

Abstract
Id proteins bind basic helix–loop–helix transcription factors and function as dominant negative inhibitors of gene expression. Id1 and Id3 are required for the recruitment of bone marrow-derived endothelial cell precursors and tumors transplanted into Id-deficient mice demonstrate impaired angiogenesis. Mouse mammary tumor virus–neu mice were bred with Id1–/–Id3+/– mice to ascertain the role of Id1 and Id3 in mammary tumorigenesis in a more physiologically relevant model. In mammary tumors from these mice, Id1 and Id3 expression was restricted to the vascular endothelium. Id1 and Id3 deficiency did not prevent or delay tumor formation but did alter tumor phenotype. The tumors that developed in the Id-deficient mice were larger and cystic with a viable rim of tumor cells surrounding a nonviable core of cellular debris. The Hsp90 chaperone protein is required for cellular survival under condition of environmental stress and for the stability of the neu oncogene. 17-Allylamino-17-demethoxygeldanamycin, an Hsp90 inhibitor, was used to treat these mice. Whereas 17-allylamino-17-demethoxygeldanamycin only modestly delayed the growth of established mammary tumors in WT mice for Id, tumor suppression was dramatically more effective in an Id1- or Id3-deficient background. These data suggest that tumorigenesis can occur in a background of defective angiogenesis but that tumors developing in such an environment may be especially sensitive to inhibitors of neu and stress-activated survival pathways. Thus angiogenesis inhibitors in combination with inhibitors of Hsp90 function should be evaluated for the treatment of advanced breast cancer.
Keywords: geldanamycin
Angiogenesis, the process by which capillaries sprout from preexisting blood vessels, is an event regulated by a balance between proangiogenic and antiangiogenic molecules (1). Id1 and Id3 proteins are important for angiogenesis during mouse development, and Id1–/–Id3–/– mice are not viable because of aberrant vascularization in the neuroectoderm (2). Id1 and Id3 are members of the Id family, composed of four related proteins that contain a highly conserved helix–loop–helix (HLH) dimerization motif that binds to basic HLH transcription factors (3), thus regulating cell-cycle progression and cellular differentiation (4). Id1 and Id3 also have a role in tumor angiogenesis, a vital process for the growth of a neoplasm. The growth of transplanted tumors in mice with partial Id1 and Id3 deficiency is delayed or in some cases abolished, and a defect in tumor angiogenesis is observed (2).
The aim of the work presented here was to determine whether HER2/neu-dependent mammary tumors develop in a Id-deficient background and, if so, whether impairment of angiogenesis alters their phenotype. HER2/neu encodes a transmembrane tyrosine kinase that is commonly amplified in human breast tumors. Amplification of HER2 correlates with a poor clinical prognosis in breast cancer patients (5), and anti-Her2 antibodies have therapeutic utility in this disease (6). Mice expressing neu under the transcriptional control of the mouse mammary tumor virus LTR develop mammary tumors after a prolonged latency (7). YD neu mice express a receptor with a 12-aa deletion in the extracellular domain that leads to its constitutive activation. In addition, four of the five tyrosine phosphorylation sites have been mutated to phenylalanine in the YD neu protein, leaving only the fourth (D) site active. These mice form multifocal solid comedo-type tumors with low metastatic potential (8).
In this article, we demonstrate that partial Id deficiency does not delay tumor development in YD neu mice but dramatically alters tumor phenotype. In contrast to tumors that developed in the WT background, tumors that developed in Id-deficient mice were larger and cystic in appearance with a viable rim of tumor cells surrounding a nonviable core of cellular debris. These cells survived in an environment in which blood supply is deficient. Under these conditions, cells depend on stress-activated signaling pathways for survival. In addition, the maintenance of high levels of neu may be required for viability. The Hsp90 chaperone is an important mediator of these pathways (9).
Geldanamycin (GM) is a natural antibiotic that binds to a conserved ATP/ADP pocket in the amino terminus of Hsp90 and inhibits its function (10, 11). This process results in the proteasomal degradation of several of the proteins that require Hsp90 for maturation or stability (12). HER2/neu is one of the most sensitive targets. GM binds tightly to a 15-Å deep, conserved pocket in the amino-terminal portions of Hsp90 (10). Hsp90-dependent folding requires the binding of ATP to this pocket, and its occupancy by GM either disrupts or regulates its chaperone function. The biological effects of GM are likely secondary to these interactions with Hsp90 and other family members. GM proved to be too toxic for human use, but a derivative, 17-allylamino-17-demethoxygeldanamycin (17-AAG) (13), has entered phase I clinical testing at several institutions, including our own. We tested whether tumors arising in an Id-deficient background were sensitive to Hsp90 inhibition. Tumor-bearing mice were treated with 17-AAG. This agent was only marginally effective in inhibiting YD neu tumors in the Id WT background. In contrast, the same treatment dramatically suppressed the growth of tumors in the Id-deficient background. These results suggest that the defect in angiogenesis caused by Id deficiency in these mice and perhaps other antiangiogenic therapeutic interventions is insufficient to completely inhibit physiologically relevant tumor growth. However, under these conditions, an inhibitor of Hsp90 completely suppresses tumor progression.
Materials and Methods
Generation of YD neu Id1- and Id3-Deficient Mice. YD neu mice in a pure FVB/NHK background were obtained from Peter Siegel (Sloan–Kettering Institute) and maintained in pressurized ventilated caging at the Sloan–Kettering Institute. All studies were performed in compliance with Institutional Animal Care and Use Committee guidelines. YD neu mice were crossed with Id1–/–Id3+/– mice in a mixed C57Bl6/129Sv background to generate YD neu Id1+/–Id3+/–mice. These mice were then crossed with Id1+/–Id3+/– siblings to generate the following: control YD neu Id1+/+Id3+/+ mice, YD neu Id1+/–Id3+/+, YD neu Id1+/+Id3+/–, YD neu Id1+/–Id3+/–, YD neu Id1–/–Id3+/+, YD neu Id1+/+Id3–/–, YD neu Id1–/–Id3+/–, and YD neu Id1+/–Id3–/– mice at the expected Mendelian frequencies.
Histological Analysis. Tumors were fixed in paraformaldehyde, and 7-μm paraffin sections were stained for hematoxylin/eosin, human CD31 (DAKO monoclonal mouse anti-human endothelial cell, CD31, 1:50), mouse CD31 (rat anti-mouse CD31 monoclonal, 2.5 μg/ml, Pharmingen), Id1 (rabbit polyclonal C-20, 1:150, Santa Cruz Biotechnology), hypoxia-inducible factor 1α (HIF-1α) (monoclonal NB-100123, 1:100, Novus Biologicals, Littleton, CO), and Ki67 (mouse monoclonal NCL-Ki67-MM1. 1:100, NovoCastra, Newcastle, U.K.). For polyclonal antibodies, we used the ABC kit (Vector Laboratories) following the manufacturer's instructions. For mAbs we used the Vector MOM kit. Sections were processed for in situ hybridization with α-33P-UTP-labeled antisense human Id1, human and mouse Id3, and mouse Ang2 RNA probes as described (14).
Immunoblotting. Tumor lysates were prepared by homogenizing tumors in SDS lysis buffer (50 mM Tris·HCl, pH 7.4/2% SDS), boiling for 10 min, and sonicating briefly. Cell lysates were cleared by centrifugation at 14,000 × g for 10 min, and supernatants were collected as the experimental samples. The protein concentration of each sample was determined by using a BCA kit (Pierce) according to the manufacturer's instructions. Samples were subjected to 7% SDS/PAGE and then transferred to a nitrocellulose membrane. Western blotting was carried out with the antibodies against Her2 (C-18, sc-284, Santa Cruz Biotechnology) and p85 subunit of phosphatidylinositol 3-kinase (Upstate Biotechnology, Waltham, MA). Bands were detected by chemiluminescence with a ECL kit (Amersham Biosciences) and quantitated by using the Gel Doc 1000 (Bio-Rad).
17-AAG Treatment. Mice were monitored weekly for mammary tumor development. They were randomly selected for treatment with either 17-AAG (75 mg/kg) three times per week or the egg phospholipids (EPL) diluent as control when the first palpable tumor reached 5 mm in size. Treatment was initiated within 1 week of randomization and continued for 8 weeks. Some treated and untreated mice were killed 12 h after the final dose of therapy. Others were monitored for tumor regrowth after discontinuation of therapy.
Statistical Analysis. Times to tumor onset as a function of Id1 and Id3 genetic profile were compared by using the Wilcoxon rank sum test. Differences in the proportion of tumors with cystic appearance were compared by using an adjusted χ2 test (15). This modification was needed because there were multiple lesions per mouse. The formula for the adjusted test statistic was
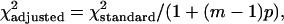 |
where m is the average cluster size and p is the correlation within mice. The frequency of abnormal vascularization or HIF-1α expression in Id WT and Id-deficient mice were compared by using the Fisher's exact test. Differences in Id expression between tumor and endothelial cells in human breast tumors were evaluated by using a McNemar's test. Tumor volumes in mice treated with 17-AAG or EPL vehicle were compared by using the Wilcoxon rank sum test. All tests with P values <0.05 were considered significant.
Results
Id Deficiency Does Not Prevent or Delay Tumor Formation in YD neu Mice, But Does Alter Tumor Phenotype. To determine whether a reduction in Id1 or Id3 expression prevents or alters the rate of spontaneous mammary tumor formation in neu transgenic mice, we bred YD neu mice with Id1–/–Id3+/– mice. The control Id WT and the various Id-deficient populations were obtained as described in Materials and Methods. YD neu Id WT mice developed their first mammary lesion with a median latency of 184 days (Fig. 1A). In the YD neu Id-deficient mice, tumor onset was not prevented or delayed. In fact, there was a small acceleration in median tumor development of 22 days in the Id1- and/or Id3-deficient mice (median tumor latency of 162 days for all Id-deficient populations, P = 0.04, Fig. 1 A). No difference in tumor latency was observed between Id1- and Id3-deficient mice or between groups missing one, two, or three copies of Id1 or Id3 (data not shown and Fig. 1 A).
Fig. 1.

Partial Id1 or Id3 deficiency does not delay tumor latency in YD neu mice, but does alter tumor phenotype. (A) The median tumor latency in YD neu Id WT mice is 184 days. The median time to tumor development in YD neu mice with one, two, or three copies of Id1 or Id3 deleted is 166, 156, and 162 days, respectively (P = 0.04 Id WT vs. Id-deficient). No statistical difference in median time to tumor onset was observed in mice deficient in Id1 versus Id3 or as a function of the number of copies of Id1 and Id3 deleted. (B) The rate of cystic tumor formation in YD neu Id-deficient mice was significantly greater than that observed in YD neu Id WT mice (64.5%, 56.6%, and 64.0% in mice missing one copy, two copies, or three copies, respectively, versus 23.5% in Id WT mice; P < 0.01 for all three Id-deficient groups versus WT).
Tumor-bearing mice were killed 8 weeks after detection of the first lesion. At death, tumors were classified as solid if >90% of the tumor volume was grossly viable and cystic if >90% of the tumor volume consisted of necrotic or cystic components (Fig. 2 A and B). Heterogeneous tumors were those that did not meet the criteria for either solid or cystic. The majority of tumors that developed in the Id WT mice were entirely solid (53%), with only minimal gross evidence of necrosis. In a subset of Id WT tumors, heterogeneous (23.5%) or cystic (23.5%) architecture was observed. In the Id-deficient mice, a significantly larger proportion of tumors were cystic in appearance (64.5%, 56.5%, and 64%, respectively, in the mice deficient in one, two, or three copies of Id, P < 0.01 for all three Id-deficient groups versus WT, Fig. 1B). No difference in the proportion of cystic lesions was observed between mice deficient in Id1 versus those deficient in Id3.
Fig. 2.

Mammary tumors arising in YD neu Id-deficient mice were cystic with a small rim of viable cells surrounding an acellular core. (A and B) Tumors dissected from a YD neu Id1+/+Id3+/+ mouse (A) and a YD neu Id1–/–Id3+/– mouse (B). (C and E) Hematoxylin/eosin staining of the YD neu Id1+/+Id3+/+ tumor seen in A.(D and F) Hematoxylin/eosin staining of the YD neu Id1–/–Id3+/– tumor seen in B. (Magnifications: E and F, ×400.)
On histological examination, the cystic lesions in Id-deficient mice consisted of a narrow rim of viable tumor cells surrounding a core of hemorrhagic fluid (Fig. 2D). In both solid and cystic lesions, the viable tumor component consisted of anaplastic tumor cells with a complete loss of glandular morphology (Fig. 2 E and F).
Id1 and Id3 Are Expressed in Tumor Vasculature. In the mammary tumors of YD neu mice, Id1 protein was expressed only in the nuclei of endothelial cells (Fig. 3 A–C). Levels of Id1 were greater in tumor vasculature in comparison to vessels contained within the surrounding normal mammary epithelium. Tumor cells themselves did not express Id1 (Fig. 3 B and C). As predicted, Id1 staining was not observed in the tumor vasculature of Id1–/– mice (Fig. 3D). Id3 expression was evaluated by in situ hybridization and observed primarily in tumor vasculature (data not shown).
Fig. 3.

Id1 was predominantly expressed in human and mouse tumor endothelium. (A–C) Immunohistochemistry of a representative YD neu Id1+/+ Id3+/+ mammary tumor for CD31 (A) and Id1 (B and C). (D) Immuhistochemistry for Id1 of a representative YD neu Id1–/–Id3+/+ mammary tumor. (E) Immunohistochemistry of Id1 in a representative human invasive breast carcinoma showing restriction of Id1 staining to the tumor endothelial cells. (F) Double staining for Id1 and CD31 in a representative human breast tumor showing the presence of Id1 (blue staining) in the nuclei of CD31-positive (brown staining) endothelial cells. Black arrows point to Id1-positive endothelial cells. White arrows point to Id1-negative endothelial cells in the YD neu Id1–/–Id3+/+ mammary tumor. (Magnifications: A and B, ×200; C and D, ×640; E, ×100; F, ×1,000.)
The pattern of Id1 and Id3 expression in human breast cancers was also characterized by using 30 primary tumors of various grades. Id1 mRNA expression by in situ hybridization was seen in the vasculature of all 30 tumors examined (Table 1). By immunohistochemistry, Id1 protein was expressed in the vessels of 83% of tumors but was never observed in tumor cells (Table 1 and Fig. 3 E and F, P < 0.01). A similar pattern was seen for Id3 mRNA expression (97% of tumors demonstrated endothelial cell expression of Id3 mRNA versus only 43% demonstrating tumor cell expression, P < 0.01).
Table 1. Id1 and Id3 expression patterns in 30 human breast carcinomas.
| Tumor cells, %
|
Endothelial cells, %
|
|||||
|---|---|---|---|---|---|---|
| Id type | Negative | Positive | Strongly positive | Negative | Positive | Strongly positive |
| Id1 (mRNA) | 67 | 20 | 13 | 0 | 20 | 80 |
| Id1 (protein) | 100 | 0 | 0 | 17 | 23 | 60 |
| Id3 (mRNA) | 57 | 30 | 13 | 3 | 23 | 73 |
By immunohistochemistry, Id1 protein expression was present in endothelial cells but never observed in the tumor cells. By in situ hybridization, Id1 expression was restricted to endothelial cells in 67% of samples. In situ for Id3 demonstrated an expression pattern similar to that observed with Id1.
Abnormal Vascularization of Id-Deficient Tumors. Tumor-infiltrating vessels were visualized by staining tumors for angiopoietin 2 (16) and CD31. In cystic tumors arising in an Id-deficient background, vessels were present in the adjacent extracellular matrix, but rarely observed inside the thin rim of viable tumor. In the subset of Id-deficient solid tumors, 66.7% had abnormal vascularization as evidenced by dilated and twisted vessels with frequent anastomoses (versus 5.9% of Id WT tumors, Fig. 4 A–D, P < 0.01). This pattern was similar to that observed in the blood vessels of nonviable Id1–/–Id3–/– embryos (2).
Fig. 4.

(A-D) Tumor vessels in YD neu Id1- and Id3-deficient mice were abnormal and dilated. (A and C) In situ hybridization for Ang2 (A dark field) and immunohistochemistry for CD31 (C) in a mammary tumor from a YD neu Id1+/+Id3+/+ mouse. (B and D) In situ hybridization for Ang2 (B) and immunohistochemistry for CD31 (D) in a tumor from a YD neu Id1+/–Id3+/+ mouse. (E and F) HIF-1α was induced in small Id1- or Id3-deficient YD neu tumors. Immunohistochemistry with an anti-HIF-1α antibody of small tumors (<0.5 cm in diameter) from a YD neu Id1+/+Id3+/+ mouse (E)and a YD neu Id1–/–Id3+/– mouse (F) demonstrating Hif-1α staining only in the Id-deficient tumor. (G and H) HIF-1α expression was observed surrounding areas of necrosis in large solid lesions but was not present in large cystic lesions. Immunohistochemistry with an anti-HIF-1α antibody of large tumors (>1.5 cm in diameter) from a YD neu Id1+/+Id3+/+ mouse (G) and a YD neu Id1–/–Id3+/– mouse (H) demonstrating HIF-1α staining next to necrotic areas in the solid tumor. (Magnifications: A–D, ×100; E–H, ×200.)
HIF-1α Is Up-Regulated in Id-Deficient Tumors. HIF-1 is a heterodimeric transcription factor regulated by oxygen concentration. The HIF-1α component is strongly induced by hypoxia, whereas it is not detectable in normoxic cells because of continuous degradation via ubiquitination (17). We used HIF-1α expression as a marker for hypoxia in YD neu Id WT and Id-deficient tumors of various sizes. In small solid lesions (<0.5 cm diameter), strong HIF-1α staining was observed in 50% (6/12) of tumors arising in an Id-deficient background, but in none of the similarly sized Id WT controls (0/12) (P < 0.01, Fig. 4 E and F). In large solid tumors (>1.5 cm in diameter), HIF-1α expression was observed surrounding areas of necrosis in both Id WT and Id-deficient mice (Fig. 4G). In large cystic lesions, HIF-1α expression was not present in the rim, suggesting that the cells in the viable component of these lesions were not under hypoxic stress (Fig. 4H).
Inhibition of Hsp90 Synergizes with Id Deficiency to Inhibit the Growth of YD neu Tumors. We hypothesize that tumors growing in an underperfused, hypoxic environment early in their development require stress-induced survival pathways to proliferate, in addition to the maintenance of the expression of the neu oncogene. As the protein chaperone Hsp90 is required for the activation of these pathways, we tested the effects of the Hsp90 inhibitor 17-AAG on the growth of these tumors. Inhibition of Hsp90 by 17-AAG results in the proteosomal degradation of its client proteins including Her2, Akt, and Raf, and treatment of YD neu mice with established mammary tumors with a single 75 mg/kg dose of 17-AAG caused a >90% reduction of neu expression in tumor cells (Fig. 5A).
Fig. 5.

Inhibition of Hsp90 cooperates with Id deficiency to inhibit the growth of YD neu tumors. (A) Immunoblot of neu demonstrating a >95% decline in neu expression in established YD neu tumors treated with one dose of 17-AAG (75 mg/kg) compared with mice treated with vehicle (EPL) only. Mice were killed 12 h after treatment. The expression of p85 phosphatidylinositol 3-kinase (PI3K), a protein that does not require Hsp90 function for stability, was unchanged after 17-AAG treatment and is included as a loading control. (B) Mean tumor volume after 8 weeks of treatment with 17-AAG (75 mg/kg) or EPL vehicle only as control. 17-AAG was significantly more effective in YD neu Id-deficient mice (two copies missing) than in Id WT mice. Error bars, SE. (C) After discontinuation of treatment, tumor regrowth was observed in all mice (each line represents change in tumor volume over time of an individual YD neu Id-deficient mouse treated with 17-AAG or EPL vehicle only as control).
Mice were treated for 3 consecutive days each week for 8 weeks. After 8 weeks of treatment, tumors in vehicle-treated YD neu Id-deficient mice were significantly larger than those in vehicle-treated YD neu Id WT mice (P = 0.04). Treatment of YD neu Id WT mice with 17-AAG resulted in only a modest growth inhibition of 48% at 8 weeks (mean tumor volume of 2,270 mm3 in vehicle-treated mice vs. 1,189 mm3 in 17-AAG-treated mice, P = 0.09, Fig. 5B). In Id-deficient mice, the activity of 17-AAG was dramatically enhanced, and 96% inhibition of growth was observed (mean tumor volume of 3,863 mm3 in vehicle-treated mice vs. 138 mm3 in 17-AAG-treated mice, P < 0.01, Fig. 5B). With discontinuation of 17-AAG, tumor progression was observed in all mice (Fig. 5C).
In solid tumors isolated from vehicle-treated mice and refractory Id WT 17-AAG-treated mice, the viable portion of the tumors was poorly differentiated, consisting of homogenous sheets of tumor cells (Fig. 6 A and C). Cells in the centers were typically quiescent whereas those at the periphery demonstrated a high proliferative index (Fig. 6 B and D). Apoptotic cells were infrequent and scattered without clear pattern (data not shown). In the vehicle-treated cystic tumors, the cells composing the viable rim possessed a similarly high proliferative index (data not shown). In contrast, in 7 of 11 17-AAG-treated tumors, a well-differentiated histological appearance was observed (4 of 4 Id-deficient tumors and 3 of 7 Id WT tumors, Fig. 6E). These tumors had a glandular morphology, a lower nuclear-to-cytoplasmic ratio, an absence of mitotic figures, and a very low proliferative index (Fig. 6 E and F). Although no clear increase in apoptotic index was observed in 17-AAG-treated mice versus vehicle only-treated mice after 8 weeks of treatment (data not shown), we cannot rule out the possibility that even slight differences in apoptosis at earlier time points during treatment can indeed contribute to the decrease in cellularity observed.
Fig. 6.

Response to 17-AAG treatment was associated with a well differentiated phenotype. (A, C, and E) Hematoxylin/eosin staining of a representative EPL-treated YD neu Id1+/+Id3+/+ mouse (A), a refractory 17-AAG-treated YD neu Id1+/+Id3+/+ mouse (C), and a responding 17-AAG-treated YD neu Id1–/–Id3+/+ mouse (E). (B, D, and F) Ki-67 staining of the same tumors demonstrating high levels of proliferation in the vehicle-treated and refractory YD neu Id1+/+Id3+/+ tumors (B and D). Seven of 11 responding 17-AAG tumors had a differentiated phenotype and a low proliferative index (F). A differentiated phenotype was not observed in any of the EPL-treated mice.
Discussion
In this article, we examined the effects of partial Id1 and Id3 deficiency on tumor development and progression in mouse mammary tumor virus YD neu transgenic mice. Those mice with activating neu mutations develop mammary carcinomas with a predictable latency period (8). In contrast to the transplantable tumor models (2), partial Id deficiency did not prevent or delay tumor onset or prolong the survival of the transgenic mice. The neu-driven tumors in these mice grew to a large size and were fatal in all cases. The vasculature of solid tumors growing in the Id-deficient mice was abnormal as compared with their Id WT littermates but distinct from the abnormal vasculature observed in the transplanted tumor model. In the neu transgenics, we did not observe a decrease in vascular density or stunted and occluded vessels, features that characterize the vascularization defect of transplanted tumors grown in an Id-deficient background. Rather, YD neu Id-deficient tumors had dilated, twisted, and highly anastomotic vessels reminiscent of the abnormalities seen within the developing neuroectoderm of Id1 and Id3 double knockout mice (2). The aberrant vascularization was observed in mice lacking one, two, or three copies of Id. This lack of a dosage effect is not uncommon with basic helix–loop–helix transcriptional factors, as they are extremely sensitive to small alterations in the balance of both activators and inhibitors (18).
Gross examination of the mammary tumors in the Id-deficient mice revealed a significantly increased proportion of cystic tumors. In these tumors a small rim of viable tumor cells was observed surrounding an avascular, nonviable core filled with hemorrhagic material. Similar phenotypes have been reported in RIP1-Tag2, VEGF-A knockout mice (19) and in tumor xenografts expressing a soluble form of Neuropilin-1 (20). This hollow core is most likely the result of necrosis or apoptosis of the central tumor cells brought about by insufficient oxygen or nutrient supply. Alternatively, cystic cavitation may be the result of vascular leakage and extravasation of fluid from the abnormal Id-deficient vasculature. In any event, the tumor cells at the periphery are viable, and their continued proliferation leads to the death of the host.
The multistage model of tumor angiogenesis predicts that tumors and metastases initially originate as small avascular masses. An angiogenic switch is then activated, leading to increased endothelial proliferation, vascular dilatation, and an acceleration of tumor growth (21). The fact that tumor growth per se is not inhibited in the YD neu Id-deficient mice suggests that these tumors are able to overcome the angiogenic defect characteristic of these mice (22). As tumors in transgenic animals develop orthotopically, they may be more adept at co-opting existing vessels from the surrounding normal mammary epithelium and stroma, allowing cells at the tumor periphery to survive. In contrast, in xenograft systems, s.c.-injected cells may depend entirely on bone marrow-derived endothelial cells for neovascularization and tumor growth. This finding has significant implications for the development of antiangiogenic therapies. It suggests that the use of xenograft models to identify agents that effectively suppress tumor angiogenesis dependent on bone marrow-derived endothelial cells may be insufficient because not all tumors are as completely reliant on bone marrow-derived precursor cells for viability. Agents with significant activity in these models may have a limited impact on tumor development or progression in transgenic models or in patients in which co-option of existing vasculature may be sufficient to allow continued tumor growth at the periphery. Further, these findings provide strong rationale for the use of antiangiogenic therapies in combination with other cytotoxic or targeted approaches.
In our study, we observed that 17-AAG, an inhibitor of Hsp90, was dramatically more effective in inhibiting tumors growing in a reduced Id background. Although this effect may not be unique to Hsp90 inhibitors, there are several reasons to suspect that inhibitors of Hsp90 would be particularly effective in this setting. Cells growing in the presence of a disrupted vasculature may depend more on Hsp90 chaperone function for survival given the low nutrient, low pH, hypoxic environment. Under these conditions, Hsp90 may play an enhanced role by stabilizing or refolding key signaling proteins required for cell survival. Inhibitors of Hsp90 also reduce HIF-1α expression (23, 24). As HIF-1α expression was prevalent in small Id-deficient tumors but not similarly sized Id WT counterparts, the enhanced activity of 17-AAG observed in the Id-deficient mice may be caused by impairment of HIF-1α-dependent angiogenesis. Finally, inhibition of Hsp90 leads to the degradation of neu, whose continued elevated expression may be required for tumor cell survival. Such dependence on continued oncogene expression has been observed in other systems (25). Further studies using novel model systems will be required to determine the proportion of the enhancement of 17-AAG activity attributable to Hsp90-specific effects.
In our study, we delayed treatment until tumors reached a minimum of 5 mm in size. At that size, Id-deficient tumors were already displaying the cystic phenotype at a higher frequency. As the total number of tumor cells in cystic tumors is less than that in similarly sized solid ones, the enhanced activity of 17-AAG in the Id-deficient setting may be caused in part by a greater tumor burden present in the Id WT tumors at treatment onset. In addition, in the solid tumors, a large fraction of the tumor interior consists of nondividing quiescent cells and these cells may be much less susceptible to agents such as 17-AAG that target the proliferating compartment.
In summary, we find that mammary tumors that developed in Id-deficient neu transgenic mice are larger and cystic with a viable rim of tumor cells surrounding a nonviable core of cellular debris. The efficacy of 17-AAG, an inhibitor of Hsp90, is dramatically enhanced in Id-deficient mice. These data suggest that Id deficiency cooperates with inhibition of Hsp90 function to inhibit tumor growth and provides a rationale for combination studies of angiogenic and Hsp90 inhibitors in patients with advanced breast cancers.
Acknowledgments
We thank K. Manova of the Molecular Cytology Core Facility (Sloan–Kettering Institute), T. Russo and P. Boccuni for helpful discussion, and S. Kerns of the Children's Blood Foundation Laboratories at Weill Medical College, Cornell University (Ithaca, NY), for technical assistance. The Ang2 plasmid was kindly provided by E. H. Lacy's laboratory (Sloan–Kettering Institute). This work was supported by the American Italian Cancer Foundation (P.d.C.), American Society of Clinical Oncology (D.B.S.), and grants from the Breast Cancer Research Foundation and the National Institutes of Health (to R.B. and N.R.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: GM, geldanamycin; 17-AAG, 17-allylamino-17-demethoxygeldanamycin; EPL, egg phospholipid; HIF-1, hypoxia-inducible factor 1.
References
- 1.Folkman, J. & D'Amore, P. A. (1996) Cell 87, 1153–1155. [DOI] [PubMed] [Google Scholar]
- 2.Lyden, D., Young, A. Z., Zagzag, D., Yan, W., Gerald, W., O'Reilly, R., Bader, B. L., Hynes, R. O., Zhuang, Y., Manova, K. & Benezra, R. (1999) Nature 401, 670–677. [DOI] [PubMed] [Google Scholar]
- 3.Benezra, R., Davis, R. L., Lockshon, D., Turner, D. L. & Weintraub, H. (1990) Cell 61, 49–59. [DOI] [PubMed] [Google Scholar]
- 4.Zebedee, Z. & Hara, E. (2001) Oncogene 20, 8317–8325. [DOI] [PubMed] [Google Scholar]
- 5.Slamon, D. J., Clark, G. M., Wong, S. G., Levin, W. J., Ullrich, A. & McGuire, W. L. (1987) Science 235, 177–182. [DOI] [PubMed] [Google Scholar]
- 6.Vogel, C. L., Cobleigh, M. A., Tripathy, D., Gutheil, J. C., Harris, L. N., Fehrenbacher, L., Slamon, D. J., Murphy, M., Novotny, W. F., Burchmore, M., et al. (2002) J. Clin. Oncol. 20, 719–726. [DOI] [PubMed] [Google Scholar]
- 7.Guy, C. T., Webster, M. A., Schaller, M., Parsons, T. J., Cardiff, R. D. & Muller, W. J. (1992) Proc. Natl. Acad. Sci. USA 89, 10578–10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dankort, D., Maslikowski, B., Warner, N., Kanno, N., Kim, H., Wang, Z., Moran, M. F., Oshima, R. G., Cardiff, R. D. & Muller, W. J. (2001) Mol. Cell. Biol. 21, 1540–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nollen, E. A. & Morimoto, R. I. (2002) J. Cell Sci. 115, 2809–2816. [DOI] [PubMed] [Google Scholar]
- 10.Stebbins, C. E., Russo, A. A., Schneider, C., Rosen, N., Hartl, F. U. & Pavletich, N. P. (1997) Cell 89, 239–250. [DOI] [PubMed] [Google Scholar]
- 11.Prodromou, C., Roe, S. M., O'Brien, R., Ladbury, J. E., Piper, P. W. & Pearl, L. H. (1997) Cell 90, 65–75. [DOI] [PubMed] [Google Scholar]
- 12.Sepp-Lorenzino, L., Ma, Z., Lebwohl, D. E., Vininsky, A. & Rosen, N. (1995) J. Biol. Chem. 270, 16580–16587. [DOI] [PubMed] [Google Scholar]
- 13.Munster, P. N., Srethapakdi, M., Moasser, M. M. & Rosen, N. (2001) Cancer Res. 61, 2945–2952. [PubMed] [Google Scholar]
- 14.Manova, K., Tomihara-Newberger, C., Wang, S., Godelman, A., Kalantry, S., Witty-Blease, K., De Leon, V., Chen, W. S., Lacy, E. & Bachvarova, R. F. (1998) Dev. Dyn. 213, 293–308. [DOI] [PubMed] [Google Scholar]
- 15.Donald, A. & Donner, A. (1987) Stat. Med. 6, 491–499. [DOI] [PubMed] [Google Scholar]
- 16.Maisonpierre, P. C., Suri, C., Jones, P. F., Bartunkova, S., Wiegand, S. J., Radziejewski, C., Compton, D., McClain, J. Aldrich, T. H., Papadopoulos, N., et al. (1997) Science 277, 55–60. [DOI] [PubMed] [Google Scholar]
- 17.Huang, L. E., Gu, J., Schau, M. & Bunn, H. F. (1998) Proc. Natl. Acad. Sci. USA 95, 7987–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herblot, S., Aplan, P. D. & Hoang, T. (2002) Mol. Cell. Biol. 22, 886–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue, M., Hager, J. H., Ferrara, N., Gerber, H. P. & Hanahan, D. (2002) Cancer Cell 1, 193–202. [DOI] [PubMed] [Google Scholar]
- 20.Gagnon, M. L., Bielenberg, D. R., Gechtman, Z., Miao, H. Q., Takashima, S., Soker, S. & Klagsbrun, M. (2000) Proc. Natl. Acad. Sci. USA 97, 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanahan, D. & Folkman, J. (1996) Cell 86, 353–364. [DOI] [PubMed] [Google Scholar]
- 22.Lyden, D., Hattori, K., Dias, S., Costa, C., Blaikie, P., Butros, L., Chadburn, A., Heissig, B., Marks, W., Witte, L., et al. (2001) Nat. Med. 7, 1194–1201. [DOI] [PubMed] [Google Scholar]
- 23.Mabjeesh, N. J., Post, D. E., Willard, M. T., Kaur, B., Van Meir, E. G., Simons, J. W. & Zhong, H. (2002) Cancer Res. 62, 2478–2482. [PubMed] [Google Scholar]
- 24.Isaacs, J. S., Jung, Y. J., Mimnaugh, E. G., Martinez, A., Cuttitta, F. & Neckers, L. M. (2002) J. Biol. Chem. 277, 29936–29944. [DOI] [PubMed] [Google Scholar]
- 25.Fisher, G. H., Wellen, S. L., Klimstra, D., Lenczowski, J. M., Tichelaar, J. W., Lizak, M. J., Whitsett, J. A., Koretsky, A. & Varmus, H. E. (2001) Genes Dev. 15, 3249–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]