

Abstract
RNA 77, derived by selection amplification, accelerates its own conversion to Phe-RNA (relative to randomized RNA) more than 6 × 107-fold, by using amino acid adenylates as substrate. A modified assay system allows measurement of slow rates of aa-RNA formation, which for disfavored amino acid substrates can be more than 104-fold slower than phenylalanine. Thus unlike previously characterized self-aminoacylators, RNA 77 catalysis is highly amino acid selective. Remarkably, both rates of aminoacyl transfer and amino acid specificities are greater for RNA 77 than measured for protein PheRS. These data experimentally support the possible existence of an ancestral amino acid-specific translation system relying entirely on RNA catalysis. RNA 77 itself embodies a possible transitional evolutionary state, in which side-chain-specific aa-RNA formation and anticodon-codon pairing were invested in the same molecule.
RNAs have been selected (1) that catalyze their own 2′(3′) aminoacylation by using amino acid adenylates as a substrate. Aminoacyl-adenylates (aa-AMPs) are the universal carboxy-activated precursors of modern aa-tRNAs. Because the products of RNA catalysis are (2′, 3′) aa-RNAs, chemically like aa-tRNAs, these RNA catalysts use biological precursors and make biological products, performing a reaction that parallels acylation of RNA by modern protein aa-tRNA synthetases. In addition, the current universality of adenylates and aa-RNAs suggests that they were used for translation by the last common ancestor of life on Earth. Thus self-aminoacylating RNAs also catalyze an essential translational reaction that has persisted at least since the last common ancestor.
Self-aminoacylating RNAs also are ribozymes that perform carbonyl chemistry rather than the phosphoryl chemistry characteristic of natural ribozymes. This characteristic means that the tetrahedral, polar transition states characteristic of nucleophilic attack at carbonyl carbon are stabilized by RNAs, and that therefore other reactions with the same transition state (e.g., acyl transfers) are likely to be found. Examples of such reactions now include slight accelerations of aminoacyl ester hydrolysis (2) and amide hydrolysis (3, 4), which occur as apparent side reactions within derivatives of group I self-splicing RNAs, as well as more rapid and specifically selected amide synthesis (5), aminoacyl ester transfer (6), and peptide formation (7). Such reactions are of more general evolutionary importance because ribosomal peptidyl transferase (8) is a related reaction.
In one respect, previously characterized RNAs from the RNA 19–RNA 29 family, coselected sequences but of independent origin (9), are unlike protein aa-tRNA synthetases. Neither the originally selected 95-mer nor a small 43-nt active RNA (10) distinguish among amino acids in their substrates, giving acylation velocities of the same order whether supplied with Phe-AMP (used in the initial selection) or other aa-AMPs. Lack of amino acid specificity would be a defect in a primordial RNA aa-RNA synthetase, as it apparently would prevent encoding of specific peptides.
We now remove this apparent limitation, showing that RNA can catalyze highly specific aa-RNA synthesis. In addition, the specificity of aa-RNA 77 synthesis has unusual characteristics, worth particular discussion.
EXPERIMENTAL PROCEDURES
Internally Labeled RNA.
RNA was transcribed in the presence of [α-32P]GTP and purified to single nucleotide resolution by gel electrophoresis (11).
3′ Terminally Labeled RNA.
RNA 77 and RNA 29 with a single 32P 5′ to the 3′ terminal nucleotide was prepared by ligating [5′-32P]pGp to an unlabeled transcript (12) from a template shortened by one 3′ nucleotide.
[5′-32P]pGp was synthesized by incubating 1 nmol 3′ GMP, 100 pmol [γ-32P]ATP (6,000 Ci/mmol, NEN) with T4 polynucleotide kinase in 7 μl of kinase buffer (3.5 units; GIBCO/BRL) at 37°C for 30 min. Polynucleotide kinase then was heat inactivated (65°C, 15 min); subsequent chromatography showed complete conversion of ATP to pGp.
RNA lacking its 3′ nucleotide was prepared by transcription of a shortened template (9).
Gel-purified, denatured, 3′ truncated RNA (200 pmol) was incubated with 100 pmol [32P]pGp and 25 units of T4 RNA ligase (Epicentre Technologies, Madison, WI) in a total of 25 μl also containing 50 mM Tris⋅HCl (pH 7.5), 10 mM MgCl2, 10 mM β-mercaptoethanol, 0.2 mM ATP, 0.1 mg/ml BSA, and 15% DMSO at 37°C for 1 hr. The RNA product was ethanol-precipitated, and the 3′ phosphate group was removed by incubation with 15 units of T4 polynucleotide kinase (GIBCO/BRL) in 5 mM MgCl2, 10 mM β-mercaptoethanol, and 20 mM Mes, pH 5.5 at 37°C for 30 min.
Acylations.
Aminoacylation reactions (0°C) have been described (10). For RNA 77, these contained 50 mM CaCl2, 50 mM NaCl, and 0.2 M Hepes or Mes at the desired pH. For RNA 29, reactions contained 20 mM CaCl2, 40 mM MgCl2, 50 mM NaCl, and 0.2 M Hepes, pH 7.25. Reactions were initiated by adding synthetic adenylates (1) and terminated by adding 30 vol of 0.1 M NaOAc, 78% (vol/vol) ethanol followed by immediate quenching in a dry ice bath. After collection of the precipitate, reactions were incubated with 2.4 units of nuclease P1 (Amersham Pharmacia, Sigma) in 3 μl of 10 mM NaOAc (pH 5.2) at 23°C for 30 min. The products were reliably single nucleotides, which were resolved by TLC on polyethyleneimine-cellulose plates (J.T. Baker) in 0.2 M NH4OAc at pH 3.5.
Curve Fitting.
Fraction aa-RNA versus time was fit to kinetics that take account of the second-order reaction of adenylates and RNA and are also correct for the instability of the adenylates, where required (10). In addition, the final extent of the reaction was made a parameter derived from fitting, so as to bring all data to bear on the measurement of reaction extents. Fits are from nonlinear matching to data by the Marquardt algorithm, in axum (version 2.0, TriMetrix, Seattle, WA).
Secondary Structure.
RNA at 0.25 μM was heated to 65° for 5 min in distilled water. Then 50 mM CaCl2 and NaCl were added and incubation was continued at room temperature for 5 min. The solution was placed on ice and made 0.1 M in Hepes, pH 7.0.
At this point Pb(OAc)2 was added to 0, 1, 2.5, and 5 mM in a final volume of 10 μl, and incubation was continued on ice for 90 min. Final RNAs were fractionated by using 10% denaturing PAGE with NaOH ladders and T1 RNase digests alongside to identify fragments. All cuts above background are shown in Fig. 1.
Figure 1.

Secondary structures of RNA 29 and RNA 77 with lead and S1 cuts shown.
Alternatively 0, 10, 25, and 50 units of S1 nuclease (GIBCO/BRL) were added to a final volume of 10 μl and ZnCl2 to a final concentration of 50 μM. After 90 min on ice, fragments were characterized as above.
RESULTS
Structure of a Specific Catalyst.
Fig. 1 compares the primary and apparent secondary structures of RNA 77 and the previous RNA 29. RNA 77 is an independent self-aminoacylating RNA derived via the same selection for self-aminoacylation with Phe-AMP that gave the RNA 29 family. The nonamino acid-specific RNA 29 has all essential catalytic elements closely grouped around the base of its two helices, particularly including the bulged active site helix on the right of the figure (9). As shown in Fig. 1, lead and S1 nuclease probing agree well on a secondary structure for RNA 77, which also closely follows the second most stable calculated (13) secondary structure. This secondary structure radiates from a larger, more complex central three-helix core rather than the simpler two-helix structures of the RNA 19–RNA 29 family.
Assay of Six aa-RNA Syntheses by Chromatography.
Fig. 2 exhibits the nucleotide products of self-acylation. RNA 77 and RNA 29 were prepared by transcription in the presence of unlabeled nucleotides, from a DNA template that lacked the 3′ terminal G. To this shortened transcript, [α-32P]pGp was added by using RNA ligase (12). After phosphatase treatment, the unacylated 3′ terminal nucleotide is marked by the only 32P as its 5′ phosphate. After digestion of such a population of acylated RNAs with P1 nuclease, one expects a single mole of labeled GMP (the 3′ terminus) per mole RNA, divided between GMP and aa-GMP.
Figure 2.

Chromatographic resolution of products of acylation with six aa-AMP–RNA 77 reactions are at pH 7.25 and 0°C with 1–5 mM adenylate. Phe and Tyr reactions are for 0.5 min, all others for 20 min. RNA 29 reactions are similar but terminated at 2 min. The 3′ [32P] nucleotide was released from aa-RNAs by P1 nuclease digestion and aminoacylated and unacylated forms resolved by cellulose polyethyleneimine chromatography.
As Fig. 2 shows, these products can be resolved by TLC and measured by PhosphorImaging. RNA 29, on incubation with Phe-AMP, Ala-AMP, and Ser-AMP yields the previously characterized products expected from 2′(3′) Phe-RNA, Ala-RNA, Ser-RNA, that is, Phe-GMP, Ala-GMP, and Ser-GMP, respectively. In addition, Fig. 2 shows the formation of the products Tyr-RNA, Ile-RNA, and Gln-RNA, each of which is necessarily 3′ terminal. These six aa-RNAs are formed at rates similar to those previously measured (see below for quantitation).
Similarities Between Reactions in RNA 29 and RNA 77.
RNA 77 gives products indistinguishable from those of RNA 29, though Ala-GMP, Ser-GMP, Ile-GMP, and Gln-GMP are formed only in relatively small amounts. Identical acylation products for the two RNAs can be seen from the similar patterns on the left and right of Fig. 2. Because RNA 29 forms 2′(3′) aa-RNA (thereby chemically similar to biological aa-tRNAs; ref. 1), the formation of six different products with chromatographic behaviors identical to products from RNA 29 argues strongly that RNA 77 forms the 2′(3′) ribose esters: Phe-RNA, Tyr-RNA, Ala-RNA, Ser-RNA, Ile-RNA, and Gln-RNA. The utilization of some minor fraction within the four very slowly reacting adenylates by RNA 77 (always a possibility when reactions markedly slower than normal are studied) thereby is virtually eliminated.
A consistent conclusion is implied by experiments showing that phosphatase treatment is required after 3′ ligation of pGp or the RNA cannot be aminoacylated (unpublished work). Thus a 3′ phosphate blocks aminoacylation. Similarly, 3′ oxidation with periodate blocks aminoacylation (9). This finding is consistent with synthesis of 3′ aa-RNA 77, chemically similar to the aa-tRNA of modern translation.
Formation of aa-RNA 77, like RNA 29, is also first order in RNA concentration to at least 4 μM (unpublished work), confirming self-acylation rather than molecular cooperation. RNA 77 therefore self-aminoacylates to form normal aa-RNAs.
The TLC assay just described is well suited to kinetics or other quantitative work on [32P]RNA, because the product is well-resolved from GMP (Fig. 2), so that low levels of reaction can be measured. In addition, there is always an internal standard for full acylation in the form of the distribution of radioactivity from the unique terminal [α-32P]GMP. We therefore have used this TLC assay to measure acylation kinetics whenever high sensitivity and resolution is required. The previous acid-gel electrophoresis assay (10) separates RNA from slightly slower aa-RNA. Comparison of the gel assay and this chromatographic one show that they measure the same rate, and therefore the same unique 3′ aminoacylation (unpublished work). However, the electrophoresis technique is both less resolved (some products, e.g., Ala-RNAs, have small gel shifts) and less sensitive (background counts trail from the unacylated into the acylated gel band).
Amino Acid-Specific Rates.
Fig. 3 shows typical aminoacylation kinetics. Reaction with Phe-AMP is faster than with Ala-AMP, Ser-AMP, Ile-AMP, or Gln-AMP. Tyr-AMP, which resembles Phe-AMP except for an added hydroxyl, is intermediate in rate. In fact, these data overemphasize the slow rates because the slowly reacting aa-AMPs have been used at concentrations about three orders of magnitude higher than the reactive ones. Tables 1 and 2 show the measured second-order rate constants for aminoacylation.
Figure 3.

Relative acylation kinetics for RNA 77 with six different aa-AMPs. Adenylate concentrations are Phe-AMP (3.3 μM), Tyr-AMP (2.6 μM), Ile-AMP (12 mM), Ala-AMP (5 mM), Gln-AMP (5 mM), and Ser-AMP (7 mM). The symbols are acylated fractions measured by PhosphorImaging, and the lines fitted second-order kinetic curves.
Table 1.
Rates for RNA 77
| Adenylate | aa-AMP conc. | Apparent 2nd-order rate constant, M−1⋅min−1 | Ratio to Phe-AMP | ΔΔG‡ |
|---|---|---|---|---|
| Phe-AMP | 3.3 μM | 60,000 ± 1,900 | 1.00 | – |
| Tyr-AMP | 2.6 μM | 40,000 ± 400 | 0.67 | 0.21 |
| Ile-AMP | 12 mM | 1.5 ± 0.05 | 2.5 10−5 | 5.7 |
| Ala-AMP | 5 mM | 3.4 ± 0.11 | 5.7 10−5 | 5.3 |
| Gln-AMP | 5 mM | 5.3 ± 0.07 | 8.8 10−5 | 5.0 |
| Ser-AMP | 7 mM | 0.9 ± 0.02 | 1.5 10−5 | 6.0 |
Table 2.
Rates for RNA 29
| Adenylate | aa-AMP conc. | Apparent 2nd-order rate constant, M−1⋅min−1 | Ratio to Phe-AMP | ΔΔG‡ |
|---|---|---|---|---|
| Phe-AMP | 1.3 mM | 140 ± 8 | 1.00 | – |
| Tyr-AMP | 1.2 mM | 220 ± 15 | 1.6 | −0.25 |
| Ile-AMP | 7.3 mM | 20 ± 0.4 | 0.14 | 0.6 |
| Ala-AMP | 0.4 mM | 1000 ± 90 | 7.1 | −1.1 |
| Gln-AMP | 0.8 mM | 650 ± 25 | 4.6 | −0.82 |
| Ser-AMP | 1.4 mM | 600 ± 40 | 4.3 | −0.79 |
Second-order rate constants are derived from kinetic data like that in Fig. 2 for reaction of adenylate and the two RNAs, correcting for decay of the adenylate during reaction where necessary (10). For RNA 77 these rate constants are little affected by adenylate concentration in this range. For RNA 29, this is also approximately true. Therefore the second-order rate constant in this range (<KM) may be taken as an estimate of kcat/KM (14). Note that these kinetic constants are derived for the first turnover, rather than multiple turnovers, as is more usual.
On this basis, RNA 77 is the most active self-aminoacylating RNA we have characterized, with a kcat/KM for Phe-AMP of 6 × 104 M−1⋅min−1. Phe-RNA from Phe-AMP incubated with RNA of randomized sequence cannot be detected, but is formed at a rate < 10−3 M−1⋅min−1 (1). Thus RNA 77 is 6 × 107-fold or more faster than the (undetectable) uncatalyzed rate for its reaction and is converted to Phe-RNA 600-fold faster than was RNA 29. This rapid rate also is accompanied by relatively greater specificity.
An Amino Acid-Unspecific RNA.
For RNA 29 and its derivatives, previously found to be relatively undiscriminating, kcat/KM for Phe-AMP is ≈102 M−1⋅min−1 (10). In Table 2 this is observed again under a new set of ionic conditions for RNA 29. Under these conditions (higher divalents), as reported before, Ser-RNA and Ala-RNA formation are faster than the Phe-RNA reaction initially selected (1). Of the three additional aa-AMPs shown here, Gln-AMP and Tyr-AMP are again better substrates than Phe-AMP, and Ile-AMP is a worse substrate. However, these unselected preferences are relatively small, and except for Ile, the range of rates is less than 8-fold. Therefore, with more data, the active site of RNA 29 still appears to accommodate virtually any amino acid side chain.
An Amino Acid-Specific RNA.
RNA 77, remarkably, though more than two orders faster than is RNA 29 with “cognate” Phe-AMP as substrate, is slower, 2-fold to 150-fold less reactive with “noncognate” adenylates Ala-AMP, Ser-AMP, Ile-AMP, and Gln-AMP. Thus RNA 77 is ≈11,000-fold to 66,000-fold selective for Phe-RNA, against Ala-RNA, Ile-RNA, Gln-RNA, and Ser-RNA. In contrast, RNA 29 remains slightly perverse, preferring the reaction for which it was selected for only one amino acid of six assayed. Only Ile-RNA synthesis is slower than Phe-RNA, and that by less than one order of magnitude. Because specific reactions are observed with the same adenylate preparations concurrently supplied to RNA 77 and RNA 29 (Fig. 2), specificity cannot be attributed to some unexpected cryptic differences between these synthetic substrates.
Meaning of Specific Rates.
We want to cast RNA 77’s specificity in terms of standard enzymological analysis. First of all, as is shown in the body of Fig. 4, RNA 77 is substantially inhibited by AMP, but not by UMP. An equivalent concentration of UMP is present as a control, which should show most of the secondary effects of AMP (e.g., chelation of calcium). Thus we interpret this data as base-specific competition by AMP at the Phe-AMP site, as previously was observed for RNA 29 (10). The apparent KI for this AMP interaction (2.5 ± 0.4 mM) resembles that previously measured for RNA 29. Thus RNA 77 is not just a second-order chemical reactant, but likely forms a Michaelis complex with its substrate, including at least contacts with the adenine base of AMP. However, similar experiments with l-Phe show no inhibition of Phe-RNA synthesis at concentrations up to 0.036 M l-Phe (not shown). We return to this observation below.
Figure 4.

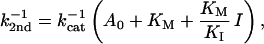 |
We also would like to measure KM and kcat for Phe-AMP. However, RNA 77 reaction is so rapid at our usual assay pH of 7.25 that this measurement cannot be done manually. We have used lower reaction pH to slow the reaction rate.
The Rate Constant Reflects Chemistry.
As shown in Fig. 5, the second-order aminoacylation rate increases linearly with [OH−] from pH 5.5–7.25 (the line in the figure has been drawn with a slope of 1.0). Thus like the RNA 29 family of self-acylating RNAs (10), RNA 77 aminoacylation appears to be base-catalyzed, with a rate constant that reflects chemistry. Linear dependence on [OH−] likely results from a requirement for deprotonation of the attacking ribose hydroxyl as a condition for the transition state.
Figure 5.

Effect of pH on the measured second-order rate constant (k2nd) of RNA 77–Hepes was used at pH 7.25, Mes was used from pH 5.0–7.0, and NaOAc was used at pH 4.8. Buffers were 0.2 M and are represented by different symbols. A line with a slope of 1 is drawn near the points.
For the purpose of studying a slower reaction, RNA 77 phenylalanylation is slowed 32-fold when performed (below) at pH 5.75 instead of the more usual pH 7.25. Below pH 5.5 in Fig. 5 the rate falls off more quickly than is consistent with base catalysis. There are no pKas in this region among RNA components, but we may be seeing a change in the conformation of the RNA because of RNA-structure-enhanced protonation of C and/or A (15, 16). However, at pH 5.75, the data of Fig. 5 make it likely that we are studying the same base-catalyzed reaction as at pH 7.25 (Fig. 5).
KM and kcat.
The reciprocal second-order rate constant, k2nd−1, should appear to vary linearly with the adenylate concentration, because of enzyme saturation with substrate:
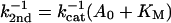 |
At pH 5.75, this variation can be measured, as shown in Fig. 6. The corresponding enzymatic constants derived from the fitted line are Km = 4.7 ± 1 mM and kcat = 14 ± 3 min−1. If we assume that the rate-pH variation is attributable to the kcat (because of ionization of an attacking ribose hydroxyl in the transition state), we calculate at pH 7.25: kcat = 430 ± 90 min−1.
Figure 6.

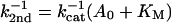 |
Accordingly, RNA 77 appears to be a very fast selected ribozyme, about 360-fold more reactive than RNA 29, which exhibits a more typical kcat of 1.2 min−1 (10). Exceptionally rapid synthesis of Phe-RNA 77 probably results from exceptionally fast chemistry (kcat is linear in [OH−], therefore probably reflects transacylation chemistry; above), rather than a low Km. Thus RNA 77 does not, as might seem plausible intuitively, become specific by using strong net binding of both the AMP and phenylalanine moieties, thereby automatically becoming selective for its amino acid. Instead it seems likely that the seryl, alanyl, isoleucyl, and glutaminyl residues are not presented for reaction with terminal ribose-OH.
Synthesis of Phe-tRNA by protein PheRS (reviewed in ref. 17) makes an interesting comparison. Aminoacyl transfer to the RNA is likely the rate-limiting reaction, just as with RNA 77. Escherichia coli and several different yeast PheRS, assayed under a variety of conditions, produce Phe-tRNA at 140–360 min−1 at 37°C. Thus comparing only transacylation, RNA 77 making Phe-RNA at 430 min−1 at the lower temperature of 0°C is a catalyst equal to or more efficient than several highly evolved PheRS proteins at this same step.
Extent of RNA 77 Reactions.
RNA 77 apparently is acylated to a low extent by substrates like Ala, Ser, Ile, or Gln, which also are slowly reacting (Fig. 7). We have investigated to see why RNA 77 cannot be quantitatively converted to aa-RNA by using poor substrates.
Figure 7.

Apparent saturation levels of aa-RNA 77 at pH 7.25 and different initial adenylate concentrations. Symbols are data and the lines are least-squares fits to kinetics that take account of the second-order RNA-adenylate reaction and the instability of the adenylates.
RNA 77 is not a hydrolase for aa-AMPs that acylate to a low level. Determinations by HPLC of the half-life of the adenylates under acylation conditions are comparable in the presence and absence of RNA 77 (unpublished work). Thus RNA 77 does not artificially limit the extent of reaction with apparently poorer adenylates by either altering or destroying poorer substrates.
The products of the limited reactions are not unstable. For example, Ala-RNA 77 and Ser-RNA 77 have stabilities comparable to Phe-RNA 77 (t½ ≥ 300 min under acylation conditions in the absence of adenylate; unpublished work). This low deacylation rate predicts quantitative formation of aa-RNA within experimental error for all amino acids. Thus for poor substrates, aa-RNAs are not recovered in low yield because of low product stability.
There are not two kinds of RNA 77, that is, a minor stable population of molecules that can be acylated only by poor substrates and a larger population acylated only as Phe-RNA. In Fig. 3, the sum of the extents of reaction with six different amino acids is 167%. In Fig. 7 the sum of four ultimate levels is 148%. All are significantly greater than 100% as a result of the use of a unique 3′ 32P label for the aa-RNA product. Because the sums would be less than 100% if there were stable subpopulations, this finding suggests that all products come from the same pool of molecules, which is supported by data (unpublished work) showing that prereaction with tyrosine depletes the population of molecules subsequently reacting with isoleucine.
Adding new aliquots of adenylate during reaction and use of high substrate concentrations (Fig. 7) also yield low apparent limits for levels of the cognate aa-RNAs. However, if the acylated RNA is recovered by gel filtration, and more adenylate is added, the aa-RNA levels can be progressively elevated above levels recovered at high initial adenylate concentrations (unpublished work). This finding suggests that with extremely low rates of aa-RNA formation, acylation is limited by the natural hydrolytic decay of the adenylate, which releases an inhibitor of aminoacylation (AMP; shown in Fig. 4).
Thus only after gel filtration removes small molecules (such as AMP) will further addition of adenylates increase acylation. This observation somewhat resembles the low extents of aa-RNA formation observed during slow acylation or misacylation of normal tRNAs by using protein aaRS, caused by hydrolysis of the aa-tRNA product (18). In our case, it is substrate, not product, instability that limits extents. Substrate hydrolysis reduces rates in two ways, yielding high concentrations of AMP inhibitor when using a poor substrate, which is necessarily present in high concentration.
Having seen the effects of spontaneous adenylate hydrolysis on the extent of slow reactions, it is also true that there will be an effect on measured rates, as in Tables 1 and 2. However, such effects partially cancel: released AMP makes the reactions appear to terminate too soon (increasing calculated rates) but also inhibits acquisition of the adenylate substrate (decreasing calculated rates). The resultant effects appear small (≤1.5-fold at maximum), theoretically and from partial measurements, and no corrections have been applied to the rate constants presented.
DISCUSSION
The Specificity of RNA 77.
RNA 77 was selected for RNA-catalyzed acceptance of phenylalanine (1). This RNA active site strongly favors the aromatic side chains of Phe and Tyr, and discriminates against large (Ile) and small (Ala) aliphatic side chains by 5.3–5.7 kcal/mol in the transition state (ΔΔG‡ = -RT ln K‡), and against large (Gln) and small (Ser) polar side chains by 5.0–6.0 kcal/mol (Tables 1 and 2). RNA 77 therefore carries out amino acid-specific, aa-RNA synthesis, as would be required for a primordial translation system relying on RNA.
To put this in perspective, RNA 77 can be more specific than the protein yeast PheRS (17). By using kcat/KM for the overall production of Phe-tRNA, which should be comparable to the rates in Tables 1 and 2, the RNA enzyme discriminates more effectively against all four noncognate amino acids (Ser, Gln, Ala, and Ile) than does the protein (17). The discrimination against Ser by RNA 77 (66,000-fold; Table 1) is greater than that for any of the 18 noncognate amino acids tested for yeast protein PheRS. This discrimination against Ser is surprising because PheRS, like many other synthetases, exploits a multistep mechanism including several proofreading functions to enhance its accuracy. The modern PheRS of yeast is self-evidently sufficiently accurate to support an evolutionarily stable biological translation system. Therefore rapid and selective aa-RNA 77 synthesis suggests that aminoacyl transfer by an RNA also could support a successful translation system.
Of course, in so saying, we are not considering facile synthesis of Tyr-RNA by RNA 77. The Phe and Tyr side chains are chemically similar and pose a discrimination problem even for protein sites. In fact, the protein yeast PheRS uses three types of rejection mechanisms for Tyr-AMP (which therefore are used after mistaken activation of tyrosine) to produce accurately formed Phe-tRNA (19). Even more germane, no selection against use of tyrosine has been applied to RNA 77, so that no conclusion can be drawn about possible discrimination against tyrosine. It therefore would be of interest to select a descendant of RNA 77 for synthesis of Phe-RNA and against Tyr-RNA.
Newly Possible Experiments.
A final way of illustrating the selectivity of RNA 77 is to note that even with only one specific ribozyme experiments now are possible that use two specifically self-acylated RNAs. In the presence of millimolar concentrations of an arbitrary aa1-AMP and micromolar concentrations of Phe-AMP, RNA 29 would be expected to exist almost entirely as aa1-RNA and RNA 77 as Phe-RNA. In such a reaction the easily misformed product, Phe-RNA 29, likely would be depressed, and aa-RNAs made more accurate, by competition with aa1-AMP. Similar competition-enhanced specificity was predicted (20) and observed (21) in the modern translation apparatus. One now might simulate specific peptide synthesis or other reactions dependent on two accurately formed aa-RNAs.
Experimental Evidence for an Evolutionary Precursor.
These results suggest a route for evolution of the modern coding apparatus. The properties of RNA 77, measured above, show that an ancestral RNA not very different in size and structure from modern tRNA could have possessed both RNA pairing function (that is, hairpin loops with coding functions) and the ability to catalyze its own aminoacylation (aaRS functions). This possibility is the more striking because smaller variants of RNA 77 (70 nt; unpublished work) are still excellent catalysts. Thus RNA 77 suggests and perhaps exemplifies a simpler ancestral state in which the modern functions of tRNA and aaRS were at least partially combined in a single molecule.
Source of RNA 77 Rate Specificity.
What is the nature of the specificity in the RNA 77 acylation reaction? Amino acid selection cannot be based on steric exclusion, because RNA 77 prefers its larger substrate Phe to smaller substrates (Ala and Ser) as well as to those of more comparable size (Gln and Ile). RNA 77 also shows no evidence of proofreading functions like those of modern proteins. Not only were such reactions unselected during the isolation of RNA 77, but as pointed out in Results, disfavored adenylates and aa-RNAs (once formed) are stable in RNA 77 reactions.
RNA 77 binds the AMP of Phe-AMP (Fig. 4), but has no similar affinity for free l-Phe, thereby paralleling RNA 29 (10). This finding suggests that binding relies mostly on AMP of AMP-Phe. RNA 77’s KI for AMP (2.5 mM, Fig. 4) is of the same order as its KM for Phe-AMP (4.7 mM, Fig. 6), suggesting again that there may be little or no net contribution of the Phe moiety to substrate binding. The KM of the unspecific RNA 29 for Phe-AMP is 8.7 mM (10), and the specific RNA 77 is similar at 4.7 mM (Fig. 6), which suggests similar substrate complexes. Nevertheless, despite these numerical similarities RNA 77 is highly selective and RNA 29 very unselective (Tables 1 and 2, Fig. 2).
To resolve this possible inconsistency, we emphasize the unusual quality we have measured: RNA 77 is 2–3 orders faster in kcat, that is, in its rate of phenylalanyl transfer (Fig. 5). We conjecture that the active site rarely adopts its active structure in the absence of the amino acid side-chain ring. Rearrangement on binding of ligands is, in fact, frequent behavior for RNA sites whose structure has been characterized (e.g., ref. 22). From independent experiments, we know that phenylalanine alone can be bound by an RNA site (23). However, in the RNA 77 active site, such phenylalanine binding in Phe-AMP must be nearly energy neutral. That is, the favorable RNA interaction with the aromatic side chain is nearly compensated by the energy requirement for rearrangement from the solution structure of the RNA to its less stable active structure. Thus even with an unexceptional KM for Phe-AMP, in the absence of the proper side chain (that is, for Ala, Ile, Ser, and Gln) the amino acid will be transferred at a very slow rate, as observed; and affinity for free phenylalanine will be minimal, as observed. Active site assembly, in this hypothesis, must be simple and not involve large conformational adjustments, to proceed much faster than chemistry at 430 min−1. We therefore suggest that this amino acid-specific self-acylating RNA transduces the differences between its substrates into active site conformational differences to perform its highly specific acylation reaction.
Acknowledgments
We thank Alex Wolfson for helpful comments on a draft manuscript. Experiments were supported by National Institutes of Health Research Grant 48080 to M.Y.
ABBREVIATION
- aa-AMP
aminoacyl-adenylate
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Illangasekare M, Sanchez G, Nickles T, Yarus M. Science. 1995;267:643–647. doi: 10.1126/science.7530860. [DOI] [PubMed] [Google Scholar]
- 2.Piccirilli J A, McConnell T S, Zaug A J, Noller H F, Cech T R. Science. 1992;256:1420–1424. doi: 10.1126/science.1604316. [DOI] [PubMed] [Google Scholar]
- 3.Dai X-C, de Mesmaeker A, Joyce G F. Science. 1995;267:237–240. doi: 10.1126/science.7809628. [DOI] [PubMed] [Google Scholar]
- 4.Joyce G F, Dai X-C, De Mesmaeker A. Science. 1996;272:18–19. [PubMed] [Google Scholar]
- 5.Lohse P A, Szostak J W. Nature (London) 1996;381:442–444. doi: 10.1038/381442a0. [DOI] [PubMed] [Google Scholar]
- 6.Jenne A, Famulok M. Chem Biol. 1998;5:23–34. doi: 10.1016/s1074-5521(98)90084-9. [DOI] [PubMed] [Google Scholar]
- 7.Zhang B, Cech T R. Nature (London) 1997;390:96–100. doi: 10.1038/36375. [DOI] [PubMed] [Google Scholar]
- 8.Noller H F, Hoffarth V, Zimniak L. Science. 1992;256:1416–1419. doi: 10.1126/science.1604315. [DOI] [PubMed] [Google Scholar]
- 9.Illangasekare M, Kovalchuke O, Yarus M. J Mol Biol. 1997;274:519–529. doi: 10.1006/jmbi.1997.1414. [DOI] [PubMed] [Google Scholar]
- 10.Illangasekare M, Yarus M. J Mol Biol. 1997;268:631–639. doi: 10.1006/jmbi.1997.0988. [DOI] [PubMed] [Google Scholar]
- 11.Ciesiolka J, Illangasekare M, Majerfeld I, Nickles T, Welch M, Yarus M, Zinnen S. Methods Enzymol. 1996;267:315–335. doi: 10.1016/s0076-6879(96)67021-9. [DOI] [PubMed] [Google Scholar]
- 12.England T E, Bruce A G, Uhlenbeck O C. Methods Enzymol. 1980;65:65–74. doi: 10.1016/s0076-6879(80)65011-3. [DOI] [PubMed] [Google Scholar]
- 13.Serra M J, Turner D H, Freier S M. Methods Enzymol. 1995;259:243–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- 14.Fersht A. Enzyme Structure and Mechanism. New York: Freeman; 1985. , chapter 12. [Google Scholar]
- 15.Legault P, Pardi A. J Am Chem Soc. 1994;116:8390–8391. [Google Scholar]
- 16.Connell G J, Yarus M. Science. 1994;264:1137–1141. doi: 10.1126/science.7513905. [DOI] [PubMed] [Google Scholar]
- 17.Friest W, Sternbach H, Cramer F. Eur J Biochem. 1996;240:526–531. doi: 10.1111/j.1432-1033.1996.0526h.x. [DOI] [PubMed] [Google Scholar]
- 18.Bonnet J, Ebel J P. Eur J Biochem. 1972;31:335–344. doi: 10.1111/j.1432-1033.1972.tb02538.x. [DOI] [PubMed] [Google Scholar]
- 19.Lin S X, Baltzinger M, Remy P. Biochemistry. 1984;23:4109–4116. doi: 10.1021/bi00313a015. [DOI] [PubMed] [Google Scholar]
- 20.Yarus M. Nat New Biol. 1972;239:106–108. doi: 10.1038/newbio239106a0. [DOI] [PubMed] [Google Scholar]
- 21.Swanson R, Hoben P, Sumner-Smith M, Uemura H, Watson L, Soll D. Science. 1988;242:1548–1551. doi: 10.1126/science.3144042. [DOI] [PubMed] [Google Scholar]
- 22.Patel D J, Suri A K, Jiang F, Jiang L, Fan P, Kumar R A, Nonin S. J Mol Biol. 1997;272:645–664. doi: 10.1006/jmbi.1997.1281. [DOI] [PubMed] [Google Scholar]
- 23.Zinnen S, Yarus M. Nucleic Acids Symp Ser. 1995;33:148–151. [PubMed] [Google Scholar]