR QGANRR QANTQR | 24 | 78 (±13) | 2,100 (±270) | 27,000 | ND |
| Optimized BCR-ABL #2 | DSPTRR QRTNIR QRNTQR | 27 | 60 (±8.5) | 1,300 (±97) | 23,000 | ND |
| Original erbB2 | RKDSVR QSGDRR DCRDAR | 2.7 | 150 (±23) | 1,000 (±120) | 6,700 | 1.0 (+0.07) |
| Optimized erbB2 #1 | RSDVAN QSSTTR ERQGKR | 8.6 | 31 (±3.1) | 1,100 (±15) | 35,000 | 2.0 (±0.27) |
| Optimized erbB2 #2 | RSDLTK QSSTTR ERQGKR | 5.9 | 65 (±3.9) | 1,100 (±81) | 17,000 | 1.3 (+0.06) |
| Original HIV | ASADTR NRSDSR TSSNKK | 1.3 | - | - | - | ND |
| Optimized HIV #1 | LRTDDR LSQTRR LRSNGR | 7.6 | 9.3 (±1.2) | 1,100 (±81) | 87,000 | ND |
| Optimized HIV #2 | NNAMVR LSQTQR MQGNSR | 7.3 | 9.3 (±0.39) | 180 (±8.8) | 19,000 | ND |
| Zif268 | 8.1 (±1.8) | 1,000 (±120) | 130,000 | ND |
-, Values that could not be determined because of weak binding of the ZFP; ND, experiments that were not performed.
Sequences of ZFP recognition helices are shown with helix residues -1, 1, 2, 3, 5, and 6 from fingers 1, 2, and 3 shown left to right.
Fold activation of a lacZ reporter gene in the bacterial two-hybrid system. Activation is calculated by comparing β-galactosidase expression in the presence and absence of the indicated ZFP. Note that only fold-activation data for the same binding site should be compared, because the values obtained for a given reporter strain were performed at different concentrations of IPTG inducer (0, 50, and 10 μM for the BCR-ABL, erbB2, and HIV reporter strains, respectively).
Dissociation constants (Kd) determined by electrophoretic mobility-shift assay (EMSA). The mean and standard error of three to seven experiments are shown.
Dissociation constant for nonspecific DNA (KdNS) determined by EMSA. The mean and standard error of three to seven experiments is shown.
Transcriptional activation of the endogenous erbB2 gene as measured by quantitative RT-PCR. The mean and standard error of three experiments are shown.
Next, we compared the in vitro DNA binding affinities and specificities of the original and optimized ZFPs. Each of these proteins (along with the naturally occurring Zif268 zinc finger domain as a control) was expressed as a maltose-binding protein fusion and purified. We quantified the affinity of each protein for its associated target DNA-binding site by determining dissociation constants (Kd) using electrophoretic mobility-shift assays (see Materials and Methods). In addition, to examine the DNA binding specificity of each protein, we used calf thymus DNA as competitor DNA and determined the dissociation constant of each protein for nonspecific DNA (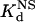 ) (see Materials and Methods). From these two parameters, we calculated a “specificity ratio,” corresponding to the ratio of the
) (see Materials and Methods). From these two parameters, we calculated a “specificity ratio,” corresponding to the ratio of the 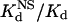 . The results of these experiments are summarized in Table 1 (note that the original HIV protein bound too weakly in our assay to determine a Kd, and therefore a
. The results of these experiments are summarized in Table 1 (note that the original HIV protein bound too weakly in our assay to determine a Kd, and therefore a 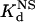 , value). All of our optimized proteins exhibit reasonable affinities for their respective target DNA sites (compare with the affinity of Zif268 for its binding site). These results also reveal that DNA binding affinity (as determined in vitro) cannot be the sole factor in determining activity in bacterial cells (compare Kd values with fold-activation results in bacteria). Interestingly, all of the optimized proteins exhibit relatively higher specificity ratios than their “original” counterpart proteins. One of our optimized HIV ZFPs actually possesses a specificity that approaches that of the naturally occurring Zif268 zinc finger domain for its target sequence.
, value). All of our optimized proteins exhibit reasonable affinities for their respective target DNA sites (compare with the affinity of Zif268 for its binding site). These results also reveal that DNA binding affinity (as determined in vitro) cannot be the sole factor in determining activity in bacterial cells (compare Kd values with fold-activation results in bacteria). Interestingly, all of the optimized proteins exhibit relatively higher specificity ratios than their “original” counterpart proteins. One of our optimized HIV ZFPs actually possesses a specificity that approaches that of the naturally occurring Zif268 zinc finger domain for its target sequence.
Finally, we performed experiments in mammalian cells to compare the DNA binding capabilities of our optimized erbB2 candidates with the original erbB2 ZFP. To do this, we fused each of the three erbB2 ZFPs with the NF-κB p65 activation domain, expressed these artificial transcription factors in human embryonic kidney 293 cells under the control of an inducible promoter, and assessed the activation of the endogenous erbB2 gene by quantitative RT-PCR. As shown in Table 1, consistent with similar published experiments (9), we find that the original erbB2 ZFP-p65 hybrid does not stimulate erbB2 gene transcription in mammalian cells (although we note this protein can stimulate erbB2 expression when covalently linked to another three-finger ZFP, thereby creating a six-finger DNA binding domain; ref. 9). By contrast, p65 fusion proteins containing our optimized erbB2 proteins can stimulate transcription of the endogenous erbB2 gene, suggesting that they occupy their intended target DNA site in vivo. Interestingly, the abilities of these proteins to activate erbB2 gene transcription in mammalian cells correlates with their abilities to activate transcription in the bacterial two-hybrid reporter cells and with the specificity ratios of these three proteins as determined in vitro.
Discussion
A Rapid Cell-Based Selection Strategy for Constructing Optimized Zinc Finger Proteins. In this report, we have validated a new rapid strategy for optimizing synthetic multifinger proteins. Like the “sequential optimization” method of Greisman and Pabo (25), our protocol yields multifinger proteins with excellent affinities and specificities for their target DNA sites, but it requires fewer libraries and fewer rounds of selection thereby making it more amenable to scale-up. Furthermore, proteins optimized by our method, when directly compared with proteins isolated by “modular assembly” or “bi-partite selection” strategies, exhibit significantly improved DNA binding affinities and/or specificities and function better in a cellular context. The relative success of our methodology is most likely due to our consideration of fingers as interdependent domains (unlike modular assembly strategies that treat them as independent modules) and to the combinatorial diversity of our finger libraries (unlike the bipartite selection approach that significantly limits randomization of the fingers). We discuss each of these issues in greater detail below but conclude overall that our optimization strategy provides a rapid and more effective alternative to existing methodologies for creating highly specific multifinger proteins.
Context-Dependent Effects on DNA Binding: Implications for the Modularity of Zinc Fingers. Our biochemical analysis of the original BCR-ABL and erbB2 ZFPs (constructed by modular assembly) reveals that these proteins are not fully optimized for DNA binding affinity or specificity despite the fact that each component finger was optimized for binding to its respective subsite. The most likely explanation for this discrepancy is that fingers selected to function optimally in one context do not necessarily do so in another context (i.e., fingers do not always function as completely modular units), an idea previously suggested by others (1, 5, 20, 21). In addition, our selection data also yield additional support for this hypothesis as fingers optimized to recognize the same DNA subsite yield different recognition helix sequences when selected in different contexts. For example, compare the different consensus recognition helix sequences of BCR-ABL finger 3 and erbB2 finger 2 (both selected to bind the GCAG subsite): QaNTqR vs. QSsTtR (Fig. 3D). Consistent with this, our optimization strategy, which selects sets of fingers that work well together, yields three-finger proteins with affinities and/or specificities superior to those produced by modular assembly. Taken together with results previously described by others, our findings strongly suggest that multifinger proteins created by modular assembly of preselected, predesigned, or naturally occurring zinc finger units may not be fully optimized for DNA binding affinity or specificity.
Importance of Combinatorial Diversity in Randomized Zinc Finger Libraries. Our new method also yields optimized ZFPs that bind to the HIV target DNA site with greater affinity and specificity than the “original” HIV protein isolated by the “bi-partite selection” method of Choo and colleagues (26). The difference in the success of the two methods most likely stems from the limited complexity of the randomized libraries used in the bipartite selections (26, 29). [The use of these restricted libraries is necessitated by the need to simultaneously randomize eight to nine recognition helix residues from one-and-a-half fingers (i.e., “half” of a three-finger protein).] We note that all of our optimized HIV proteins contain at least one recognition helix residue that was not present in the restricted libraries used in the bipartite selections. In addition, many of the candidates identified in the BCR-ABL and erbB2 optimizations also contain fingers that could not have been present in the bipartite selection libraries. Our results therefore strongly suggest that restricting amino acid possibilities in randomized libraries can potentially exclude optimal zinc fingers. One of the significant advantages of our approach over all other previously described methods is that it provides a means to sample a much larger “sequence space” for all fingers in the protein and therefore does not require significant compromises in the diversity of the initial randomized finger libraries used.
Affinity and Specificity Requirements for Cellular Function. Our results demonstrate that the bacterial two-hybrid system can be used to select DNA-binding proteins that possess both high affinity and high specificity for their target sites. One might naively assume that the occupancy of a specific target DNA sequence in the two-hybrid system would correlate primarily with the dissociation constant of the ZFP-DNA interaction. However, our data show that activation correlates not only with affinity but also with the DNA binding specificity of a protein (i.e., proteins that possess a high specificity ratio activate efficiently in the bacterial two-hybrid whereas proteins with a lower specificity ratio activate only weakly). Our data show that both DNA binding affinity and specificity influence how efficiently a ZFP can occupy its target DNA sequence in the environment of a bacterial cell. This result makes sense as high specificity will be necessary for a protein to find its single copy target DNA sequence amidst the vast excess of chromosomal DNA in a bacterial cell.
We have also demonstrated for the original and optimized erbB2 ZFPs that a correlation exists between their abilities to activate transcription in bacterial and human cells. This observation suggests that our bacterial cell-based strategy may provide a simple method to optimize proteins destined for use in mammalian cells. There are several reasons to believe that many of the factors governing how well a ZFP occupies its target DNA sequence are similar in the two different cellular environments. In both cases, a DNA-binding protein faces the challenge of having sufficient affinity and specificity to bind its target sequence without becoming “diverted” by nonspecific binding to other competing DNA sequences in the genome. We suspect that the sequence content of the E. coli chromosome might provide a reasonable surrogate for mammalian genomic DNA as our optimized proteins (which are selected in the presence of E. coli chromosomal “competitor” DNA) demonstrate high specificity ratios when characterized in vitro with mammalian (calf thymus) competitor DNA. In addition, although a great disparity in size exists between the E. coli chromosome (≈4 × 106 bp) and the human genome (≈3 × 109 bp), the difference in the effective concentration of genomic DNA in the two cases may be somewhat equalized by two factors: (i) the larger size of a mammalian nucleus relative to the nucleoid of the smaller bacterial cell (30) and (ii) the fact that <1% of DNA in a mammalian cell is accessible because of the presence of chromatin (4). Consistent with this hypothesis, we note that proteins evolved to function in bacteria (e.g., lexA repressor, tetracycline repressor) can also bind their DNA target sequence in mammalian cells. Thus, we suggest that the bacterial two-hybrid system may provide a rapid method to identify optimized ZFPs that function well in mammalian cells.
Optimizing Zinc Finger Proteins for Any 10-bp DNA Sequence. Using our strategy and the three master libraries described in this report, it should be possible to isolate optimized three-finger proteins that bind to any 10-bp target sequence matching the consensus 5′-GNNGNNGNNN-3′. Three bases within this consensus sequence are restricted to guanines because the anchor fingers used at the finger 1, finger 2, and finger 3 positions in the randomized libraries bind to subsites of the form GNNN, GNNG, and GNNG, respectively, and because these adjacent finger subsites overlap each other by 1 bp. By constructing additional master libraries harboring anchor fingers that recognize different four base pair subsites (e.g., ANNN subsites; ref. 12), our strategy may be able to isolate three finger proteins that bind to any 10-bp sequence of interest.
Importance of DNA Binding Specificity for Artificial Transcription Factors. We have validated a method that rapidly isolates synthetic multifinger proteins that bind with excellent affinity and specificity to desired target DNA sequences. Our studies reemphasize the complexity of zinc finger-DNA binding and reinforce the idea that finger units are not always fully modular. In addition, our results reveal the importance of affinity and, perhaps more importantly, of specificity for the effective occupancy of a DNA target site in a cellular environment. The issue of specificity is one of tremendous importance especially if engineered ZFPs are to be used to target functional domains [e.g., transcriptional activator or repressor domains, restriction endonuclease domains (31–34), site-specific recombinases (35, 36)] to a gene of interest. Methods for optimizing DNA binding specificity will be of critical importance for expanding the potential applications of designer zinc fingers in biological research and gene therapy.
Acknowledgments
We thank R. Fang, S. Wolfe, and J. Miller for helpful discussions, D. Sgroi for support with the quantitative RT-PCR experiments, S. Fay-Richard for advice on cell culture, S. Wolfe and C. Case for comments on the manuscript, and R. Colvin and D. Louis for support and encouragement. J.K.J. is partially supported by National Institutes of Health Grant K08 DK02883, J.A.H. and C.O.P. were supported by the Howard Hughes Medical Institute, and A.S.H. is supported by National Institutes of Health Grant 5T32CA09216. C.O.P. is a consultant for and Chairman of the Scientific Advisory Board of Sangamo Biosciences, Inc. (Richmond, CA). J.K.J. is an occasional consultant to Sangamo Biosciences, Inc.
Abbreviations: ZFP, zinc finger protein; 3-AT, 3-aminotriazole; IPTG, isopropyl β-d-thiogalactoside.
References
- 1.Pabo, C. O., Peisach, E. & Grant, R. A. (2001) Annu. Rev. Biochem. 70, 313–340. [DOI] [PubMed] [Google Scholar]
- 2.Beerli, R. R. & Barbas, C. F., III (2002) Nat. Biotechnol. 20, 135–141. [DOI] [PubMed] [Google Scholar]
- 3.Lee, D. K., Seol, W. & Kim, J. S. (2003) Curr. Top. Med. Chem. 3, 645–657. [DOI] [PubMed] [Google Scholar]
- 4.Jamieson, A. C., Miller, J. C. & Pabo, C. O. (2003) Nat. Rev. Drug Discovery 2, 361–368. [DOI] [PubMed] [Google Scholar]
- 5.Wolfe, S. A., Nekludova, L. & Pabo, C. O. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 183–212. [DOI] [PubMed] [Google Scholar]
- 6.Segal, D. J. & Barbas, C. F., III (2001) Curr. Opin. Biotechnol. 12, 632–637. [DOI] [PubMed] [Google Scholar]
- 7.Choo, Y. & Klug, A. (1997) Curr. Opin. Struct. Biol. 7, 117–125. [DOI] [PubMed] [Google Scholar]
- 8.Choo, Y. & Klug, A. (1995) Curr. Opin. Biotechnol. 6, 431–436. [DOI] [PubMed] [Google Scholar]
- 9.Beerli, R. R., Dreier, B. & Barbas, C. F., III (2000) Proc. Natl. Acad. Sci. USA 97, 1495–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beerli, R. R., Segal, D. J., Dreier, B. & Barbas, C. F., III (1998) Proc. Natl. Acad. Sci. USA 95, 14628–14633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blancafort, P., Magnenat, L. & Barbas, C. F. (2003) Nat. Biotechnol. 21, 269–274. [DOI] [PubMed] [Google Scholar]
- 12.Dreier, B., Beerli, R. R., Segal, D. J., Flippin, J. D. & Barbas, C. F., III (2001) J. Biol. Chem. 276, 29466–29478. [DOI] [PubMed] [Google Scholar]
- 13.Liu, Q., Segal, D. J., Ghiara, J. B. & Barbas, C. F., III (1997) Proc. Natl. Acad. Sci. USA 94, 5525–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu, P. Q., Rebar, E. J., Zhang, L., Liu, Q., Jamieson, A. C., Liang, Y., Qi, H., Li, P. X., Chen, B., Mendel, M. C., et al. (2001) J. Biol. Chem. 276, 11323–11334. [DOI] [PubMed] [Google Scholar]
- 15.Liu, Q., Xia, Z., Zhong, X. & Case, C. C. (2002) J. Biol. Chem. 277, 3850–3856. [DOI] [PubMed] [Google Scholar]
- 16.Bae, K. H., Do Kwon, Y., Shin, H. C., Hwang, M. S., Ryu, E. H., Park, K. S., Yang, H. Y., Lee, D. K., Lee, Y., Park, J., et al. (2003) Nat. Biotechnol. 21, 275–280. [DOI] [PubMed] [Google Scholar]
- 17.Ren, D., Collingwood, T. N., Rebar, E. J., Wolffe, A. P. & Camp, H. S. (2002) Genes Dev. 16, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang, L., Spratt, S. K., Liu, Q., Johnstone, B., Qi, H., Raschke, E. E., Jamieson, A. C., Rebar, E. J., Wolffe, A. P. & Case, C. C. (2000) J. Biol. Chem. 275, 33850–33860. [DOI] [PubMed] [Google Scholar]
- 19.Rebar, E. J., Huang, Y., Hickey, R., Nath, A. K., Meoli, D., Nath, S., Chen, B., Xu, L., Liang, Y., Jamieson, A. C., et al. (2002) Nat. Med. 8, 1427–1432. [DOI] [PubMed] [Google Scholar]
- 20.Isalan, M., Choo, Y. & Klug, A. (1997) Proc. Natl. Acad. Sci. USA 94, 5617–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isalan, M., Klug, A. & Choo, Y. (1998) Biochemistry 37, 12026–12033. [DOI] [PubMed] [Google Scholar]
- 22.Elrod-Erickson, M., Rould, M. A., Nekludova, L. & Pabo, C. O. (1996) Structure (London) 4, 1171–1180. [DOI] [PubMed] [Google Scholar]
- 23.Wolfe, S. A., Grant, R. A., Elrod-Erickson, M. & Pabo, C. O. (2001) Structure (London) 9, 717–723. [DOI] [PubMed] [Google Scholar]
- 24.Joung, J. K., Ramm, E. I. & Pabo, C. O. (2000) Proc. Natl. Acad. Sci. USA 97, 7382–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greisman, H. A. & Pabo, C. O. (1997) Science 275, 657–661. [DOI] [PubMed] [Google Scholar]
- 26.Isalan, M., Klug, A. & Choo, Y. (2001) Nat. Biotechnol. 19, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choo, Y., Sanchez-Garcia, I. & Klug, A. (1994) Nature 372, 642–645. [DOI] [PubMed] [Google Scholar]
- 28.Elrod-Erickson, M. & Pabo, C. O. (1999) J. Biol. Chem. 274, 19281–19285. [DOI] [PubMed] [Google Scholar]
- 29.Isalan, M. & Choo, Y. (2001) Methods Enzymol. 340, 593–609. [DOI] [PubMed] [Google Scholar]
- 30.Lin, S. & Riggs, A. D. (1975) Cell 4, 107–111. [DOI] [PubMed] [Google Scholar]
- 31.Bibikova, M., Beumer, K., Trautman, J. K. & Carroll, D. (2003) Science 300, 764. [DOI] [PubMed] [Google Scholar]
- 32.Bibikova, M., Golic, M., Golic, K. G. & Carroll, D. (2002) Genetics 161, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bibikova, M., Carroll, D., Segal, D. J., Trautman, J. K., Smith, J., Kim, Y. G. & Chandrasegaran, S. (2001) Mol. Cell. Biol. 21, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porteus, M. H. & Baltimore, D. (2003) Science 300, 763. [DOI] [PubMed] [Google Scholar]
- 35.Peng, W. J., Chang, C. M. & Lin, T. H. (2002) J. Biomed. Sci. 9, 171–184. [DOI] [PubMed] [Google Scholar]
- 36.Akopian, A., He, J., Boocock, M. R. & Stark, W. M. (2003) Proc. Natl. Acad. Sci. USA 100, 8688–8691. [DOI] [PMC free article] [PubMed] [Google Scholar]