

Abstract
Light-induced electron transfer reactions leading to the fully reduced, catalytically competent state of the flavin adenine dinucleotide (FAD) cofactor have been studied by flash absorption spectroscopy in DNA photolyase from Anacystis nidulans. The protein, overproduced in Escherichia coli, was devoid of the antenna cofactor, and the FAD chromophore was present in the semireduced form, FADH⋅, which is inactive for DNA repair. We show that after selective excitation of FADH⋅ by a 7-ns laser flash, fully reduced FAD (FADH−) is formed in less than 500 ns by electron abstraction from a tryptophan residue. Subsequently, a tyrosine residue is oxidized by the tryptophanyl radical with t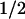 = 50 μs. The amino acid radicals were identified by their characteristic absorption spectra, with maxima at 520 nm for Trp⋅ and 410 nm for TyrO⋅. The newly discovered electron transfer between tyrosine and tryptophan occurred for ≈40% of the tryptophanyl radicals, whereas 60% decayed by charge recombination with FADH− (t
= 50 μs. The amino acid radicals were identified by their characteristic absorption spectra, with maxima at 520 nm for Trp⋅ and 410 nm for TyrO⋅. The newly discovered electron transfer between tyrosine and tryptophan occurred for ≈40% of the tryptophanyl radicals, whereas 60% decayed by charge recombination with FADH− (t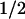 = 1 ms). The tyrosyl radical can also recombine with FADH− but at a much slower rate (t
= 1 ms). The tyrosyl radical can also recombine with FADH− but at a much slower rate (t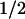 = 76 ms) than Trp⋅. In the presence of an external electron donor, however, TyrO⋅ is rereduced efficiently in a bimolecular reaction that leaves FAD in the fully reduced state FADH−. These results show that electron transfer from tyrosine to Trp⋅ is an essential step in the process leading to the active form of photolyase. They provide direct evidence that electron transfer between tyrosine and tryptophan occurs in a native biological reaction.
= 76 ms) than Trp⋅. In the presence of an external electron donor, however, TyrO⋅ is rereduced efficiently in a bimolecular reaction that leaves FAD in the fully reduced state FADH−. These results show that electron transfer from tyrosine to Trp⋅ is an essential step in the process leading to the active form of photolyase. They provide direct evidence that electron transfer between tyrosine and tryptophan occurs in a native biological reaction.
The role of amino acid radicals as intermediates in electron and hydrogen atom transfer is a topic of growing interest and research activity in enzymology (1, 2). Prominent examples include a catalytically essential tyrosyl radical in ribonucleotide reductase (3, 4), tyrosine YZ in photosynthetic water-oxidizing enzyme (5–7), and a tryptophan that can be oxidized by a photoexcited flavin semiquinone in DNA photolyase (PL) from Escherichia coli (8–10). It has even been suggested that tryptophan and tyrosine could act as sequential redox components in long-range electron transfer in proteins (11–13). The feasibility of such an electron transfer has been demonstrated in aqueous amino acids solutions (14), in model peptides (12, 15, 16), and in proteins after artificial oxidation of tryptophan (11, 17), but it has never been demonstrated in a native biological reaction.
DNA PLs use blue or near-UV light to repair UV-induced DNA lesions [refs. 18 and 19; either cyclobutane pyrimidine dimers or (6-4) photoproducts] in a variety of organisms, ranging from bacteria to multicellular eukaryotes (reviewed in ref. 20). This enzyme is a single polypeptide of 50–65 kDa. It contains a flavin adenine dinucleotide (FAD) as the essential catalytic cofactor and a second chromophore (either a reduced folate or a 8-OH-5-deazaflavin), which has the sole function of absorbing light and transferring excitation energy to the FAD cofactor (21). The FAD cofactor must be in the two-electron reduced form (FADH−) for enzymatic activity. In the purified enzyme, the FAD is typically present in the blue semiquinone form FADH⋅. This radical is stable in the dark but can be reduced to FADH− by illumination with visible light. According to studies on cyclobutane pyrimidine dimer PL from E. coli, the excited state of FADH⋅ abstracts an electron from a tryptophan residue in ≈1 μs (8). FADH− formed in this way back-reacts with the oxidized tryptophan in ≈10 ms, unless an external electron donor is present, which efficiently reduces the oxidized tryptophan and thus stabilizes FADH− (8).
Cyclobutane pyrimidine dimer PL from Anacystis nidulans has 52% sequence homology with that from E. coli and a similar three-dimensional structure as determined by x-ray crystallography (22, 23). Recombinant protein obtained by overexpression in E. coli host cells contains the FAD cofactor in the semiquinone form FADH⋅ but is completely devoid of the second chromophore (24). This apo-PL is active in DNA repair (25). We studied flavin photoreduction by exciting FADH⋅ selectively by a short laser flash and monitored the absorbance changes accompanying the subsequent reactions in the recombinant enzyme on a time scale from 10 ns to 2 s after the flash. We show that the light-induced tryptophanyl radical oxidizes a tyrosine residue, demonstrating that intraprotein electron transfer between tyrosine and tryptophan residues can be induced by a natural stimulus (visible light) and fulfills a biological function.
MATERIALS AND METHODS
Sample Preparation.
A. nidulans PL was overproduced in E. coli (24), purified as described by Eker et al. (26), and stored at −80°C in the presence of 2-mercaptoethanol (ME). The reducing agent was removed by buffer exchange with polyacrylamide chromatography microcolumns immediately before starting measurements. For transient absorption spectroscopy, we typically used 90 μM of enzyme in 0.2 M NaCl/20 mM Tris⋅HCl, pH 7.4/15% (vol/vol) glycerol in a cuvette with an optical path of 10 mm for the measuring beam and 4 mm for the excitation beam. The temperature of the sample was 10°C for all measurements. The redox state of the flavin cofactor was checked systematically by measuring the ground-state absorption spectrum from 400 nm to 700 nm. For some experiments, ME was added in the cuvette at the concentration indicated. PL deficient in the antenna chromophore was used throughout this study.
Transient Absorption Spectroscopy.
Flash-induced absorbance changes were measured with laboratory-built spectrometers (27, 28). The sample was excited typically by a 7-ns dye laser pulse at 635 nm with an energy of ≈30 mJ/cm2. For the measurement of absorbance changes at wavelengths greater than 610 nm, the excitation was tuned to 675 nm (≈20 mJ/cm2); this excitation induced about 10 times less photoreduction of FADH⋅ than the excitation at 635 nm, mainly because of the lower absorption of FADH⋅ at 675 nm. For measurements with a time resolution of 2 μs, a continuous monochromatic (spectral bandwidth of ≈5 nm) measuring-light beam, obtained by appropriate filtering of a 100-W tungsten-halogen lamp, passed through the sample perpendicular to the excitation beam. The measuring-light intensity and its flash-induced changes were monitored behind the sample by a Si photodiode connected to an amplifier (40 dB, DC-300 kHz) and a digital storage oscilloscope (DSA 602A from Tektronix). Stray light from the excitation flash and fluorescence were blocked by appropriate filters in front of the detector.
For measurements with a time resolution of 10 ns, a faster detection system was used. The tungsten-halogen lamp was replaced by a 50-μs Xe flash, and the relatively flat top of the temporal flash profile was used as a quasicontinuous measuring light (28).
To improve the signal-to-noise ratio, between 4 and 64 transients (measured at intervals of at least 1 s) were averaged at each wavelength; however, only a single excitation flash was used in the presence of ME to avoid photoaccumulation of FADH−.
Amplitudes (ai,λ) and half-times (t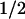 ,i = ln2/ki) of the different kinetic phases (i) of the flash-induced absorbance changes (ΔAλ) were obtained by globally fitting the transients at all wavelengths (λ) to a sum of exponential phases:
,i = ln2/ki) of the different kinetic phases (i) of the flash-induced absorbance changes (ΔAλ) were obtained by globally fitting the transients at all wavelengths (λ) to a sum of exponential phases:
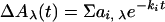 |
To compensate for differences in sample concentration, for differences in excitation conditions, and for some degradation of the samples during the series of experiments, the amplitudes presented in the figures were rescaled to standard conditions (90 μM of fresh enzyme, excitation with 30 mJ/cm2 at 635 nm), based on regular control measurements at reference wavelengths and on measurements on overlapping time scales. The rate of sample degradation (measured as the loss in flash-induced signal amplitude) was typically 7% per 100 excitation flashes of 30 mJ/cm2 at 635 nm and ≈2.5% per 100 excitation flashes of 20 mJ/cm2 at 675 nm. As judged from the evolution of the sample absorption spectrum, there is probably some oxidation of FADH· to fully oxidized FAD. The signal amplitude decreases accordingly, because fully oxidized FAD has no absorption at our excitation wavelength. The sample was replaced by a fresh one regularly so that degradation never exceeded 15%.
Flavin Photoreduction by Continuous Light.
The FADH− − FADH⋅ difference spectrum in PL from A. nidulans was obtained by subtracting absorption spectra recorded in a Cary (model 5E) spectrophotometer (Varian, Melbourne) before and after saturating illumination by continuous light (500–650 nm) in the presence of 5 mM ME.
RESULTS AND DISCUSSION
The earliest transient state observed was characterized by a bleaching in the visible absorption bands of FADH⋅; ≈40% of this bleaching recovered with t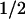 = 55 ns (data not shown). This phase has not been studied in detail in this work. It might be caused by the decay of an excited state (possibly a quartet state; ref. 29) of FADH⋅ to the (doublet) ground state, presumably in competition with the formation of FADH− by electron abstraction from tryptophan. The absorbance changes remaining after completion of the 55-ns decay (Fig. 1, •; measured 500 ns after excitation) agree well with those observed in E. coli PL at 4 μs after excitation (8). They are attributed to the reduction of FADH⋅ and the oxidation of a tryptophan (TrpH) residue. This attribution is evident from the comparison of the 500 ns spectrum (Fig. 1, •) with the FADH− minus FADH⋅ difference spectrum (Fig. 1, dotted line) obtained by photoreduction with continuous light (see Materials and Methods). The difference between these two spectra (squares) shows a broad positive contribution centered at 520 nm, which is characteristic of the formation of the neutral (deprotonated) tryptophanyl radical (Trp⋅; ref. 30). The relative amplitudes of the spectra in Fig. 1 are consistent with the formation of one Trp⋅ (ɛ520 ≈ 1,900 M−1⋅cm−1; ref. 30) per FADH⋅ (ɛ585 ≈ 5,000 M−1⋅cm−1; ref. 26) reduced.
= 55 ns (data not shown). This phase has not been studied in detail in this work. It might be caused by the decay of an excited state (possibly a quartet state; ref. 29) of FADH⋅ to the (doublet) ground state, presumably in competition with the formation of FADH− by electron abstraction from tryptophan. The absorbance changes remaining after completion of the 55-ns decay (Fig. 1, •; measured 500 ns after excitation) agree well with those observed in E. coli PL at 4 μs after excitation (8). They are attributed to the reduction of FADH⋅ and the oxidation of a tryptophan (TrpH) residue. This attribution is evident from the comparison of the 500 ns spectrum (Fig. 1, •) with the FADH− minus FADH⋅ difference spectrum (Fig. 1, dotted line) obtained by photoreduction with continuous light (see Materials and Methods). The difference between these two spectra (squares) shows a broad positive contribution centered at 520 nm, which is characteristic of the formation of the neutral (deprotonated) tryptophanyl radical (Trp⋅; ref. 30). The relative amplitudes of the spectra in Fig. 1 are consistent with the formation of one Trp⋅ (ɛ520 ≈ 1,900 M−1⋅cm−1; ref. 30) per FADH⋅ (ɛ585 ≈ 5,000 M−1⋅cm−1; ref. 26) reduced.
Figure 1.

Spectrum of the absorbance changes measured 500 ns after the excitation flash (●) and difference (▫) between that spectrum and the PL-FADH− − PL-FADH⋅ spectrum (dotted line; normalized to the 500-ns spectrum in the range from 590 nm to 660 nm).
The subsequent evolution of the absorbance changes was studied on different time scales in the microsecond and millisecond range (see Fig. 2 for examples at three key wavelengths). The step-like absorbance changes at t = 0 are instrument-limited (2-μs time resolution), and they agree well with the absorbance changes at 500 ns after excitation (Fig. 1, •). After these initial absorbance changes, we observed a kinetic phase with t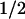 ≈ 50 μs, which has not been reported for the PL from E. coli. This phase was most pronounced at 410 nm, where it corresponds to an absorbance increase (Fig. 2, Top). At 520 nm (center of the Trp⋅ absorption band), we observed a bleaching on a time range of ≈50 μs (Fig. 2, Middle). In the absorption maximum of FADH⋅ at 590 nm (Fig. 2, Bottom), however, only a minor absorbance change occurred with t
≈ 50 μs, which has not been reported for the PL from E. coli. This phase was most pronounced at 410 nm, where it corresponds to an absorbance increase (Fig. 2, Top). At 520 nm (center of the Trp⋅ absorption band), we observed a bleaching on a time range of ≈50 μs (Fig. 2, Middle). In the absorption maximum of FADH⋅ at 590 nm (Fig. 2, Bottom), however, only a minor absorbance change occurred with t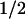 ≈ 50 μs (the analysis into exponential components shows that most of the recovery of the 590-nm bleaching between 0 ms and 0.3 ms is caused by a 1-ms phase, described below), indicating that FADH⋅ is not involved significantly in the 50-μs phase either as a reactant or as a product. These features suggest that the 50-μs phase reflects reduction of the tryptophanyl radical by electron transfer from a previously undetected third species that gives rise to an absorbance increase at 410 nm on oxidation.
≈ 50 μs (the analysis into exponential components shows that most of the recovery of the 590-nm bleaching between 0 ms and 0.3 ms is caused by a 1-ms phase, described below), indicating that FADH⋅ is not involved significantly in the 50-μs phase either as a reactant or as a product. These features suggest that the 50-μs phase reflects reduction of the tryptophanyl radical by electron transfer from a previously undetected third species that gives rise to an absorbance increase at 410 nm on oxidation.
Figure 2.

Kinetics of flash-induced absorbance changes at three selected wavelengths. The excitation flash was at t = 0. Time resolution = 2 μs. Three different time scales are used for a complete presentation of the kinetics.
An obvious candidate for this third species is tyrosine, because the tyrosyl radical is known to have an absorption band centered at ≈410 nm (30) and because the reduction potential of the tyrosyl radical in neutral aqueous solution (0.93 V vs. normal hydrogen electrode; ref. 31) is lower than that of the tryptophanyl radical (1.015 V; ref. 31). The complete difference spectrum of the 50-μs phase corresponds to a superposition of published difference spectra (32, 33) for the oxidation of tyrosine and for the reduction of the neutral tryptophanyl radical Trp⋅ (see Fig. 3). Tyrosine (TyrOH) is likely to deprotonate very rapidly on oxidation (the pKa of the phenoxyl radical is ≈−2; ref. 34), forming the neutral tyrosyl radical TyrO⋅. The spectral features of the 50-μs phase are very similar to those observed for the reaction Trp⋅⋅⋅⋅TyrOH → TrpH⋅⋅⋅TyrO⋅ in model peptides (12, 15, 16). We considered alternative assignments for the species, which is oxidized by the tryptophanyl radical during the 50-μs phase. However, published absorbance difference spectra (30) and potentials (35) for the oxidation of amino acids do not identify any possible candidate other than tyrosine. Because our recombinant protein contains no cofactor apart from FAD (which is not involved in this reaction), we conclude that the 50-μs phase in PL from A. nidulans reflects electron transfer from a tyrosine residue to the tryptophanyl radical.
Figure 3.

Spectrum of the t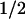 = 50-μs phase (left-hand scale; ●) attributed to electron transfer from a tyrosine residue to a tryptophanyl radical. The data result from a fit to transients like those shown in Fig. 2. Difference spectra for the oxidation of tyrosine (×) and for reduction of the neutral tryptophan radical (+) were obtained from refs. 30 and 31, respectively (right-hand scale).
= 50-μs phase (left-hand scale; ●) attributed to electron transfer from a tyrosine residue to a tryptophanyl radical. The data result from a fit to transients like those shown in Fig. 2. Difference spectra for the oxidation of tyrosine (×) and for reduction of the neutral tryptophan radical (+) were obtained from refs. 30 and 31, respectively (right-hand scale).
Comparison of the amplitudes of the 50-μs phase (Fig. 3, •) with the data presented in Fig. 1 shows that only ≈40% of the tryptophanyl radicals were reduced by tyrosine (the remaining tryptophanyl radicals were reduced finally through a back-reaction with FADH−; see below). This behavior may be caused by some heterogeneity in our protein or by an as yet undetected competing reaction that renders the reduction of the tryptophanyl radical by tyrosine less favorable.
The absorbance change kinetics in the millisecond range (Fig. 2, Right) show that within 500 ms, the sample recovered to the same state as before excitation. This recovery is well described by two exponential phases with half-times of 1 ms and 76 ms. The difference spectra of the two phases (Fig. 4) are readily interpretable as being caused by back-reactions of FADH− with Trp⋅ (t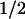 = 1 ms; 58% of the recovery of FADH⋅) and with TyrO⋅ (t
= 1 ms; 58% of the recovery of FADH⋅) and with TyrO⋅ (t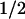 = 76 ms; 42% of the recovery of FADH⋅). These percentages are consistent with the above estimate that ≈40% of the tryptophanyl radicals were reduced by tyrosine during the 50-μs phase.
= 76 ms; 42% of the recovery of FADH⋅). These percentages are consistent with the above estimate that ≈40% of the tryptophanyl radicals were reduced by tyrosine during the 50-μs phase.
Figure 4.

Absorbance difference spectra of the kinetic phases with t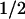 = 1 ms (▪) and t
= 1 ms (▪) and t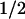 = 76 ms (●), compared with the PL-FADH− − PL-FADH⋅ spectrum (dotted lines; normalized to the other spectra in the range from 590 nm to 660 nm).
= 76 ms (●), compared with the PL-FADH− − PL-FADH⋅ spectrum (dotted lines; normalized to the other spectra in the range from 590 nm to 660 nm).
The addition of ME (1–5 mM) as an external electron donor affected neither the formation of FADH−, Trp⋅, and TyrO⋅ nor the back-reaction between FADH− and Trp⋅. The reduction of the tyrosyl radical, however, was largely modified in that the slowest decay phase of the absorbance change at 400 nm (Fig. 5a) was accelerated from t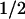 = 76 ms (without addition) to 7 ms (with 5 mM ME). The spectrum of this 7-ms phase matched that of the tyrosyl radical well (data not shown). The rate of the absorbance decay at 400 nm increased linearly with the concentration of ME (Fig. 5a Inset). On the other hand, the extent of recovery of FADH⋅ (monitored at 610 nm; Fig. 5b) decreased dramatically because of the addition of ME. We conclude from these observations that ME reduced the tyrosyl radical in an apparently bimolecular reaction (rate constant of ≈2 × 104 M−1⋅s−1), thus preventing the back-reaction between FADH− and TyrO⋅ and leaving FADH− as a stable reaction product.
= 76 ms (without addition) to 7 ms (with 5 mM ME). The spectrum of this 7-ms phase matched that of the tyrosyl radical well (data not shown). The rate of the absorbance decay at 400 nm increased linearly with the concentration of ME (Fig. 5a Inset). On the other hand, the extent of recovery of FADH⋅ (monitored at 610 nm; Fig. 5b) decreased dramatically because of the addition of ME. We conclude from these observations that ME reduced the tyrosyl radical in an apparently bimolecular reaction (rate constant of ≈2 × 104 M−1⋅s−1), thus preventing the back-reaction between FADH− and TyrO⋅ and leaving FADH− as a stable reaction product.
Figure 5.

Kinetics of the flash-induced absorbance changes at 400 nm (a) and at 610 nm (b) in the absence and presence of ME (5 mM). (Inset) Dependence of the decay rate of the 400 nm absorbance changes on the concentration of ME (with linear regression line).
Taken together, our results show that two amino acids, tryptophan and tyrosine, are involved in the photoreduction of the flavin cofactor in A. nidulans DNA PL. The electron transfer pathway is as follows:excited FADH⋅ ← tryptophan ← tyrosine ← external reductant. Future work will aim at identifying these two redox-active amino acids in the recently determined structure of the protein (22). The electron transfer pathway suggests that the flavin is further away from the redox-active tyrosine and closer to the tryptophan. The same spatial arrangement is expected from the different back-reaction rates of FADH− with TyrO⋅ (t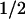 = 76 ms) and with Trp⋅ (t
= 76 ms) and with Trp⋅ (t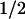 = 1 ms). The relatively slow back-reaction with the tyrosyl radical favors the reduction of the latter by external reductants—hence the accumulation of FADH−, which is required for the DNA repair function of the enzyme. In contrast to our present results on the PL from A. nidulans, only tryptophan oxidation reportedly occurs in the PL from E. coli (8–10). It is not clear whether the mechanism of flavin photoreduction is different in these two enzymes or whether tyrosine oxidation has been overlooked in previous work.
= 1 ms). The relatively slow back-reaction with the tyrosyl radical favors the reduction of the latter by external reductants—hence the accumulation of FADH−, which is required for the DNA repair function of the enzyme. In contrast to our present results on the PL from A. nidulans, only tryptophan oxidation reportedly occurs in the PL from E. coli (8–10). It is not clear whether the mechanism of flavin photoreduction is different in these two enzymes or whether tyrosine oxidation has been overlooked in previous work.
Other important points to be addressed in future work are the deprotonation and protonation events accompanying electron transfer during photoreduction of FADH⋅. A detailed study of the pH dependence of the electron transfer kinetics and the associated absorbance difference spectra is necessary. As the tryptophanyl radical seems to be deprotonated, one may speculate that electron transfer from tyrosine to the tryptophanyl radical could be coupled to a proton transfer between the two residues, such that the overall reaction would be a hydrogen atom transfer. Recently, such a mechanism has been suggested to be relevant when redox-active amino acids are connected by a hydrogen-bonded pathway (36). It will be possible to verify the existence of such a pathway in the PL from A. nidulans once the redox-active tryptophan and tyrosine residues are identified.
Intraprotein electron transfer between tyrosine and tryptophan has been observed under artificial conditions (11, 17). This study provides direct evidence for such an electron transfer in a native biological reaction. A similar reaction may take place in the assembly of the tyrosyl radical-diiron(III) cofactor of E. coli ribonucleotide reductase under limiting Fe2+ conditions, where it has been proposed that a tryptophan radical cation generates the tyrosyl radical (37). Electron transfer between tyrosine and tryptophan may be involved more generally in radical enzymes and in electron transfer proteins.
Acknowledgments
We thank A. Ivancich, T. A. Mattioli, and A. W. Rutherford for discussion and comments on the manuscript, A. Yasui for providing us the A. nidulans PL expression construct, and P. Setif for the data analysis software.
ABBREVIATIONS
- FAD
flavin adenine dinucleotide
- FADH⋅
FAD in its semireduced state
- FADH−
FAD in its fully reduced (deprotonated) state
- PL
photolyase
- ME
2-mercaptoethanol
References
- 1.Sigel H D, Sigel A, editors. Metalloenzymes Involving Amino Acid Residue and Related Radicals, Metals Ions in Biological Systems. Vol. 30. New York: Dekker; 1994. [Google Scholar]
- 2.Stubbe J, van der Donk W A. Chem Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 3.Reichard P, Ehrenberg A. Science. 1983;221:514–519. doi: 10.1126/science.6306767. [DOI] [PubMed] [Google Scholar]
- 4.Gräslund A, Sahlin M. Annu Rev Biophys Biomol Struct. 1996;25:259–286. doi: 10.1146/annurev.bb.25.060196.001355. [DOI] [PubMed] [Google Scholar]
- 5.Barry B A, Babcock G T. Proc Natl Acad Sci USA. 1987;84:7099–7103. doi: 10.1073/pnas.84.20.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerken S, Brettel K, Schlodder E, Witt H T. FEBS Lett. 1988;237:69–75. [Google Scholar]
- 7.Hoganson C W, Babcock G T. Science. 1997;277:1953–1956. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- 8.Heelis P F, Okamura T, Sancar A. Biochemistry. 1990;29:5694–5698. doi: 10.1021/bi00476a008. [DOI] [PubMed] [Google Scholar]
- 9.Kim S T, Heelis P F, Sancar A. Methods Enzymol. 1995;258:319–343. doi: 10.1016/0076-6879(95)58054-9. [DOI] [PubMed] [Google Scholar]
- 10.Kim S T, Sancar A, Essenmacher C, Babcock G T. Proc Natl Acad Sci USA. 1993;90:8023–8027. doi: 10.1073/pnas.90.17.8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butler J, Land E J, Prütz W A, Swallow A J. Biochim Biophys Acta. 1982;705:150–162. [Google Scholar]
- 12.Faraggi M, DeFilippis M R, Klapper M H. J Am Chem Soc. 1989;111:5141–5145. [Google Scholar]
- 13.Lee C-Y. FEBS Lett. 1992;299:119–123. doi: 10.1016/0014-5793(92)80228-9. [DOI] [PubMed] [Google Scholar]
- 14.Jovanovic S V, Harriman A, Simic M G. J Phys Chem. 1986;90:1935–1939. [Google Scholar]
- 15.Prütz W A, Land E J. Int J Radiat Biol. 1979;36:513–520. doi: 10.1080/09553007914551301. [DOI] [PubMed] [Google Scholar]
- 16.Prütz W A, Land E J, Sloper R W. J Chem Soc Faraday Trans 1. 1979;77:281–292. [Google Scholar]
- 17.Prütz W A, Butler J, Land E J, Swallow A J. Biochem Biophys Res Commun. 1980;96:408–414. doi: 10.1016/0006-291x(80)91230-9. [DOI] [PubMed] [Google Scholar]
- 18.Wulff D L, Rupert C S. Biochem Biophys Res Commun. 1962;7:237–240. doi: 10.1016/0006-291x(62)90181-x. [DOI] [PubMed] [Google Scholar]
- 19.Setlow J K, Setlow R B. Nature (London) 1963;197:560–562. [Google Scholar]
- 20.Yasui A, Eker A P M. In: DNA Repair in Higher Eukaryotes. Nickoloff J A, Hoekstra M F, editors. Vol. 2. Totowa, NJ: Humana; 1998. pp. 9–32. [Google Scholar]
- 21.Sancar A. Science. 1996;272:48–49. doi: 10.1126/science.272.5258.48. [DOI] [PubMed] [Google Scholar]
- 22.Tamada T, Kitadokoro K, Higuchi Y, Inaka K, Yasui A, de Ruiter P E, Eker A P M, Miki K. Nat Struct Biol. 1997;4:887–891. doi: 10.1038/nsb1197-887. [DOI] [PubMed] [Google Scholar]
- 23.Park H W, Kim S T, Sancar A, Deisenhofer J. Science. 1995;268:1866–1872. doi: 10.1126/science.7604260. [DOI] [PubMed] [Google Scholar]
- 24.Takao M, Oikawa A, Eker A P M, Yasui A. Photochem Photobiol. 1989;50:633–637. doi: 10.1111/j.1751-1097.1989.tb04319.x. [DOI] [PubMed] [Google Scholar]
- 25.Eker, A. P. M. & Yasui, A. (1991) Photochem. Photobiol.53, Suppl., 17S–18S.
- 26.Eker A P M, Kooiman P, Hessels J K C, Yasui A. J Biol Chem. 1990;265:8009–8015. [PubMed] [Google Scholar]
- 27.Sétif P Q Y, Bottin H. Biochemistry. 1994;33:8495–8504. doi: 10.1021/bi00194a014. [DOI] [PubMed] [Google Scholar]
- 28.Brettel K, Leibl W, Liebl U. Biochim Biophys Acta. 1998;1363:175–181. doi: 10.1016/s0005-2728(98)00010-3. [DOI] [PubMed] [Google Scholar]
- 29.Li Y F, Heelis P F, Sancar A. Biochemistry. 1991;30:6322–6329. doi: 10.1021/bi00239a034. [DOI] [PubMed] [Google Scholar]
- 30.Bensasson R V, Land E J, Truscott T G. Flash Photolysis and Pulse Radiolysis: Contributions to the Chemistry of Biology and Medicine. Oxford: Pergamon; 1983. [Google Scholar]
- 31.Harriman A. J Phys Chem. 1987;91:6102–6104. [Google Scholar]
- 32.Bent D V, Hayon E. J Am Chem Soc. 1975;97:2599–2606. doi: 10.1021/ja00843a002. [DOI] [PubMed] [Google Scholar]
- 33.Posener M L, Adams G E, Wardam P. J Chem Soc Faraday Trans 1. 1976;72:2231–2239. [Google Scholar]
- 34.Dixon W T, Murphy D. J Chem Soc Faraday Trans 2. 1976;72:1221–1230. [Google Scholar]
- 35.Brabec V, Mornstein V. Biophys Chem. 1980;12:159–165. doi: 10.1016/0301-4622(80)80048-2. [DOI] [PubMed] [Google Scholar]
- 36.Siegbahn E M, Blomberg M R A, Crabtree R H. Theor Chem Accounts. 1997;97:289–300. [Google Scholar]
- 37.Bollinger J M, Tong W H, Ravi N, Huynh B H, Edmondson D E, Stubbe J. J Am Chem Soc. 1994;116:8024–8032. [Google Scholar]