

Abstract
Release of proteins through the outer mitochondrial membrane can be a critical step in apoptosis, and the localization of apoptosis-regulating Bcl-2 family members there suggests they control this process. We used planar phospholipid membranes to test the effect of full-length Bax and Bcl-xL synthesized in vitro and native Bax purified from bovine thymocytes. Instead of forming pores with reproducible conductance levels expected for ionic channels, Bax, but not Bcl-xL, created arbitrary and continuously variable changes in membrane permeability and decreased the stability of the membrane, regardless of whether the source of the protein was synthetic or native. This breakdown of the membrane permeability barrier and destabilization of the bilayer was quantified by using membrane lifetime measurements. Bax decreased membrane lifetime in a voltage- and concentration-dependent manner. Bcl-xL did not protect against Bax-induced membrane destabilization, supporting the idea that these two proteins function independently. Corresponding to a physical theory for lipidic pore formation, Bax potently diminished the linear tension of the membrane (i.e., the energy required to form the edge of a new pore). We suggest that Bax acts directly by destabilizing the lipid bilayer structure of the outer mitochondrial membrane, promoting the formation of a pore—the apoptotic pore—large enough to allow mitochondrial proteins such as cytochrome c to be released into the cytosol. Bax could then enter and permeabilize the inner mitochondrial membrane through the same hole.
Despite widespread interest in apoptosis, the cellular mechanism by which programmed cell death proceeds, there is considerable controversy about the precise pathway by which cell death is irreversibly committed. However, in many systems studied to date, proteins of the Bcl-2 family have consistently proven either to promote apoptosis or to protect against it. One of these proteins, Bcl-xL, shares a specific structural homology with the diphtheria toxin translocation domain that mediates both toxin passage through cellular membranes and channel activity in lipid bilayers (1). It has been recently shown that Bcl-xL, Bcl-2, and Bax also form channels in artificial lipid systems (2–5), but it is not clear how this activity could regulate apoptosis. These ionic channels are similar to each other, which is surprising for proteins with opposite effects on apoptosis, and they are too small for translocation of critical proteins (cytochrome c, apoptosis-inducing factor) currently thought to mediate the next step in the apoptotic pathway, caspase activation (6–9). Although the outer mitochondrial membrane has been less studied than the inner membrane, it is known to be thoroughly fenestrated with a much larger channel, the voltage-dependent anion channel (10). Thus, it is not plausible that the addition of a smaller conductance pathway should materially affect the permeability properties of that membrane.
Bax, Bcl-2, and Bcl-xL contain a carboxyl-terminal hydrophobic domain that has been presumed to function as a membrane anchor domain (11). However, we have found that Bax translocates from the cytoplasm to the mitochondrial membrane on activation of apoptosis (12). Thus, Bax is a good candidate to mediate release of mitochondrial proteins. In addition, removal of the carboxyl-terminal membrane anchor domain abrogates Bax redistribution and inhibits its death-promoting activity (12). To date, all experiments reporting channel activity of Bcl-2 family members used bacterial systems to express truncated forms of these proteins (2–5). In the present work, we tested the membrane-perturbing effect of full-length Bax and Bcl-xL synthesized in vitro and in vivo.
MATERIALS AND METHODS
Expression of Bax and Bcl-xL in Bacteria and by the Reticulocyte Lysate System.
The truncated form of Bax, lacking 22 carboxyl-terminal amino acids, was expressed in bacteria by using the pET system (Novagen) and purified to near homogeneity by conventional and monoclonal antibody affinity chromatography. The full-length bax gene was expressed under the control of the T7 and cytomegalovirus promoters (13) in a TNT coupled reticulocyte lysate system (Promega). Two μg of the bax plasmid DNA were incubated in a 200-μl reaction mixture for 90 min. After expression, samples were filtered in a 100-kDa Centricon (Amicon) to remove high molecular mass components. The same strategy was used to obtain full-length Bcl-xL. Empty pcDNA 3 vector plasmid (Invitrogen) incubated under identical conditions and filtered was used as negative control. In the coexpression experiments, 2 μg of bax plasmid and 2 μg of Bcl-xL plasmid were incubated together in 200 μl of reticulocyte lysate.
Quantification of Protein Expression and Immunoprecipitation Analysis.
Expression of Bax and Bcl-xL by the reticulocyte lysate was determined by SDS/PAGE (15% polyacrylamide gels) and Western blotting by using the monoclonal antibodies α hBax 1F6 and α uBcl-xL 2H12, as previously reported (13). Protein concentration was determined by comparing immunoblots of the reticulocyte lysate products with known amounts of purified truncated Bax and Bcl-xL bacterially expressed, that contained the same epitopes. For the immunoprecipitation studies, 30 μl of the reticulocyte lysate were first diluted into 970 μl of 150 mM NaCl/10 mM Hepes (pH = 7.4) and then mixed with 50 μl of hBax 1F6 agarose beads and allowed to incubate for 2 h at 4°C. After brief centrifugation (5 min, 5,000 × g), the supernatant was recovered and the protein concentrated by chloroform:methanol precipitation for further analysis. The beads were washed three times with 1.5 ml of 150 mM NaCl/10 mM Hepes (pH = 7.4). To the resulting pellet, 90 μl of 0.1 M acetic acid, 0.2% TritonX-100 were added to remove the bound proteins from the beads, followed by neutralization with 15 μl of 1 M Tris (pH = 8.0).
Purification of Endogenous Bax from Bovine Thymus.
Bovine Bax was purified from the soluble lysate of thymus by a modification of the method previously described to purify murine thymus Bax (14). Briefly, bovine thymus was homogenized (100 g tissue/500 ml homogenization buffer), and the soluble protein fraction was prepared by differential centrifugation. The lysate was first passed through a trimethylammoniumethyl column (15 ml lysate/ml beads). The flowthrough was then loaded onto a green agarose column (25 ml lysate/ml beads). The subsequent flowthrough was loaded onto an anti-human Bax 2D2 antibody column (13). After the column was washed with the washing buffer, bovine thymus Bax was eluted off the beads by incubation in presence of 0.2 mg/ml of a synthetic peptide corresponding to the epitope of the antibody. Resultant purity was 80%–90% as judged by Coomassie blue on SDS/PAGE, and yield was approximately 80 μg from 100 g of thymus.
Planar Phospholipid Bilayer Studies.
Phospholipids were purchased from Avanti Polar Lipids. Squalene and salts were from Sigma. “Solvent-free” bilayer membranes were formed by either a modified Mueller-Rudin technique (200-μm diameter hole) or the Montal-Mueller technique (230-μm diameter hole), as previously described (15, 16). Unless otherwise mentioned, Mueller-Rudin membranes were used throughout with a membrane-forming solution of dioleoylphosphatidylcholine, dioleoylphosphatidylethanolamine, and dioleoylphosphatidylserine (DOPC/DOPE/DOPS, 1:1:1 mol ratio) at a concentration of 30 mg/ml in squalene. For both types of membranes, purified squalene was used to prepaint the hole, and the solution bathing the membrane contained 100 mM KCl/10 mM Hepes/1 mM MgCl2/1 mM EGTA (pH = 7.0). Protein was added to one side of the bilayer (defined as cis) and the solution was stirred for 30 sec to ensure good mixing. Conductance was measured through a voltage clamp consisting of an operational amplifier and a feedback resistor, then recorded on a chart recorder or digitized and stored. For lifetime measurements, a software program (browse, available on request) was modified to apply pulses and facilitate measurements of membrane lifetime.
RESULTS
Full-Length Bax and Bcl-xL Are Produced in Micromolar Quantities by the Reticulocyte Lysate System.
To study the membrane-perturbing ability of wild-type proteins, we synthesized them using the reticulocyte lysate system. Synthesis of full-length and truncated Bax and Bcl-xL was verified by SDS/PAGE followed by Western blot analysis (Fig. 1A). The samples containing full-length Bax, truncated Bax, and full-length Bcl-xL showed prominent bands at 21 kDa, 19 kDa, and 31 kDa, respectively, in accordance with the expected molecular masses of those proteins. The protein concentration was estimated by using known standards of purified bacterially expressed Bax and Bcl-xL as explained in Methods (Fig. 1B). Under appropriate conditions, synthesis of full-length Bax and Bcl-xL was robust and highly reproducible. Routinely, fresh batches of protein were prepared weekly and stored at −86°C until they were used.
Figure 1.

Expression and quantification of Bax and Bcl-xL. (A) Immunoblots showing expression of full-length Bax (Left) and full-length Bcl-xL (Right) by reticulocyte lysate. (Left) Lane 1, filtered reticulocyte lysate with empty vector plasmid added (control); lane 2, filtered reticulocyte lysate after full-length bax plasmid addition; lane 3, truncated Bax, expressed in Escherichia coli, lacking the 22 carboxyl-terminal amino acids. (Right) Lane 1, control; lanes 2 and 3 correspond to two different samples of filtered reticulocyte lysate after Bcl-xL plasmid addition. (B) Quantification of Bax and Bcl-xL expressed by reticulocyte lysate. (Left) A SDS/PAGE gel loaded with known amounts of purified truncated Bax (ΔC22-Bax) and a sample of full-length Bax (F.L. Bax) obtained from the reticulocyte lysate was incubated with a monoclonal antibody (1F6) against the amino terminus of the protein. Full-length Bax was estimated by densitometry using truncated Bax as a standard. (Right) Quantification of full-length Bcl-xL (F.L. Bcl-xL) produced from reticulocyte lysate. Bacterially expressed purified Bcl-xL was used as a standard as above.
Bax, but Not Bcl-xL, Destabilizes Planar Phospholipid Bilayer Membranes.
Changes in membrane conductance were recorded in the presence of truncated or full-length proteins with a low transmembrane potential. In most experiments, addition of bacterially expressed truncated Bax and Bcl-xL caused channel-like fluctuations with several conductance states (Montal-Mueller bilayers; data not shown). These results are in agreement with previous studies showing complex multiconductance states induced by Bcl-xL and Bax lacking the carboxyl terminus (2, 4, 5). Interestingly, in some experiments, when truncated Bax was added, noisy current increases without discrete conductance levels were recorded, and membranes became unstable. In the course of these studies, we noted that the activity of Bax diminished with time on ice. Thus, we decided to prepare Bax using the reticulocyte lysate system, as this approach permits fast in vitro protein expression. Fresh full-length Bax had a ≈100-fold more potent ability to perturb these lipid bilayers. In 90% of the experiments (n = 39), Bax induced monotonic increases in membrane conductance that usually led to membrane rupture. At concentrations of ≈0.1–0.3 nM of full-length Bax, membranes typically ruptured after some tens of minutes (Fig. 2A), but at higher concentrations rupture occurred faster (Fig. 2B). Full-length Bcl-xL expressed in the reticulocyte lysate did not induce any noticeable changes (Fig. 2C) until the concentration was increased (Fig. 2D). Then, at concentrations above 1 nM, fluctuations of conductance between various levels on the scale of 10–300 pS were seen, but the membranes did not become unstable. At any given concentration of Bcl-xL, activity increased at pH = 4.0 (data not shown). These results obtained with full-length Bcl-xL are similar to those previously reported for bacterially expressed truncated Bcl-xL (2). In other control experiments (no exogenous DNA added to the reticulocyte lysate system), we found that the lysate protein mixture had minimal effects on conductance (Fig. 2E).
Figure 2.

Effect of full-length Bax and Bcl-xL on planar phospholipid bilayer membranes. (A and B) Conductance changes induced by adding filtered reticulocyte lysate to planar phospholipid bilayer membranes formed from a solution of DOPC:DOPE:DOPS (1:1:1) in squalene. Full-length Bax at 0.12 nM (A) and 1.3 nM (B), final concentration. Arrows indicate the time of protein addition. (C and D) Effect of full-length Bcl-xL at two concentrations. Note the different scales. (E) Recording obtained in the presence of the filtered reticulocyte lysate with no Bax or Bcl-xL expressed (control). In all the cases, the holding potential was 40 mV.
Bax Synthesized in Vitro and in Vivo Dominantly Decreases Membrane Lifetime.
The above results show that at relatively low transmembrane potentials (40 mV), Bax, but not Bcl-xL, destabilizes membranes and promotes membrane rupture. To quantify this effect, the voltage across the bilayer was varied and membrane lifetime measurements were performed. Membrane lifetime at 250 mV decreased approximately 10-fold when Bax was added to 100-pM final concentration, whereas Bcl-xL had no significant effect (Fig. 3A). Membrane lifetime progressively diminished as the amount of Bax varied from 0.1 nM to 1 nM (Fig. 3B). The presence of the hydrophobic tail per se was not the source of destabilization, because the truncated form of Bax, missing this part of the protein, still decreased membrane lifetime, although its activity was reduced 7- to 10-fold compared with the full-length protein. Furthermore, full-length Bcl-xL, which also contains a carboxyl-terminal hydrophobic domain, did not affect membrane stability even at nanomolar concentrations.
Figure 3.

Bax, but not Bcl-xL, decreases membrane lifetime. (A) Lifetimes of DOPC:DOPE:DOPS (1:1:1) bilayers were measured at 250 mV, in the presence of full-length Bax, full-length Bcl-xL, and reticulocyte control. The final concentration of Bax and Bcl-xL was 150 pM. Standard errors are shown for 10–15 experiments. There was no systematic change in lifetime between membranes painted in the presence of Bax and those to which Bax was freshly added. (B) Membrane lifetime as a function of full-length Bax (F.L. Bax), full-length Bcl-xL (F.L. Bcl-xL), and truncated Bax (ΔC22-Bax).
Similar results (Bax, but not Bcl-xL, decreased membrane lifetime) were also found in membranes composed of 2:1 DOPC/DOPE, bovine brain phosphatidylserine, and, in an attempt to mimic the lipid composition of the outer mitochondrial membrane (17), 6:3:1 DOPC/DOPE/phosphatidylinositol, regardless of the use of squalene or decane as a solvent for the membrane-forming solution. The destabilization of membranes by Bax in single-component membranes suggests that this effect is not caused by a phase separation in all mixtures containing DOPE, an important caveat because this lipid at room temperature spontaneously forms an inverted hexagonal phase in aqueous suspension.
In an attempt to study the activity of in vivo-synthesized Bax and as a control for the possibility that the destabilizing effect of Bax required its expression in the reticulocyte lysate system, we purified Bax from the cytosol of bovine thymus (Fig. 4A). This endogenous Bax was then added to the solution bathing a phospholipid bilayer membrane. A decrease in membrane lifetime was seen, albeit higher concentrations of thymus Bax were required compared with fresh Bax (Fig. 4B).
Figure 4.

Endogenous Bax from bovine thymus decreases membrane lifetime. (A) Purification of Bax from bovine thymocytes. Bovine Bax was purified from thymocyte soluble protein fraction by conventional column chromatography and α Bax 2D2 immunoaffinity chromatography followed by elution by using a peptide containing the antibody epitope. The resultant eluent was analyzed by SDS/PAGE and silver staining (S.S.) or by Western blotting by using α uBax 2D2 monoclonal antibody (W.B.). (B) Effect of bovine Bax on DOPC:DOPE:DOPS (1:1:1) membrane lifetime. The holding potential was 250 mV.
Interactions between Bcl-2 family members have been proposed to regulate their function. To address this issue, we measured membrane lifetime in the presence of both Bax and Bcl-xL. Bax and Bcl-xL were preincubated together before addition, sequentially added to the solution bathing the bilayer, or coexpressed in the reticulocyte lysate system (Fig. 5A). In all cases, the dominant effect on the membrane conductance and stability was that of Bax (Table 1 and Fig. 5B). There was no evidence for protection of membrane stability by Bcl-xL. To directly test for dimerization between Bax and Bcl-xL, immunoprecipitation was performed on the coexpression mixture by using a monoclonal antibody to Bax. No Bcl-xL was precipitated with this antibody, rather it all stayed in the supernatant, as seen in cell homogenates (ref. 13; Fig. 5A). Thus, no evidence was found for either functional or physical interaction between these two proteins.
Figure 5.

Bcl-xL does not protect against Bax-induced membrane destabilization. (A) Bax and Bcl-xL do not form stable complexes in the reticulocyte lysate. Bax and Bcl-xL were coexpressed in the reticulocyte, and a sample of the reaction was immunoprecipitated with a monoclonal antibody against Bax. The presence of Bax and Bcl-xL in the reticulocyte lysate (lane 1), immunoprecipitate (lane 2) and in the supernatant (lane 3) was checked by SDS/PAGE and Western blotting, by using monoclonal antibodies against both proteins. (B) Decrease in the lifetime induced by Bax expressed alone or together with Bcl-xL. Bax and Bcl-xL concentrations were 150 pM.
Table 1.
Effect of Bcl-xL on Bax-induced membrane destabilization
| Protein | Concentration, nM | Lifetime, s |
|---|---|---|
| None | NA | 12.8 ± 3.5 |
| Bcl-xL | 0.25 | 11.7 ± 2.9 |
| Bax | 0.25 | 0.41 ± 0.14 |
| *Bcl-xL/Bax | 0.25/0.25 | 0.47 ± 0.13 |
| †Bcl-xL + Bax | 0.25 + 0.25 | 0.25 ± 0.09 |
| †Bcl-xL + Bax | 0.50 + 0.25 | 0.34 ± 0.17 |
| †Bcl-xL + Bax | 1.00 + 0.25 | 0.15 ± 0.04 |
| ‡Bcl-xL + Bax | 2.50 + 0.25 | 0.41 ± 0.10 |
Bcl-xL and Bax coexpressed in the reticulocyte lysate.
Bcl-xL and Bax expressed separately in the reticulocyte lysate. Bcl-xL was first incubated for 5 min in the chamber followed by Bax addition.
As in † except that Bcl-xL expressed in bacteria.
Full-Length Bax Potently Reduces the Linear Tension of the Membrane.
Because Bax is targeted to the outer mitochondrial membrane, and the outer mitochondrial membrane is a fluid mosaic of protein and lipid bilayer membranes, it is reasonable to assume that Bax would act on the lipid bilayer of the outer mitochondrial membrane in a similar fashion. Because outer mitochondrial membrane destabilization and poration would promote apoptosis, it is essential to determine the physical mechanism for this effect. There is a general theory for lipidic pore formation and phospholipid bilayer rupture under high electrical fields that has been applied to electroporation of cell membranes and extended to fusion pore formation in enveloped viral infection (18, 19). In purely lipidic systems, voltages applied across a membrane facilitate a localized infolding of the phospholipids in both apposing monolayers to form and expand lipidic hydrophilic pores. Once such a pore exceeds a critical radius, it enlarges indefinitely, yielding irreversible breakdown. The dependence of the lifetime of the membrane (τ) on the applied voltage (V) can be described by the theoretical expression
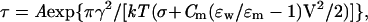 |
1 |
where A is a preexponential factor dependent on additional model assumptions, γ is the linear tension of the pore, k is the Boltzmann constant, T is temperature in K, σ is the bilayer tension, Cm is the specific capacitance of the membrane, and ɛw= 80 and ɛm= 2 are the dielectric constants of water and the hydrophobic core of the membrane, respectively (18). After obtaining values for τ, σ, and Cm experimentally, the linear tension of the pore (γ) can be calculated from Eq. 1. Linear tension is a key parameter in lipidic pore development as it quantifies the work needed to form a unit of pore perimeter and gives a measure of the membrane’s resistance to rupture.
Bax addition had no significant effect on either the specific capacitance (Fig. 6A) or the surface tension of the membrane (Fig. 6B). To obtain linear tension values (γ), the experimental dependence of mean lifetime (n = 10–15) on the applied voltages were fitted to Eq. 1 (Fig. 6C). Linear tension decreased from 9.13 pN for a bilayer not exposed to Bax to 7.73 pN in the presence of very low amounts of Bax (100 pM). Using the parameters from this fit, we could then estimate linear tension as a function of Bax concentration (Fig. 6D). Bax altered linear tension in a dose-dependent fashion decreasing its original value by approximately 50% at the highest concentration tested (1 nM). It is important to note that as γ enters the above equation exponentially, even small variations of linear tension should have a significant effect on pore promotion. For example, the amphipathic peptide of HIV gp41, which also disrupted both phospholipid and cell membranes, reduces linear tension by 3.2 pN at 2 μM (15). These results indicate that full-length Bax is very potent at decreasing membrane stability by lowering the energetic barrier for pore development.
Figure 6.

Bax reduces the linear tension of the membrane. (A) Measurement of specific capacitance in the absence and presence of Bax. (B) Effect of Bax on surface tension. Surface tension was found by measuring the change in membrane capacitance that resulted from application of hydrostatic pressure differences between bathing solutions (15). (C) Fitting of the membrane lifetime voltage dependence to Eq. 1, in the presence and absence of Bax (100 pM). The values for A were 0.027 and 0.039 in the absence and presence of Bax, respectively. (D) Linear tension as a function of Bax concentration.
DISCUSSION
Apoptosis is promoted by the cytosolic protein Bax (20). On stimulation of apoptosis, Bax changes from a soluble cytoplasmic protein to become bound to the mitochondrial membrane (12), possibly because of targeting or retention mechanisms that may involve phosphorylation (21). Our finding that Bax decreases the stability of phospholipid membranes leads us to suggest that this protein acts directly on the phospholipid bilayer of the outer mitochondrial membrane, decreasing membrane stability and causing a breakdown of the permeability barrier of that membrane to macromolecules. This lesion would allow intermembrane proteins such as cytochrome c to diffuse into the cytosol. If the crucial step in the apoptotic pathway is the loss of the permeability barrier of the outer mitochondrial membrane, but not of the inner membrane, then cytochrome c could be released with no changes in the mitochondrial membrane potential. Moreover, once the outer membrane is disrupted, Bax would rapidly have access to the inner mitochondrial membrane, where a similar activity could lead to inner membrane leakage, a decrease in mitochondrial membrane potential, and mitochondrial swelling, which can augment the apoptotic cascade.
The method of expression of protein does not seem to qualitatively affect or contaminate their activity, because bacterially expressed Bax, reticulocyte lysate-expressed Bax, and bovine thymus-purified Bax all destabilize planar phospholipid bilayer membranes, albeit at different concentrations. Nor do we think that the reticulocyte lysate-expressed Bcl-xL is denatured or misfolded, because it changes the permeability of the membrane in ways reported for active Bcl–xL that have been interpreted as the formation of ionic channels (2). Most Bcl-2 family members contain a hydrophobic stretch of amino acids located at their carboxyl terminus (22), but the functional significance of this region is still a matter of debate. To address this issue, we compared the activity of full-length Bax and truncated Bax, which lack the hydrophobic domain. We found that both truncated and full-length forms of Bax decreased membrane lifetime. The main effect of adding the hydrophobic tail of Bax seems to be to increase its potency. Thus, our results indicate that the membrane-destabilizing activity of Bax is a property of the entire molecule, not of the hydrophobic tail alone. In support of this view, note that full-length Bcl-xL has a similar tail but does not cause destabilization. One simple explanation for the increased potency of full-length Bax is that the activity of these molecules is a function of their surface density. With the addition of the hydrophobic tail, there may be a higher partitioning of the full-length protein to the surface of the bilayer membrane. Because targeting of Bax on apoptosis increases its surface density, we will need to devise methods to compare the surface density of Bax on the planar bilayer membrane and compare it to that of the outer mitochondrial membrane at different stages of apoptosis. Until then, we are reporting the final concentrations of Bax and Bcl-xL as calculated from the mols of protein added and the volume of the solution bathing the membrane.
There was no protective effect of Bcl-xL on the destabilization of membranes caused by Bax, as might be expected from reports of their dimerization. There is some controversy on the conditions and stabilities of heterodimers of Bax and Bcl-xL. Recently it was shown that dimerization does not occur between these proteins unless detergent is added (13, 14). Indeed, endogenous Bax from thymocytes is present as a monomer in the cytosol (14). We have now shown by immunoprecipitation experiments that reticulocyte-lysate expressed Bax and Bcl-xL do not form dimers. Thus, it is unlikely that binding of Bcl-xL to Bax would decrease the active concentration of Bax by mass action and inhibit the ability of the latter protein to perturb membranes. In accordance to this view, we saw no evidence for inhibition after incubation of both full-length proteins in solution or on the membrane. Rather, our results are consistent with the idea that the proapoptotic and antiapoptotic members of the Bcl-2 family function independently to regulate cell death (23–25).
The Apoptotic Pore.
Taken together, the variability of the membrane conductance increments in Bax-treated membranes, the lack of effect of Bax on membrane thickness, and the fit of the effect of Bax on the voltage dependence of membrane lifetime are consistent with the hypothesis that Bax induces changes in intrinsic membrane monolayer curvature promoting the formation of a pore that is at least partially lipidic. Our proposal is that the activity of Bax to destabilize planar phospholipid bilayer membranes is also an activity of Bax on the lipid bilayer of the outer mitochondrial membrane to promote the formation of an apoptotic pore. We do not know whether this apoptotic pore is purely lipidic or a protein/lipid complex, but the present data are inconsistent with a purely proteinaceous ionic channel. This hypothesis is consistent with recent data showing that substantial cytochrome c is lost relatively soon after induction of apoptosis, concomitant with large regions of outer mitochondrial membrane rupture and without any changes in the inner mitochondrial membrane potential (26). Furthermore, overexpression of Bax has been shown to cause cytochrome c release from mitochondria of yeast and mammalian cells (27–29). Also, addition of Bax to isolated mitochondria in vitro causes cytochrome c release sufficient for caspase activation (30). On the other hand, there is increasing evidence showing that inner mitochondrial membrane permeability is increased during apoptosis (31), and Bax has been implicated in this mechanism (29, 32). We see no conflict in this view, because once the permeability barrier (to macromolecules) of the outer mitochondrial membrane has been breached, Bax would be available to destabilize the inner mitochondrial membrane, leading to a loss of mitochondrial electrochemical potential, activation of the permeability transition pore, and mitochondrial swelling. In addition, mitochondrial swelling by itself can also lead to increased tension of the outer mitochondrial membrane, membrane rupture, and release of protein (26, 33). The effect of tension and that of Bax should be additive (see Eq. 1). Mitochondrial outer membrane rupture, then, would be a common intermediate between different pathways of apoptosis.
Bax is not unique in causing membrane instability and decreasing the lifetime and linear tension of phospholipid bilayers. A peptide made from the sequence of an internal amphipathic helix on the carboxyl-terminal cytosolic domain of the HIV envelope protein gp41 has a similar effect (15). The significance of this finding relates to current models for membrane fusion in which a cluster of fusion proteins provides the positive membrane curvature needed to cause the actual opening of the fusion pore during viral infection, syncytia formation, and exocytosis (19, 34). These mechanisms may also relate to protein translocation. The diphtheria toxin (DT) translocation domain mediates the passage of a 20-kDa protein, the diphtheria toxin A chain, across cellular membranes into the cytosol (35). Although there are no conclusive data to explain the translocation mechanism, some studies suggest that this process could be related to the bilayer destabilization induced by the concerted action of several DT molecules (35, 36). Proteins may have evolved domains that manipulate local lipid membrane curvature in similar ways to produce toxin translocation pores, fusion pores, and apoptotic pores, phenomena that provide for macromolecular or quantal transport of substances across membranes.
Acknowledgments
We thank Drs. Leonid V. Chernomordik and Pierre Henkart for useful conversations and expert advice. G.B. was a recipient of a postdoctoral fellowship from the Basque Government.
ABBREVIATIONS
- DOPC
1,2-dioleoylphosphatidylcholine
- DOPE
1,2-dioleoylphosphatidylethanolamine
- DOPS
dioleoylphosphatidylserine
References
- 1.Muchmore S W, Sattler M, Liang H, Meadows R P, Harlan J E, Yoon H S, Nettesheim D, Chang B S, Thompson C B, Wong S L, et al. Nature (London) 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 2.Minn J A, Velez P, Schendel S L, Liang H, Muchmore S W, Fesik S W, Fill M, Thompson C B. Nature (London) 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- 3.Schendel S L, Xie Z, Montal M O, Matsuyama S, Montal M, Reed J C. Proc Natl Acad Sci USA. 1997;94:5113–5118. doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod J J, Mazzei G, et al. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- 5.Schlesinger P H, Gross A, Yin X-M, Yamamoto K, Saito M, Waksman G, Korsmeyer S. Proc Natl Acad Sci USA. 1997;94:11357–11362. doi: 10.1073/pnas.94.21.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X, Kim C N, Yang J, Jemmerson R, Wang X. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 7.Kluck R M, Bossy-Wetzel E, Green D R, Newmeyer D D. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Liu X, Bhalla K, Kim N C, Ibrado A M, Cai J, Peng T-I, Jones D P, Wang X. Science. 1997;275:1129–1131. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 9.Susin S A, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. J Exp Med. 1996;184:1331–1342. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zimmerberg J, Parsegian V A. Nature (London) 1986;323:36–39. doi: 10.1038/323036a0. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen M, Millar D G, Yong V W, Korsmeyer S J, Shore G C. J Biol Chem. 1993;269:16521–16524. [Google Scholar]
- 12.Wolter K H, Hsu Y-T, Smith C L, Nechushtan A, Xi X-G, Youle R J. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu Y-T, Youle R J. J Biol Chem. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- 14.Hsu Y-T, Youle R J. J Biol Chem. 1998;273:10777–10783. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- 15.Chernomordik L V, Chanturiya A N, Suss-Toby E, Nora E, Zimmerberg J. J Virol. 1994;68:7115–7123. doi: 10.1128/jvi.68.11.7115-7123.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chanturiya A, Chernomordik L V, Zimmerberg J. Proc Natl Acad Sci USA. 1997;94:14423–14428. doi: 10.1073/pnas.94.26.14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Kroon A I P M, Dolis D, Mayer A, Lill R, de Kruijff B. Biochim Biophys Acta. 1997;1325:108–116. doi: 10.1016/s0005-2736(96)00240-4. [DOI] [PubMed] [Google Scholar]
- 18.Chernomordik L V, Melikyan G B, Chizmadhzev Y A. Biochim Biophys Acta. 1985;812:643–655. doi: 10.1016/0304-4157(87)90016-5. [DOI] [PubMed] [Google Scholar]
- 19.Zimmerberg J, Vogel S S, Chernomordik L V. Annu Rev Biophys Biomol Struct. 1993;22:433–466. doi: 10.1146/annurev.bb.22.060193.002245. [DOI] [PubMed] [Google Scholar]
- 20.Oltvai Z N, Milliman C L, Korsmeyer S J. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 21.Zha J, Harada H, Yang E, Jockel J, Korsmeyer S J. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 22.Adams J M, Cory S. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 23.Cheng E H-Y, Levine B, Boise L H, Thompson C B, Hardwick J M. Nature (London) 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- 24.Zha H, Reed J C. J Biol Chem. 1997;272:31482–31488. doi: 10.1074/jbc.272.50.31482. [DOI] [PubMed] [Google Scholar]
- 25.Knudson C M, Korsmeyer S J. Nat Genet. 1997;16:358–363. doi: 10.1038/ng0897-358. [DOI] [PubMed] [Google Scholar]
- 26.Vander Heiden M G, Chandel N S, Williamson E K, Schumacker P T, Thompson C B. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 27.Manon S, Chauduri B, Guerin M. FEBS Lett. 1997;415:29–32. doi: 10.1016/s0014-5793(97)01087-9. [DOI] [PubMed] [Google Scholar]
- 28.Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Nature (London) 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- 29.Pastorino J G, Chen S- T, Tafani M, Snyder J W, Farber J L. J Biol Chem. 1998;273:7770–7775. doi: 10.1074/jbc.273.13.7770. [DOI] [PubMed] [Google Scholar]
- 30.Jurgensmeier J M, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed J C. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroemer G, Zamzani N, Susin S A. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 32.Marzo I, Brenner C, Zamzani N, Jurgensmeier J H, Susin S A, Vieira H L A, Prevost M C, Xie Z H, Matsuyama S, Reed J C, et al. Science. 1999;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 33.Skulachev V P. FEBS Lett. 1996;397:7–10. doi: 10.1016/0014-5793(96)00989-1. [DOI] [PubMed] [Google Scholar]
- 34.Chernomordik L V, Frolov V, Leikina E, Bronk P, Zimmerberg J. J Cell Biol. 1998;23:1369–1382. doi: 10.1083/jcb.140.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.London E. Biochim Biophys Acta. 1992;1113:25–51. doi: 10.1016/0304-4157(92)90033-7. [DOI] [PubMed] [Google Scholar]
- 36.Jiang G S, Solow R, Hu V W. J Biol Chem. 1989;23:13424–13429. [PubMed] [Google Scholar]