

Abstract
Pyramidal neurons of the mammalian cerebral cortex are generated in the ventricular zone of the pallium whereas the subpallium provides the cortex with inhibitory interneurons. The marginal zone contains a subpial stream of migratory interneurons and two different classes of transient neurons, the pioneer neurons provided with corticofugal axons, and the reelin-expressing Cajal–Retzius cells. We found in cultured slices that the medial ganglionic eminence provides the reelin-negative pioneer neurons of the marginal zone. Pioneer neurons sent long projection axons that went through the cortical plate and reached the subplate and the lateral border of the lateral ganglionic eminence. In the cultured slices, pioneer neurons were functionally mature: they displayed a voltage-gated sodium current, expressed functional α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, and showed γ-aminobutyric acid type A (GABAA) postsynaptic events that were modulated by presynaptic AMPA receptors. Pioneer neurons expressed the adhesion molecules L1 and TAG-1; the latter has been reported to control tangential migrations to the neocortex [Denaxa, M., Chan, C.-H., Schachner, M., Parnavelas, J. & Karagogeos, D. (2001) Development (Cambridge, U.K.) 128, 4635–4644], and we show here that the pioneer neurons of the marginal zone are the cellular substrate of such a function. Finally, we show that, in early corticogenesis, reelin controls both the tangential migration of cortical interneurons toward the cortical plate and the tangential migration of pioneer neurons toward the marginal zone.
Two germinative zones of the telencephalon, the dorsal (pallial) and the ventral (subpallial) ventricular zones (VZ), provide new neurons to the cerebral cortex. Postmitotic cells from the dorsal VZ differentiate into excitatory projection neurons after a radial migration whereas the ventral part of the VZ, including the lateral (LGE), medial (MGE), and caudal ganglionic eminences, generates neurons of the basal ganglia and cortical interneurons (1–7). The cortical neurons, born early to form the preplate, are thought to function as a scaffold for the assembly of the cortical architecture (8–12). Other cortical neurons, born later, migrate radially and insert themselves into the preplate to generate the cortical plate (CP), which splits the preplate into the marginal zone (MZ) and the subplate (SP) (8, 11).
Cajal–Retzius cells, which reside in the preplate and thereafter in the MZ, are considered to be the earliest neurons to differentiate in the developing neocortex (8). These cells secrete reelin (13–19), an extracellular matrix glycoprotein that is crucial for cortical lamination (20, 21). Pioneer neurons are also preplate-derived; they guide thalamocortical afferent axons into the cortex and cortical efferent axons to their subcortical targets (10, 11). Recently, we described a population of pioneer neurons in the MZ (17, 18, 22, 23), but their origins, their molecular properties, and their functions remained unexplored (12, 24). We here characterize the MZ pioneer neurons by combining cell tracers and cell-specific markers with patch-clamp recordings. We found that the adhesion molecules TAG-1 and L1 are reliable markers of such neurons. We show that MZ pioneer neurons migrate tangentially to the MZ from subpallial sources. We demonstrate that such a migration is reelin-dependent and, in addition, that reelin controls the arrival of MGE-derived interneurons to the CP during early corticogenesis.
Materials and Methods
Organotypic Slices. Brains were obtained from embryos of pregnant OF1 mice at embryonic day 14 (E14) (Iffa Credo; n = 64; date of plug = E0). All animals used in this study were maintained and treated according to protocols approved by the authors' institutions. Dams were killed by cervical dislocation under Isoflurane anesthesia (Belamont, Neuilly-sur-Seine, France). The uterus and embryos were removed and rapidly isolated in cold oxygenated (5% CO2 and 95% O2) artificial corticospinal fluid (in mM): 124 NaCl, 3 KCl, 1.3 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, and 2 CaCl2. Brains were embedded in 4% agar and coronally sliced (300 μm) with a vibrating microtome (VT-1000, Leica, Argenteuil, France). Slices were placed onto Millicell-CM membranes (Millipore, Bedford, MA) in 35-mm Petri dishes containing 1 ml of the following medium (Life Technologies, Cergy Pontoise, France): 50% basal medium with Eagle's salts, 25% horse normal serum, 25% Hanks' balanced salt solution, 4.5 mg/ml d-glucose, and 0.1 mM l-glutamine (25). The medium had been heated previously at 56°C for 30 min to inactivate the complement.
Adenovirus Vector. We previously described the construction, purification, and storage of CAVGFP (canine adenovirus-GFP; refs. 26 and 27). CAVGFP is an E1-deleted vector derived from the canine adenovirus serotype 2 (CAV-2), and harbors a cytomegalovirus GFP (eGFP, Clontech) expression cassette. The stock concentration was 2.5 × 1012 particles per ml with a particle to infectious unit ratio of 3- to 10:1.
MGE Injections. CAVGFP and the membrane-permeant dyes CellTracker Green (CMFDA) and CellTracker Orange (CMTMR) (Molecular Probes; see ref. 28) were both pressure injected with a glass micropipette into the LGE or MGE of one hemisphere of E14 slices, and kept for two days (2 D.I.V.). Crystals of DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate, Molecular Probes) were inserted in the MGE. Slices were then either used for electrophysiological experiments or fixed in 4% paraformaldehyde for immunohistochemistry.
Immunohistochemistry. Brains of E12–E16 OF1 mice, E12 Nkx2.1 knockout mice, and E16 and early postnatal reeler Orleans mice (including control littermates) were fixed overnight in 4% paraformaldehyde (PFA). Forty-micrometer-thick vibratome sections were obtained. Cultured slices were also sectioned into 40-μm-thick slices after PFA fixation. Primary antibodies included monoclonal antibodies CR-50 (1:500; ref. 20) and G10 (1:1,000; ref. 29) to reelin, 4D7/TAG-1 (1:100; refs. 30 and 31) to TAG-1 and 12E3 (1:1,000; ref. 32) to PSA-NCAM (polysialic acid neural cell adhesion molecule; 1:100; T. Seki, Juntendo University School of Medicine, Tokyo), and polyclonal antibodies to TAG-1 (1:1,000; K. Takeuchi, Nagoya, Japan), L1 (1:1,000; F. G. Rathjen, Berlin), Dlx (1:40; ref. 33); calretinin (1:2,000; Swant, Bellinzona, Switzerland), and calbindin (1:5,000; Swant). Secondary antibodies were conjugated to Alexa Fluor 488 or Alexa Fluor 568 (Molecular Probes) (for details, see Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org.).
Electrophysiological Recordings. Whole-cell patch-clamp recordings were performed by using an RK 300 patch-clamp amplifier (Biologic, Claix, France). GFP-expressing cells were identified under visual control by using an upright Zeiss Axioskop microscope equipped with fluorescence and infrared differential interference contrast (DIC) video-microscopy. Neurons were imaged with a CoolSnap camera (Photometrics, Tucson, AZ). The morphology of the recorded cells was visualized by adding 0.4% dextran-rhodamine (Sigma) in the pipette solution. Cells labeled both with GFP and rhodamine were the only ones taken into account. The internal solution contained (in mM): 135 KCl, 8 NaCl, 0.2 K-EGTA, 10 Na-Hepes, 10 d-glucose, 0.3 GTP, 2 Mg-ATP, 0.2 AMPc, 0.1 spermine; pH 7.3 (280 mOsm). Synaptic responses were filtered at 1–5 kHz with an eight-pole Bessel filter, digitized at 4 kHz on a TL-1 interface (Axon Instruments, Foster City, CA), and collected on an IBM-compatible computer. On- and off-line data analyses were carried out with Acquis-1 (Gérard Sadoc, Centre National de la Recherche Scientifique–Agence Nationale de Valorisation de la Recherche, France). Drug solutions were bath-applied by using a gravity-driven perfusion system. N-methyl-d-aspartate (NMDA) and d-serine were purchased from Tocris (Ellisville, MO); all other drugs and salts were obtained from Sigma.
Blocking Experiments and Controls. E14 slices received a CMTMR or DiI in the MGE and were cultured for 2 D.I.V. with blocking monoclonal antibodies (30, 31, 34–36): CR-50 (IgG subclass) to reelin (20) diluted 1:50, and 4D7/TAG-1 (IgM) to TAG-1 (30, 31) diluted 1:100. Non-blocking monoclonal antibodies to reelin and TAG-1, and to PSA-NCAM (an adhesion molecule that is expressed by migrating interneurons; see ref. 32) were used as controls: 142 (IgG) to reelin (29) diluted 1:100; 3.1C12 (IgG1) to TAG-1 (DSHB; see ref. 30) diluted 1:100; 12E3 (IgM) to PSA-NCAM (32) diluted 1:100.
Results
Identification of MZ Pioneer Neurons Derived from the Subpallium. Injections of CAVGFP (26, 27) into the MGE provided a robust cell labeling that allowed whole-cell recordings of GFP-labeled neurons (n = 52) in the MZ after 2 D.I.V. Dextran-rhodamine intracellular filling revealed that some of these neurons (n = 23) corresponded to the MZ pioneer neurons, as described elsewhere (17, 18, 22, 23). They possessed a large soma, either fusiform and oriented radially below the pia (Figs. 1A and 4 A–C) or multipolar (Fig. 1B). At E14 plus 2 D.I.V., their axon-like processes descended into the CP with collaterals in the CP and the SP.
Fig. 1.

Morphology of pioneer neurons in the MZ labeled after CAV injections in the MGE and intracellular filling with Dextran-rhodamine. (A) A neuron with a fusiform, vertically oriented perikaryon. The axon is descending and gives rise to a collateral branch that extends toward the lateral side of the slice (arrow) and some descending branches. Dendrites are not shown. (B) A large multipolar neuron endowed with a descending axon that collateralizes in the CP and the SP. (Scale bar = 50 μm.)
Fig. 4.

Electrophysiological properties of putative pioneer neurons in the MZ (E14 + 2 D.I.V.). (A) Infrared differential interference contrast (DIC) image showing a cell body located in the MZ, perpendicular to the border of the slice. (B) The same cell expresses GFP. (C) Morphology of the same cell after filling it with rhodamine. (D) Voltage steps (±20 mV, 50 ms) elicit sodium current. (E) Sample recordings of spontaneous IPSCs. Note that, under our conditions, IPSCs were seen as inward at -60 mV. Bath application of 20 μM bicuculline methiodide blocked all events. The average of 37 IPSCs is shown in the Lower Right (the decay was fitted with a single exponential). (F) In a different MZ cell, application of 5 μM AMPA increased the frequency of miniature IPSCs recorded in the presence of 1 μM tetrodotoxin and 100 μM d,l-APV. The histogram (Lower; bins of 10 s) shows the reversible increase in mIPSC frequency. (G) Cumulative probability plots of mIPSC frequency (Left) and amplitude (Right), corresponding to the cell presented in F. These plots were constructed from 110-s samples of continuous recording (58, 150, and 36 events in Ctrl, AMPA, and Wash, respectively). (H) Averaged mIPSCs before (Ctrl, 41 events) and during (AMPA, 62 events) AMPA application. Traces are superimposed (Upper) and scaled (Lower) to illustrate that AMPA has no effect on the time course of mIPSCs. (I) Summary graph showing the normalized increase in mIPSC frequency after different treatments (AMPA, KA). The effect of glutamate receptor agonists was still observable when 200–300 μM cadmium was added to the bath whereas it was not in the presence of 1 μM NBQX. (J) Summary graph showing the effect of AMPA receptor agonists (AMPA ago.) on the mean mIPSC amplitude (mIPSC amp changes). From D to J, the potential was held at -60 mV.
The large pioneer neurons labeled by dyes injected into the MGE did not express reelin (Fig. 7 G and H, which is published as supporting information on the PNAS web site), the marker for Cajal–Retzius cells (13–19) or γ-aminobutyric acid (GABA, not shown). To define the phenotype of MZ pioneer neurons in vivo, we immunostained control E16 sections for the adhesion molecule TAG-1, a marker of pioneer neurons in different areas of the developing CNS (30, 31, 36, 37). The monoclonal 4D7/TAG-1 antibody stained the axons of pioneer neurons that project to the LGE (Fig. 2 A–C), but not their cell bodies. The axons fasciculated tightly in the intermediate zone (IZ) and diverged on arrival to the LGE, where they intermingled with calbindin-positive migrating interneurons (Fig. 2 B and C). This pathway of descending axons from MZ pioneer neurons was physically separated (Fig. 2D) from the subpopulation of ascending thalamocortical fibers that are immunoreactive for calretinin and for the adhesion molecule L1 (Fig. 2E). By E16, TAG-1-immunoreactive fibers went through the CP vertically (Fig. 2E); the polyclonal antibody revealed the cell bodies in the upper CP (Fig. 2F; ref. 18).
Fig. 2.

(A) Immunohistochemistry for TAG-1 and calbindin. The calbindin antibody revealed part of the population of tangentially migrating neurons (green). Note the absence of calbindin-immunoreactive neurons in the mantle of the LGE (*). Shown are TAG-1-labeled cortical axonal projections (red), which end in the LGE. (B) High magnification of the boxed area in A, illustrating TAG-1-positive axons entering the LGE. (C) From boxed area in A: calbindin-positive interneurons are in close contact with TAG-1-positive fibers in the IZ. (D) Immunohistochemistry for TAG-1 (red) and calretinin (green) revealed two different axonal pathways that do not coincide. Calretinin is expressed by a subset of thalamocortical axons (TCAx) and TAG-1 by the corticofugal axons (CAx). (E) L1 (green) labeled a larger population of thalamocortical axons. These axons and TAG-1-positive axons only partially overlap. (F) An area similar to that boxed in D; TAG-1 is present in cell bodies in the upper CP; see also E. (Scale bars: A, 200 μm; B,60 μm; C,10 μm; D,40 μm; E, 30 μm; and F, 20 μm.)
At E12, TAG-1 was detected in numerous cell bodies of the mantle layers of the MGE (Fig. 8, which is published as supporting information on the PNAS web site) and the LGE (Fig. 3A and Fig. 9A, which is published as supporting information on the PNAS web site). Thus, TAG-1 is a marker of pioneer cell bodies in the MZ and of their axonal projection. However, TAG-1 did not seem to be an exclusive marker of pioneer neurons, because a small population of reelin-positive cells of the upper preplate colocalized TAG-1 (Fig. 3C). In addition, the adhesion molecule L1 was found in preplate cell bodies at E12 (Fig. 3D) but not at later stages. Compared with TAG-1 immunoreactive cells, the L1-labeled cells seemed more densely packed in the preplate. The LGE was densely populated by L1-immunoreactive cells at this stage (Fig. 3E; Fig. 9B). L1 also was present in MZ axons that terminated in growth cones in the LGE (Fig. 3F, arrows) and colocalized with reelin in certain cases (Fig. 3 G–I). Overall, the data indicated that L1 is a transient marker of pioneer neurons of the preplate.
Fig. 3.

Immunohistochemical experiments at E12 support the subcortical origin of MZ pioneer neurons. (A) Cell bodies immunoreactive for TAG-1 populate the mantle of the LGE and form a continuum toward a putative pioneer migratory stream to the neocortex. (B) TAG-1-immunoreactive cell bodies in the neocortical preplate (PP). (C) TAG-1-immunoreactive cell bodies and dendrites (green) lie deep in the preplate, deeper than reelin-immunoreactive neurons (red). A red arrow points to a reelin-immunoreactive cell in the MZ; in rare instances, reelin and TAG-1 colocalize (yellow arrow). (D) Cell bodies immunoreactive for L1 in the preplate (PP). Note the immunoreactive neurites descending toward the VZ. (E) L1-immunoreactive cell bodies populate the mantle of the MGE. (F) Conspicuous growth cones (arrows) of cortical efferent axons (CAx) in the LGE, immunoreactive for L1. (G–I) L1-immunoreactive cell bodies lie deep in the preplate (PP). Reelin-immunoreactive cells are at the pial surface, and, in rare instances, reelin colocalizes with L1 (arrows). [Scale bars: A, 50 μm; B, 25 μm; C, 15 μm; D, 40 μm; E, 30 μm; F, 20 μm; and G–I, 10 μm (bar in I).]
The distribution of these cells in the MGE and the LGE suggested a pioneer migratory stream to the neocortical preplate (Figs. 3 A and E and 9 A and B). Our results pointed at the MGE as a possible source of the MZ pioneer neurons. Next, we explored Nkx2.1 knockout mice at E12 for the presence of such cells in the subpallium and in the MZ and found no alterations in the distribution of TAG-1 immunoreactive cells (Fig. 10, which is published as supporting information on the PNAS web site). Therefore, we searched for a more caudal origin of the migratory stream and found that the thalamic eminence (38), the MGE, the LGE, and the cortical MZ (Fig. 8) contained a continuum of abundant TAG-1-immunoreactive cells.
Patch-Clamp Recordings of Pioneer Neurons of the MZ. CAVGFP-labeled pioneer neurons (Fig. 4 A–C) were recorded in the whole-cell configuration. The average resting membrane potential, input membrane resistance, and cell membrane capacitance of these cells were -49.6 ± 2.4 mV, 1.07 ± 0.15 GΩ, and 20.8 ± 1.7 pF, respectively (n = 22), which is not significantly different from other immature neurons (P > 0.05). However, most of these neurons (16/22 tested cells) displayed large enough sodium current to trigger spikes (Fig. 4D). NMDA (100 μM) coapplied with d-serine (20 μM) generated an inward current of -30 ± 10 pA at -60 mV in 3 of 5 neurons (data not shown). The presence of functional non-NMDA receptors was revealed by bath application of 25 μM kainate (or 5 μM AMPA) that induced a current of -52 ± 7 pA at -60 mV (n = 12/12, not shown). These receptors belonged mainly to the AMPA receptor subclass because 1 μM 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f]quinoxaline (NBQX) blocked all evoked responses (n = 4, data not shown), and 100 μM cyclothiazide, known to slow down the desensitization of AMPA receptors, increased this current (n = 5, a 3.2-fold increase, data not shown). In the majority of recorded neurons (16 of 20), spontaneous inhibitory postsynaptic currents (sIPSCs) or miniature inhibitory postsynaptic currents (mIPSCs) were detected. mIPSCs had mean amplitude of -15.6 ± 1.5 pA, and their decay time was fitted with a single exponential (time constant of 19.1 ± 1.9 ms; n = 15, data not shown). Fig. 4E shows a typical recording where sIPSCs recorded at -60 mV were totally blocked by a specific GABAA receptor antagonist, bicuculline methiodide (20 μM, n = 8). In addition, bath application of 25 μM muscimol revealed the presence of extrasynaptic GABAA receptors in cells that did not exhibit IPSCs (-354 ± 152 pA at -60 mV; n = 5).
MZ pioneer neurons displayed IPSCs that were modulated by ionotropic glutamate receptor activation. Fig. 4F shows examples of mIPSCs recorded in a cell under control conditions (1 μM tetrodotoxin and 100 μM d,l-APV), in the presence of 5 μM AMPA and after drug washout. AMPA application increased the frequency of mIPSCs in this cell (Fig. 4 F, G, and I), as well as in all other tested cells (n = 12). The mean mIPSC frequency was 4.24 ± 1.35 Hz before and 40.33 ± 0.08 Hz during AMPA treatment (Fig. 4I; n = 12; P < 0.004 with the Wilcoxon test). In addition, the mean mIPSC amplitude (-15.7 ± 1.5 pA) was also significantly increased after AMPA treatment (-28.5 ± 3.3 pA, n = 12; P < 0.005 with the Wilcoxon test) (Fig. 4 F, H, and J). However, AMPA had no effect on the kinetics of the mIPSCs (Fig. 4H). The AMPA-induced effect on mIPSCs frequency persisted throughout the AMPA treatment (Fig. 4F), was dose-dependent (not shown), was mimicked by kainate (Fig. 4I), and was fully reversible after washout (Fig. 4 F and G). Kainate (25 μM) increased the mean mIPSC frequency from 0.60 ± 0.17 Hz to 5.65 ± 3.23 Hz (n = 4; P < 0.004). At 50 μM kainate, the mean frequency increased from 0.18 ± 0.05 Hz to 6.32 ± 3.02 Hz (n = 3; P < 0.004). To further characterize the pharmacology of this modulation induced by activation of non-NMDA receptors, the AMPA/kainate receptor antagonist NBQX (1 μM) was applied before kainate treatment. At this concentration, this antagonist blocks virtually all AMPA receptors without affecting kainate receptors (see ref. 39). Under such conditions, kainate was unable to modulate the frequency of GABA release, indicating that AMPA receptors have a specific role in the regulation of GABA release (Fig. 4I; n = 3). The effect of AMPA/kainate receptors was not mediated by calcium influx through voltage-sensitive calcium channels. When cadmium (200–300 μM) was used to block voltage-dependent calcium channels, AMPA (5 μM) or kainate (25–50 μM) still increased the frequency of mIPSCs (from 0.35 ± 0.16 Hz to 3.82 ± 1.97 Hz; n = 5; P < 0.05 with the Wilcoxon test; Fig. 4I). This finding demonstrates that the enhancement of GABA release from terminals impinging onto pioneer neurons involves a direct action of presynaptic calcium-permeable AMPA receptors. An alternative explanation is that this effect results from metabotropic action of AMPA receptor activation as it has been suggested for other synapses.
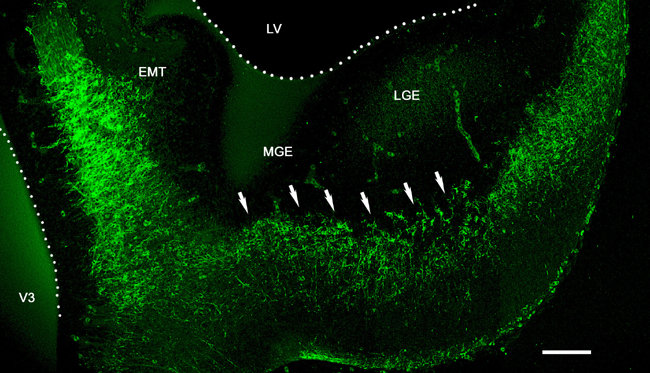
Blocking Experiments Indicated That Reelin Controls Tangential Migrations to the Neocortex. Slices were incubated with blocking monoclonal antibodies 4D7/TAG-1 to TAG-1 (36) and CR–50 to reelin. When CMTMR was injected into the MGE of control slice cultures (Fig. 5A), labeled cells scattered in the SP, CP, and MZ of the neocortex (Fig. 5B). Slices treated during 48 h either with CR–50 (10/10 slices; Fig. 5C) or with 4D7/TAG-1 (10/12 slices; Fig. 5D) showed no migration. The effect of CR–50 on the distances traveled by the migrating cells, assayed with CMTMR or DiI, was stronger than that of 4D7/TAG-1. In CR–50-treated slices, the cells never reached the neocortex (Fig. 5C) whereas cells entered the IZ in the presence of 4D7/TAG-1 antibody (Fig. 5D). Interestingly, both blocking antibodies completely arrested the migration of MZ pioneer neurons and of CP interneurons. As a control, a 48-h treatment with a different, non-blocking monoclonal antibody to reelin (mAb 142, 8/8 slices) did not reduce the population of migrating cells in the neocortex (Fig. 5E). Similarly, incubation with different non-blocking monoclonal antibodies to TAG-1 (3.1C12; 8/9 slices; data not shown), or with the IgM mAb 12E3 (32) to PSA-NCAM (6/6 slices, Fig. 5F), even at a higher titer (1/100), did not arrest the migrations. Because the possible role of reelin in the regulation of the tangential migration was an unexpected finding, we explored embryos and early postnatal reeler Orleans mice and control littermates for MGE-derived interneurons, which express the transcription factor Dlx (1, 2). Differences were again dramatic: in E16 reeler mice (Fig. 6 A and B), we found abundant labeled cell nuclei in the superficial tier of the cortex (corresponding to the superplate) and a net decrease in the packing density of labeled cells in the IZ as compared with controls. The difference was even more evident at P0 (Fig. 6 C and D): whereas in normal mice Dlx-immunoreactive cells were densely packed in the MZ, in the lower CP and SP and in the IZ, reeler littermates showed densely packed Dlx-positive cells in the superplate, and a lower packing density in lower levels of the cortex.
Fig. 5.

Reelin and TAG-1 are both required for the tangential migration of MGE-derived neurons. (A) Diagram of a forebrain slice illustrating the injection point (gray) of CMTMR or DiI in the MGE. (B) Migrating neurons invade the cortex in a control slice; CMTMR. (C) A cultured slice, labeled with DiI and incubated with the blocking antibody CR-50, showed arrest of the migration at the basal ganglia. (D–F) CMTMR. (D) Incubation with the blocking antibody 4D7/TAG-1 showed a milder arrest of the migration, as compared with C. Incubation with the monoclonal antibody 142 to reelin (E) or the monoclonal antibody 12E3 to PSA-NCAM (F) did not impair the migration. (Scale bars: B, 100 μm; C and D, 200 μm; and E and F, 50 μm.)
Fig. 6.

Distribution of Dlx-immunoreactive cortical interneurons in reeler mice. (A and B) Dlx-immunoreactive neurons are more densely packed in the cortex of E16 control mice (A) than in their reeler littermates (B). (C and D) The difference in packing density of Dlx-immunoreactive interneurons is even more dramatic at P0: control mice show immunoreactive interneurons in the MZ and in the lower tiers of the CP and in the SP, as well as in the IZ (C), whereas most immunoreactive interneurons concentrate in the superplate (SPP) of reeler littermates (D). (Scale bar = 100 μm.)
Discussion
Using slice cultures of embryonic mouse brains and neurochemical markers, we demonstrated that the pioneer neurons of the neocortical MZ are of subpallial origin. We show that these MZ pioneer neurons express functional characteristics of mature neurons. They are endowed with functional ionotropic receptors such as glutamate NMDA and non-NMDA receptors and GABAA receptors. They exhibit a sodium conductance, generate action potentials, and receive synaptic inputs. Furthermore, we found that MZ pioneer neurons express the adhesion molecules L1 and TAG1. Based on the use of blocking antibodies, we propose that reelin controls both the tangential migration of pioneer neurons to the neocortical MZ and the tangential migration of cortical interneurons to the cortical plate during early corticogenesis.
Subpallial Origins of MZ Pioneer Neurons. Initially, MZ pioneer neurons were postulated to originate in the pallial VZ (17). The present experiments using CAVGFP and CellTrackers injections in the MGE unambiguously pointed to their subpallial origins. However, the fact that the TAG-1-labeled pioneer migratory stream remained unaltered in Nkx2.1 knockout mice, where the MGE is compromised, made us examine more closely the distribution of TAG-1-immunoreactive cells in the early stages of corticogenesis. The finding of a continuum of TAG-1-immunoreactive cells, from the thalamic eminence to the ganglionic eminences and to the MZ, confirmed the subpallial origin of this neuronal population and pointed at the thalamic eminence as a possible source of the migration.
Neurophysiological Properties of MZ Pioneer Neurons. At midgestation, MZ pioneer neurons receive functional GABAA-mediated synaptic inputs. These inputs most likely originate from GABAergic fibers located in the MZ (6, 40) although their exact origin cannot be elucidated unequivocally. These GABAA-mediated events may have a depolarizing action as previously suggested during early cortical development (41, 42). Although the low frequency and moderate amplitude of IPSCs indicate a small number of synapses and a low density of associated channels, the high input resistance of MZ pioneer neurons should amplify the efficiency of these synapses. Furthermore, the activation of presynaptic AMPA receptors located on GABAergic terminals seems also to represent a highly efficient mechanism by which GABA release could be enhanced. In this respect, glutamate has been shown to be involved in several developmental processes, such as cell proliferation (43) and radial migration (44). Although embryonic cortical cells express a variety of glutamate receptor subunits (45–47), the distribution of most glutamate receptor subunits in the developing cortex remains elusive. Our data indicate that, because presynaptic AMPA receptors are at least partly permeable to calcium, they either lack the specific GluR2 subunit or express a nonedited form (48).
Does Reelin Control Tangential Migrations? Our results demonstrate an unexpected function of reelin in corticogenesis (see ref. 21). The incubation of E14 slices with the blocking CR-50 antibody caused a failure in the tangential migration of virtually all classes of MGE-derived neurons, including MZ pioneer neurons, and a descent of CP interneurons was observed in mutant reeler mice at prenatal and early postnatal stages. The underlying mechanism remains unknown, and it will be fruitful to analyze tangential migrations in reeler mice. In Tbr-1 mutants, where expression of reelin is compromised, the distribution of cortical GABAergic cells is not strictly normal (24). In addition, a recent study (49) has presented evidence that Emx1/2 double mutant cerebral cortex has no Cajal–Retzius cells and shows an altered tangential migration. Migrating interneurons do not express molecules of the reelin signaling cascade (50). However, the intracellular adaptor protein Dab1 is expressed by large neurons located in the MZ (50) that are morphologically comparable to the pioneer neurons described in the present study.
Our blocking experiments confirmed, in accordance with previous results (36), that 4D7/TAG-1 mAb interfered with tangential migrations to the cortex. We have further found evidence suggesting that the pioneer neurons in the MZ are the cellular substrate of TAG-1 functions in regulating migrations. TAG-1 is present in cell body and processes of MZ cells before it is detected in descending axonal projections from that layer. Thus, we show that TAG-1 is expressed by the pioneer neurons of the MZ that derive from the MGE. In addition, we provide clear-cut evidence that no MGE-derived neurons arrive to the MZ under TAG-1-blocking conditions and that blocking TAG-1 arrests both migrations of interneurons and pioneer neurons to this layer. Altogether, these findings point to an unexpected degree of complexity of the tangential migrations of neurons toward the neocortex.
Supplementary Material
Acknowledgments
We thank A. Goffinet, M. Ogawa, T. Seki, K. Takeuchi, F. G. Rathjen, and J. Kohtz for antibodies; C. Soudais and S. Boutin for assistance using the virus; C. Gil-Sanz for assistance with immunocytochemistry; J.-L. Guenet for breeding pairs of reeler mice; and O. Marín for Nkx2.1 knockout mice tissues. Monoclonal antibodies 4D7/TAG1 and 3.1C12 were obtained from the Developmental Studies Hybridoma Bank, University of Iowa, Iowa City. We thank A. Alvarez-Buylla, O. Marín, S. Rétaux, and J. Rubenstein for comments on previous versions of this manuscript. This work was supported by the Fondation Annette Gruner-Schlumberger, the French Ministry of Research and Education (Action Concertée Incitative Biologie du Développement et Physiologie Intégrative 2000), and the Pasteur Institute (P.-M.L.), the Centre National de la Recherche Scientifique (P.-M.L. and E.J.K.), the Association Française contre les Myopathies (E.J.K.), and Spanish Ministry of Science and Technology Grants PB97-0582-CO2-01, PGC2000-2756-E, and BFI2001-1504 (to A.F.).
Abbreviations: CAVGFP, canine adenovirus-GFP; CP, cortical plate; DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; D.I.V., days in vitro;En, embryonic day n; GABA, γ-aminobutyric acid; LGE, lateral ganglionic eminence; IPSC, inhibitory postsynaptic current; MZ, marginal zone; MGE, medial ganglionic eminence; NBQX, 1,2,3,4-tetrahydro-6-nitro-2,3-dioxobenzo[f]quinoxaline-7-sulfonamide; mIPSC, miniature IPSC; sIPSC, spontaneous IPSC; SP, subplate; VZ, ventricular zone; NMDA, N-methyl-d-aspartate; CMTMR, CellTracker orange; IZ, intermediate zone; PSA-NCAM, polysialic acid neural cell adhesion molecule.
References
- 1.Anderson, S. A., Eisenstat, D. D., Shi, L. & Rubenstein, J. L. (1997) Science 278, 474-476. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, S. A., Marin, O., Horn, C., Jennings, K. & Rubenstein, J. L. (2001) Development (Cambridge, U.K.) 128, 353-363. [DOI] [PubMed] [Google Scholar]
- 3.Lavdas, A. A., Grigoriou, M., Pachnis, V. & Parnavelas, J. G. (1999) J. Neurosci. 19, 7881-7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He, W., Ingraham, C., Rising, L., Goderie, S. & Temple, S. (2001) J. Neurosci. 21, 8854-8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marín, O., Yaron, A., Bagri, A., Tessier-Lavigne, M. & Rubenstein, J. L. (2001) Science 293, 872-875. [DOI] [PubMed] [Google Scholar]
- 6.Wichterle, H., Turnbull, D. H., Nery, S., Fishell, G. & Alvarez-Buylla, A. (2001) Development (Cambridge, U.K.) 128, 3759-3771. [DOI] [PubMed] [Google Scholar]
- 7.Nery, S., Fishell, G. & Corbin, J. G. (2002) Nat. Neurosci. 5, 1279-1287. [DOI] [PubMed] [Google Scholar]
- 8.Marín-Padilla, M. (1971) Z. Anat. Entwicklungs-gesch. 134, 117-145. [DOI] [PubMed] [Google Scholar]
- 9.McConnell, S. K., Ghosh, A. & Shatz, C. J. (1989) Science 245, 978-982. [DOI] [PubMed] [Google Scholar]
- 10.de Carlos, J. A. & O'Leary, D. D. (1992) J. Neurosci. 12, 1194-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allendoerfer, K. L. & Shatz, C. J. (1994) Annu. Rev. Neurosci. 17, 185-218. [DOI] [PubMed] [Google Scholar]
- 12.Landry, C. F., Pribyl, T. M., Ellison, J. A., Givogri, M. I., Kampf, K., Campagnoni, C. W. & Campagnoni, A. T. (1998) J. Neurosci. 18, 7315-7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Arcangelo, G., Miao, G. G., Chen, S. C., Soares, H. D., Morgan, J. I. & Curran, T. (1995) Nature 374, 719-723. [DOI] [PubMed] [Google Scholar]
- 14.D'Arcangelo, G., Nakajima, K., Miyata, T., Ogawa, M., Mikoshiba, K. & Curran, T. (1997) J. Neurosci. 17, 23-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schiffmann, S. N., Bernier, B. & Goffinet, A. M. (1997) Eur. J. Neurosci. 9, 1055-1071. [DOI] [PubMed] [Google Scholar]
- 16.Alcántara, S., Ruiz, M., D'Arcangelo, G., Ezan, F., de Lecea, L., Curran, T., Sotelo, C. & Soriano, E. (1998) J. Neurosci. 18, 7779-7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer, G., Soria, J. M., Martinez-Galan, J. R., Martin-Clemente, B. & Fairén, A. (1998) J. Comp. Neurol. 397, 493-518. [PubMed] [Google Scholar]
- 18.Meyer, G., Goffinet, A. M. & Fairén, A. (1999) Cereb. Cortex 9, 765-775. [DOI] [PubMed] [Google Scholar]
- 19.Hevner, R. F., Neogi, T., Englund, C., Daza, R. A. M. & Fink, A. (2003) Dev. Brain Res. 141, 39-53. [DOI] [PubMed] [Google Scholar]
- 20.Ogawa, M., Miyata, T., Nakajima, K., Yagyu, K., Seike, M., Ikenaka, K., Yamamoto, H. & Mikoshiba, K. (1995) Neuron 14, 899-912. [DOI] [PubMed] [Google Scholar]
- 21.Rice, D. S. & Curran, T. (2001) Annu. Rev. Neurosci. 24, 10005-10039. [DOI] [PubMed] [Google Scholar]
- 22.Soria, J. M. & Fairén, A. (2000) Cereb. Cortex 10, 400-412. [DOI] [PubMed] [Google Scholar]
- 23.Fairén, A., Morante-Oria, J. & Frassoni, C. (2002) Prog. Brain Res. 136, 281-291. [DOI] [PubMed] [Google Scholar]
- 24.Hevner, R. F., Shi, L., Justice, N., Hsueh, Y., Sheng, M., Smiga, S., Bulfone, A., Goffinet, A. M., Campagnoni, A. T. & Rubenstein, J. L. (2001) Neuron 29, 353-366. [DOI] [PubMed] [Google Scholar]
- 25.Gähwiler, B. H., Thompson, S. M., McKinney, R. A., Debanne, D. & Robertson, R. T. (1998) in Culturing Nerve Cells, eds. Banker, G. & Goslin, K. (MIT Press, Cambridge, MA).
- 26.Kremer, E. J., Boutin, S., Chillon, M. & Danos, O. (2000) J. Virol. 74, 505-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soudais, C., Laplace-Builhe, C., Kissa, K. & Kremer, E. J. (2001) FASEB J. 15, 2283-2285. [DOI] [PubMed] [Google Scholar]
- 28.Nadarajah, B., Alifragis, P., Wong, R. O. & Parnavelas, J. G. (2002) Nat. Neurosci. 5, 218-224. [DOI] [PubMed] [Google Scholar]
- 29.de Bergeyck, V., Naerhuyzen, B., Goffinet, A. M. & Lambert de Rouvroit, C. (1998) J. Neurosci. Methods 82, 17-24. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto, M., Boyer, A. M., Crandall, J. E., Edwards, M. & Tanaka, H. (1986) J. Neurosci. 6, 3576-3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dodd, J., Morton, S. B., Karagogeos, D., Yamamoto, M. & Jessell, T. M. (1988) Neuron 1, 105-116. [DOI] [PubMed] [Google Scholar]
- 32.Seki, T. & Arai, Y. (1991) Anat. Embryol. 184, 395-401. [DOI] [PubMed] [Google Scholar]
- 33.Kohtz, J. D., Lee, H. Y., Gaiano, N., Segal, J. D., Ng, E., Larson, T. A., Baker, D. P., Garber, E. A., Williams, K. P. & Fishell, G. (2001) Development (Cambridge, U.K.) 128, 2351-2363. [DOI] [PubMed] [Google Scholar]
- 34.Nakajima, K., Mikoshiba, K., Miyata, T., Kudo, C. & Ogawa, M. (1997) Proc. Natl. Acad. Sci. USA 94, 8196-8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yip, J. W., Yip, Y. P., Nakajima, K. & Capriotti, C. (2000) Proc. Natl. Acad. Sci. USA 97, 8612-8616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Denaxa, M., Chan, C.-H., Schachner, M., Parnavelas, J. & Karagogeos, D. (2001) Development (Cambridge, U.K.) 128, 4635-4644. [DOI] [PubMed] [Google Scholar]
- 37.Wolfer, D. P., Henehan-Beatty, A., Stoeckli, E. T., Sonderegger, P. & Lipp, H. P. (1994) J. Comp. Neurol. 345, 1-32. [DOI] [PubMed] [Google Scholar]
- 38.Kim, A. S., Anderson, S. A., Rubenstein, J. L., Lowenstein, D. H. & Pleasure, S. J. (2001) J. Neurosci. 21, RC132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bureau, I., Bischoff, S., Heinemann, S. F. & Mulle, C. (1999) J. Neurosci. 19, 653-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dammerman, R. S., Flint, A. C., Noctor, S. C. & Kriegstein, A. R. (2000) J. Neurophysiol. 84, 428-434. [DOI] [PubMed] [Google Scholar]
- 41.Cherubini, E., Gaiarsa, J. L. & Ben-Ari, Y. (1991) Trends Neurosci. 14, 515-519. [DOI] [PubMed] [Google Scholar]
- 42.Ben-Ari, Y. (2002) Nat. Rev. Neurosci. 3, 728-739. [DOI] [PubMed] [Google Scholar]
- 43.LoTurco, J. J., Owens, D. F., Heath, M. J., Davis, M. B. & Kriegstein, A. R. (1995) Neuron 15, 1287-1298. [DOI] [PubMed] [Google Scholar]
- 44.Komuro, H. & Rakic, P. (1993) Science 260, 95-97. [DOI] [PubMed] [Google Scholar]
- 45.Hollmann, M. & Heinemann, S. (1994) Annu. Rev. Neurosci. 17, 31-108. [DOI] [PubMed] [Google Scholar]
- 46.Bochet, P., Audinat, E., Lambolez, B., Crepel, F., Rossier, J., Iino, M., Tsuzuki, K. & Ozawa, S. (1994) Neuron 12, 383-388. [DOI] [PubMed] [Google Scholar]
- 47.López-Bendito, G., Shigemoto, R., Fairén, A. & Luján, R. (2002) Cereb. Cortex 12, 625-638. [DOI] [PubMed] [Google Scholar]
- 48.Métin, C., Denizot, J. P. & Ropert, N. (2000) J. Neurosci. 20, 696-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shinozaki, K., Miyagi, T., Yoshida, M., Miyata, T., Ogawa, M., Aizawa, S. & Suda, Y. (2002) Development (Cambridge, U.K.) 129, 3479-3492. [DOI] [PubMed] [Google Scholar]
- 50.Luque, J. M., Morante-Oria, J. & Fairén, A. (2003) Dev. Brain Res. 140, 195-203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}