

Abstract
Endoplasmic reticulum (ER)-associated degradation (ERAD) is required for ubiquitin-mediated destruction of numerous proteins. ERAD occurs by processes on both sides of the ER membrane, including lumenal substrate scanning and cytosolic destruction by the proteasome. The ER resident membrane proteins Hrd1p and Hrd3p play central roles in ERAD. We show that these two proteins directly interact through the Hrd1p transmembrane domain, allowing Hrd1p stability by Hrd3p-dependent control of the Hrd1p RING-H2 domain activity. Rigorous reevaluation of Hrd1p topology demonstrated that the Hrd1p RING-H2 domain is located and functions in the cytosol. An engineered, completely lumenal, truncated version of Hrd3p functioned normally in both ERAD and Hrd1p stabilization, indicating that the lumenal domain of Hrd3p regulates the cytosolic Hrd1p RING-H2 domain by signaling through the Hrd1p transmembrane domain. Additionally, we identified a lumenal region of Hrd3p dispensable for regulation of Hrd1p stability, but absolutely required for normal ERAD. Our studies show that Hrd1p and Hrd3p form a stoichiometric complex with ERAD determinants in both the lumen and the cytosol. The HRD complex engages in lumen to cytosol communication required for regulation of Hrd1p stability and the coordination of ERAD events on both sides of the ER membrane.
Keywords: proteasome, endoplasmic reticulum, ubiquitin ligase, transmembrane signaling, topology
Introduction
A significant component of eukaryotic cellular protein degradation occurs at the ER. ER-associated degradation (ERAD) is widely employed in cellular quality control, resulting in the destruction of numerous misfolded or mutant proteins present in the lumen or at the surface of the organelle (Ciccarelli et al. 1993; Finger et al. 1993; Ward et al. 1995; Biederer et al. 1996). ERAD continuously occurs in normal cells to minimize levels of unfolded proteins in the ER lumen (Travers et al. 2000). ERAD is also used to regulate the steady state levels of specific proteins in response to physiological cues (Edwards et al. 1983; Nakanishi et al. 1988; Chun et al. 1990; Hampton and Rine 1994; Yeung et al. 1996; Fisher et al. 1997; Zhou et al. 1998). For instance, production of sterols by the mevalonate pathway is controlled, in part, by the regulated, ER-associated destruction of hydroxymethylglutaryl-coenzyme A reductase (HMGR), a key enzyme of the mevalonate pathway.
Genes involved in ERAD have been identified by several genetic analyses in yeast (Hampton et al. 1996a; Hiller et al. 1996; Knop et al. 1996; Hampton and Bhakta 1997; Bordallo et al. 1998; Plemper et al. 1999). These studies indicate that the ER-associated ubiquitin-conjugating enzyme Ubc7p is centrally involved in the degradation of ER substrates (Biederer et al. 1996; Hiller et al. 1996; Hampton and Bhakta 1997; Wilhovsky et al. 2000; and our unpublished results), sometimes in conjunction with other ubiquitin-conjugating enzymes (Biederer et al. 1996; Friedlander et al. 2000; and our unpublished results). These studies also revealed two previously unknown membrane proteins, Hrd1p (also isolated as Der3p) and Hrd3p, required for the ER degradation of numerous proteins (Hampton et al. 1996a; Bordallo et al. 1998; Plemper et al. 1999). Unlike Ubc7p, the functions of Hrd1p and Hrd3p are not as clear. Our recent work demonstrates that Hrd1p provides the core ubiquitin ligase (E3) activity in ERAD (our unpublished results), recruiting Ubc7p for substrate ubiquitination. However, the interplay and specific actions of Hrd1p/Hrd3p are poorly defined in current models of ERAD.
A key, but poorly understood feature of ERAD is the coordination of molecular processes on separate sides of the ER membrane. The degradation process begins in the interior of the ER, where ERAD substrates partially or completely present in the lumen are recognized for degradation by an unknown mechanism. Many proteins required for ERAD have critical determinants located in the lumen (Hampton et al. 1996a; Bordallo et al. 1998; Plemper et al. 1999; Saito et al. 1999; Zhou and Schekman 1999). However, ubiquitination and degradation of ERAD substrates occur on the cytosolic face of the ER by the action of ER-associated ubiquitin-conjugating enzymes and the 26S proteasome (Hampton et al. 1996a; Hiller et al. 1996; Hampton and Bhakta 1997; Plemper et al. 1998). How these membrane-separated events are coordinated is not clear. In particular, it is unknown if Hrd1p and Hrd3p function to allow communication across the ER membrane.
Hrd3p is an ER resident glycoprotein with a single membrane span near the COOH terminus and a large NH2-terminal region in the lumen of the ER (Hampton et al. 1996a; Saito et al. 1999; Plemper et al. 1999). Hrd3p has widely conserved sequence motifs and full-length orthologues in a variety of species (Hampton et al. 1996a; Donoviel et al. 1998), including the Caenorhabditis elegans SEL-1 protein, which may participate in membrane protein degradation (Grant and Greenwald 1997). However, the exact role of Hrd3p in ER degradation has yet to be elucidated.
Hrd1p is also an ER-associated protein with two distinct domains: an NH2-terminal, hydrophobic region with multiple predicted transmembrane spans and a COOH-terminal, hydrophilic region containing a RING-H2 motif required for the function of Hrd1p in ER degradation (Hampton et al. 1996a; Bordallo et al. 1998). The Hrd1p RING-H2 motif is conserved in a group of ubiquitin ligases or E3 proteins, including Ubr1p (Xie and Varshavsky 1999), Hrt1p/Rbx1p (Ohta et al. 1999a; Seol et al. 1999; Skowyra et al. 1999), Apc11p (Peters 1999), and c-Cbl (Joazeiro et al. 1999). In fact, we have recently demonstrated that Hrd1p is a ubiquitin ligase, catalyzing the transfer of ubiquitin from Ubc7p to ERAD substrates by the activity of its RING-H2 motif, which is required for a direct in vivo physical interaction between Hrd1p and Ubc7p (our unpublished results). Unlike other family members, the Hrd1p RING-H2 domain is anchored to the ER membrane by an NH2-terminal, multimembrane spanning domain. Although the Hrd1p NH2-terminal transmembrane domain contains conserved sequences, no function has been suggested for this anchor.
In this paper, we have performed an extensive study of the functional interplay between Hrd1p and Hrd3p. From these studies, we have demonstrated for the first time a direct physical interaction between the two proteins, defined domain-specific functions for each protein, ascertained the correct topology of Hrd1p, demonstrated transmembrane communication between Hrd1p and Hrd3p, and evaluated the importance of the stoichiometry of the Hrd1p–Hrd3p complex. Importantly, we have clearly shown that the Hrd1p transmembrane domain alone mediates the interaction between Hrd1p and Hrd3p. This interaction regulates the activity of the Hrd1p cytosolic RING-H2 domain to allow ERAD by preventing Hrd1p degradation. Hrd3p-mediated regulation of the cytosolic Hrd1p RING-H2 domain occurs across the ER membrane, as a completely lumenal version of Hrd3p allowed ERAD through normal regulation of Hrd1p stability. These studies further demonstrated that the lumenal functions of Hrd3p include both transmembrane regulation of Hrd1p RING-H2 domain activity and as yet undefined aspects of ERAD separate from control of Hrd1p stability.
Materials and Methods
Materials and Reagents
Rabbit anti-hemagglutinin (HA) polyclonal antiserum was obtained from Babco. Rabbit polyclonal anti–green fluorescent protein (GFP) antiserum was obtained from Dr. Suresh Subramani (University of California at San Diego). Rabbit polyclonal antiserum to the COOH-terminal residues 348–551 of Hrd1p or the NH2-terminal residues 220–417 of Hrd3p was made in collaboration with Scantibodies, Inc. All other materials and reagents were obtained from previously described sources (Gardner et al. 1998).
Plasmids and DNA Methods
All DNA manipulations were performed as described (Gardner et al. 1998), using the reagents described therein. For PCR amplifications, primer sequences will be provided upon request.
Plasmids expressing epitope-tagged versions of the HRD genes were constructed as follows. pRH1196 (YIp/TRPI) expressed Hrd1p with a triple HA epitope sequence fused in frame to the immediate COOH terminus. Integration at the HRD1 genomic locus resulted in expression of only 3HA-Hrd1p. The triple HA epitope sequence was fused to the HRD1 sequence through the splicing by overlap extension (SOEing) PCR technique (Horton et al. 1989). The 3HA-HRD1 gene complemented an hrd1Δ mutation when integrated as a single copy behind the native HRD1 promoter. pRH1263 contained a triple HA epitope sequence inserted between codons 20 and 21 of the HRD3 sequence cloned from pYS14 (Saito et al. 1999). Integration of pRH1263 at the HRD3 genomic locus resulted in expression of only 3HA-Hrd3p, which complemented an hrd3Δ mutation when expressed from the native promoter. pRH1048 allowed expression of 3HA-Hrd3p from the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (TDH3) promoter (Bitter and Egan 1984). The plasmid was constructed by subcloning a PCR-generated DNA fragment encoding the 5′ sequence of the 3HA-HRD3 allele between the XhoI and NheI sites in pRH632 (YIp/LEU2/PTDH3-HRD3), thereby replacing the corresponding normal HRD3 sequence.
Plasmids expressing the Hrd1p NH2-terminal transmembrane domain, called hemi-Hrd1p, were constructed as follows. pRH936 (YIp/TRP1) allowed hemi-Hrd1p expression from the GAPDH promoter. The hemi-Hrd1p coding region was prepared by PCR amplification of the 5′ end of HRD1 between the HRD1 start codon and the NcoI site just distal to the end of the transmembrane domain coding region. The PCR product was cloned into a TRP1 variant of pRH98-2 (Hampton et al. 1996b), placing hemi-HRD1 behind the GAPDH promoter, yielding pRH936. pRH986 (YIp/TRP1), which allowed expression of 1myc-hemi-Hrd1p from the GAPDH promoter, was prepared from pRH936 by addition of a PCR-generated DNA fragment encoding a single myc epitope sequence (EQKLISEEDL) to the 3′ end of hemi-HRD1. The DNA fragment replaced the sequence between the NsiI and KpnI sites in pRH936. pRH1297 (YIp/TRP1) allowed expression of hemi-Hrd1p-GFP from the GAPDH promoter. hemi-Hrd1p-GFP contained a replacement of the Hrd1p COOH-terminal domain with the autofluorescent GFP protein. The GFP fusion sequence was created by SOEing PCR amplification of the GFP coding sequence from pRH469 (Gardner et al. 1998), and replaced the sequence between the NsiI and KpnI sites in pRH936.
Plasmids expressing the isolated COOH-terminal RING-H2 domain of Hrd1p, termed RING-Hrd1p, were constructed as follows. The plasmid pRH730 (YIp/TRP1), which contained 3HA-HRD1 behind the GAPDH promoter, was digested with NcoI, the ends were filled in, and the resulting plasmid was religated to form pRH1227. The PTDH3-RING-HRD1 allele was subcloned into pRH1153 (YIp/LEU2), which contained the normal HRD1 gene, to yield pRH1303. The GFP coding sequence was fused to the 5′ end of HRD1 through the PCR SOEing method, and the resulting plasmid was named pRH684 (YIp/TRP1). The GFP fusion portion of this construct was subcloned into the RING-HRD1 plasmid to yield pRH1231 (YIp/TRP1/RING-HRD1-GFP).
The C399S Hrd1p point mutation was generated by PCR SOEing and introduced into the HRD1 coding region of the plasmids described above to yield pRH1245 (YIp/TRPI/C399S-HRD1) and pRH1314 (YIp/TRPI/C399S-RING-HRD1).
The HRD3 truncation plasmids were constructed as follows. Deletion of codons 768–833 was accomplished by generating a DNA fragment containing a stop codon and XbaI site placed after codon 767 in the HRD3 coding region by PCR SOEing and introducing the DNA into pRH508. The subsequent plasmid was digested with XbaI to remove the DNA encoding the transmembrane domain and cytosolic extension, and the resulting plasmid was named pRH1244. Deletion of codons 393–833 was accomplished by digesting pRH508 with NdeI and religating the vector fragment to yield the plasmid pRH1219. Deletion of codons 24–356 was accomplished by generating a DNA fragment containing an SalI site placed after codon 24 in the HRD3 coding region by PCR SOEing and introducing the DNA into pRH508 (YIp/LEU2). The subsequent plasmid was digested with SalI to remove the intervening DNA, and the resulting plasmid was named pRH1242. The various HRD3 truncation alleles, HRD3 1–767, HRD3 1–390, and HRD3 357–833, were cloned by the TDH3 promoter in pRH632 to yield pRH1249, pRH1252, and pRH1248, respectively.
Strains and Transformations
Yeast and bacteria were cultured as described (Gardner et al. 1998). Harvest, lysis, and transformation protocols were all standard and identical to those described (Gardner et al. 1998).
All yeast strains were derived from the same genetic background used in all our previous work (Hampton and Rine 1994; Hampton et al. 1996b). The parent strain from which every subsequent strain was derived had the following genotype: his3D200 lys2-801 ade2-101 ura3-52 met2 hmg1::LYS2 hmg2::HIS3. The subsequent parent strains used for each appropriate experiment had the additional following genotypes and phenotypes: RHY853 (ura3-52::HMG2cd:: hmg2GFP trp1::hisG leu2Δ) expressed Hmg2p-GFP and the catalytic domain of Hmg2p as the sole source of Hmg2p; RHY1383 (ura3-52::1MYCHMG2:: hmg2GFP trp1::hisG leu2Δ) expressed normal, mevalonate pathway–regulated 1myc-Hmg2p and Hmg2p-GFP; RHY1886 (HMG2) expressed native Hmg2p; and RHY1914 (HMG2 ura3-52::1MYC-HMG2) expressed normal, mevalonate pathway–regulated 1myc-Hmg2p and native Hmg2p.
Flow Cytometry
Flow cytometric analyses were performed as described (Gardner et al. 1998). Living cells were analyzed by flow microfluorimetry using a FACSCalibur™ (Becton Dickinson) flow microfluorimeter with settings for fluorescein-labeled antibody analysis. Histograms were produced from 10,000 individual, log-phase cells.
Cycloheximide Chase Degradation Assay
Degradation of expressed, epitope-tagged proteins was evaluated by treatment of log phase strains with cycloheximide as described (Gardner et al. 1998). In brief, log phase cells were treated with 50 μg/ml cycloheximide to halt protein synthesis, and then lysed and immunoblotted after various incubation times to evaluate degradation as described (Gardner et al. 1998). myc-tagged or HA-tagged proteins were detected using the 9E10 or 12CA5 mAbs as described (Gardner et al. 1998).
In Vivo Cross-Linking
The in vivo cross-linking assay was modified from the in vitro assay described (Marcusson et al. 1994). Cells expressing the appropriate epitope-tagged proteins were grown to log phase (OD600 = 1.0) in 30 ml minimal medium, harvested by centrifugation, and resuspended in XL buffer (Marcusson et al. 1994) to a density of 6.0 OD600 units. Zymolase was added (final concentration 50 μg/ml) and dithiobis(succinimidyl-propionate) (DSP) was added in varying concentrations up to 400 μg/ml. Cells were incubated for 30 min at 30°C. Spheroplasts were harvested by centrifugation and lysed by resuspension in 300 μl SUME (Hampton and Bhakta 1997), plus the detergents deoxycholate (0.5%) and Triton X-100 (1%), which were in addition to SDS (1%), and 20 μM hydroxylamine to quench the cross-linking reaction. Supernatants were clarified by centrifugation for 15 min. Supernatants were added to 1 ml immunoprecipitation buffer (IP buffer) (Hampton and Bhakta 1997), containing 20–30 μl of the appropriate antisera. Samples were incubated for 16 h at 4°C. Protein A–sepharose CL-4B beads (100 μl of a 10% wt/vol solution) were added, and the samples were incubated for 2 h. Beads were harvested by centrifugation and washed once with IP buffer and once with wash buffer (Hampton and Bhakta 1997). Proteins were removed from the beads by addition of 50 μl 2× USB (Hampton and Bhakta 1997) and incubation at 55°C for 10 min.
Immunoblotting of precipitated proteins was performed as described (Gardner et al. 1998). In brief, separate 5- and 35-μl volumes of each precipitated sample were resolved on 8% SDS-PAGE gels and transferred to nitrocellulose. The 12CA5 anti-HA antibody was used to detect HA-tagged proteins, and a polyclonal anti-GFP antisera was used to detect GFP fusion proteins.
Native Coimmunoprecipitation
The native immunoprecipitation was adapted from Stack et al. 1993. Log phase yeast cells (0.75 OD600 units) were harvested by centrifugation and washed once with distilled water. Cells were resuspended in 0.5 ml zymolyase buffer (Marcusson et al. 1994). Zymolase (10 μg) was added to the cells, which were then incubated for 45 min at 30°C. Cells were washed once with 0.5 ml of ice-cold IP buffer minus detergent, then resuspended in 1 ml IP buffer. The resuspended cells were lysed by incubation on ice for 5 min. The supernatant was clarified by centrifugation for 5 min at 4°C, and again by centrifugation for 30 min at 4°C. For evaluation of total lysate protein, 20 μl of the final supernatant was removed and added to 20 μl of 2× USB. 7 μl rabbit anti-HA antisera was added to the remaining lysate to immunoprecipitate HA-tagged proteins, and the mixture was incubated overnight at 4°C. Protein A–sepharose CL-4B bead suspension (100 μl of a 10% wt/vol solution) was added to the mixture, which was then incubated for 2 h at 4°C. Beads were harvested by centrifugation and washed twice with IP buffer. Proteins were removed from the beads by addition of 40 μl 2× USB and incubation at 55°C for 10 min. Immunoblotting of HA-tagged proteins (5 μl of sample) or coprecipitated myc-tagged proteins (20 μl of sample) was performed with the 12CA5 or 9E10 mAbs, respectively.
Protease Protection Assay
Microsomes were prepared as described previously (Marcusson et al. 1994). Increasing amounts of protease were added to isolated microsomes, which were incubated on ice for 30 min. Reactions were stopped, and proteins were denatured by addition of an equal volume of 2× USB containing PMSF, followed by incubation at 65°C for 15 min. Samples were resolved by SDS-PAGE and immunoblotted with the appropriate antisera to assess levels of each protein.
Results
Hrd3p Interacted with the Hrd1p Transmembrane Domain
Previous studies indicated that Hrd1p and Hrd3p functioned in the same ER-associated degradation pathway and showed genetic interactions (Hampton et al. 1996a; Bordallo et al. 1998; Plemper et al. 1999; Wilhovsky et al. 2000). Therefore, we first examined if Hrd1p and Hrd3p physically interacted by chemical cross-linking and native immunoprecipitation assays.
Physical interactions between Hrd1p and Hrd3p were determined using an in vivo cross-linking assay modified from a procedure for cell lysates (Marcusson et al. 1994). The strains used for the cross-linking assays expressed fully functional, triple HA epitope–tagged versions of Hrd1p and/or Hrd3p from their native promoters. Each modified protein completely complemented the respective null alleles for ERAD when expressed in single copy from their native promoters (data not shown). When lysates were derived from cells treated with increasing amounts of cross-linker and Hrd1p was immunoprecipitated using Hrd1p-specific antisera, Hrd3p coimmunoprecipitated in a cross-linker concentration–dependent fashion (Fig. 1 a, top left). In the absence of cellular Hrd1p, no cross-linker–dependent Hrd3p coimmunoprecipitation was observed (Fig. 1 a, top right). The small amount of Hrd3p that coimmunoprecipitated with Hrd1p in the absence of cross-linker was roughly equivalent to that seen in the absence of Hrd1p, indicating that this amount was nonspecific.
Figure 1.
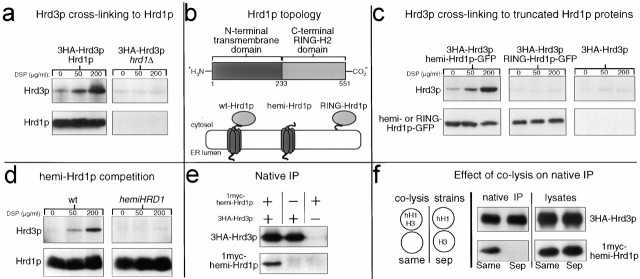
Hrd1p and Hrd3p interacted via the Hrd1p NH2-terminal transmembrane domain. (a) Hrd3p cross-linked to Hrd1p. Log phase cells expressing the indicated 3HA epitope–tagged proteins were treated with the indicated concentrations of DSP, lysed, and immunoprecipitated with anti-Hrd1p antisera. Precipitated proteins were next immunoblotted with an anti-HA mAb to detect coimmunoprecipitated 3HA-Hrd3p (top) or with an anti-Hrd1p polyclonal antisera to verify equal amounts of immunoprecipitated Hrd1p (bottom). (b) Cartoon depicting Hrd1p topology. Top row, linear representation of Hrd1p domains. The start and end of each domain are indicated by the number of the corresponding amino acid residue below. Bottom row, cartoon representations of the various Hrd1p constructs including wt-Hrd1p, only the Hrd1p transmembrane domain (hemi-Hrd1p), and only the COOH-terminal RING-H2 domain (termed RING-Hrd1p). (c) Hrd3p cross-linked to hemi-Hrd1p-GFP, but not RING-Hrd1p-GFP. Cells coexpressing 3HA-Hrd3p and the indicated Hrd1p-GFP fusion were subject to the cross-linking assay using anti-GFP antisera to immunoprecipitate the Hrd1p-GFP fusions from the lysates. Precipitated proteins were immunoblotted with an anti-HA mAb to detect coimmunoprecipitated 3HA-Hrd3p (top), or with an anti-GFP mAb to detect immunoprecipitated Hrd1p-GFP fusions (bottom). (d) hemi-Hrd1p expression inhibited Hrd3p cross-linking Hrd1p. The same cross-linking assay in panel a was performed with cells expressing 3HA-Hrd3p with or without the PTDH3-hemi-HRD1 allele. To compensate for lower Hrd1p in the hemi-Hrd1p cells (see Fig. 4 a), four times less lysate was used in the control lanes so that all lanes had identical amounts of immunoprecipitated Hrd1p. (e) Native coimmunoprecipitation of hemi-Hrd1p with Hrd3p. Cells expressing either 1myc-hemi-Hrd1p, 3HA-Hrd3p, or both proteins were lysed under nondenaturing conditions and immunoprecipitated with anti-HA polyclonal antisera. Immunoprecipitates were immunoblotted with the appropriate mAb to detect coimmunoprecipitated 1myc-hemi-Hrd1p (bottom) or immunoprecipitated 3HA-Hrd3p (top). (f) hemi-Hrd1p coimmunoprecipitated with Hrd3p only when expressed in the same cell. Same experiment as in panel d, except 1myc-hemi-Hrd1p and 3HA-Hrd3p were expressed either in the same cell or in separate cells (sep). Cells expressing each protein individually were mixed in equal quantities and lysed. Cells expressing both proteins were mixed with an equal number of empty cells to ensure an equal protein load and lysed (cartoon). Each lysate (right) was immunoblotted with an anti-myc mAb to detect total 1myc-hemi-Hrd1p (bottom) or with an anti-HA mAb to detect total Hrd3p (top).
We evaluated the importance of each Hrd1p domain in the Hrd3p interaction by testing Hrd1p truncation mutants and fusion proteins. We first tested if the Hrd1p NH2-terminal transmembrane domain, defined by residues 1–233, cross-linked with Hrd3p. In these and the following genetic studies, we refer to versions of Hrd1p that contained only the NH2-terminal transmembrane domain as hemi-Hrd1p because this domain is roughly one half the size of normal Hrd1p (Fig. 1 b). The Hrd1p/Hrd3p cross-linking experiment was repeated in a strain coexpressing hemi-Hrd1p-GFP, a fusion protein consisting of the NH2-terminal domain of Hrd1p fused to GFP, and 3HA-Hrd3p. Immunoprecipitation of hemi-Hrd1p-GFP with anti-GFP antisera resulted in coimmunoprecipitation of Hrd3p in a cross-linker–dependent manner (Fig. 1 c, top left). When the same experiment was performed on cells that did not express hemi-Hrd1p-GFP, no cross-linker–dependent Hrd3p coimmunoprecipitation was observed (Fig. 1 c, top right).
We also tested if the Hrd1p COOH-terminal RING-H2 domain cross-linked with Hrd3p when expressed as an isolated protein encompassing residues 224–551 of Hrd1p. We refer to the 327-residue COOH-terminal domain as RING-Hrd1p because it contains the RING-H2 motif (Fig. 1 b). When the same cross-linking assay was performed in strains coexpressing 3HA-Hrd3p and RING-Hrd1p-GFP, which contained GFP fused in frame to the extreme COOH terminus of RING-Hrd1p, no cross-linker–dependent Hrd3p coimmunoprecipitation was observed (Fig. 1 c, top middle). Similarly, the isolated RING-Hrd1p without GFP also did not cross-link to Hrd3p (data not shown). Thus, the isolated COOH-terminal RING-H2 domain did not appear to interact with Hrd3p, despite it having a dominant negative ERAD phenotype and ER membrane localization (see below).
These mapping experiments indicated that the Hrd1p NH2-terminal transmembrane domain was necessary and sufficient for the Hrd1p–Hrd3p interaction. Accordingly, we tested if overexpression of hemi-Hrd1p could compete with native Hrd1p for Hrd3p cross-linking. When hemi-Hrd1p was coexpressed from the strong TDH3 promoter (also known as the GAPDH promoter; Bitter and Egan 1984) in the strain used to examine the Hrd1p–Hrd3p interaction, Hrd3p cross-linking to Hrd1p was eliminated (Fig. 1 d, compare top panels). It is important to point out that hemi-Hrd1p expression resulted in decreased Hrd1p levels in the cell (see below, Fig. 3). To account for this, the same amount of total Hrd1p immunoprecipitated was loaded in each lane so that the amount of cross-linked Hrd3p could be directly compared.
Figure 3.

Characteristics of Hrd1p stability and degradation. (a) hemi-Hrd1p destabilized Hrd1p by Hrd3p sequestration. Stability of 3HA-Hrd1p was assessed by cycloheximide chase assay in isogenic strains with either the hrd3Δ allele, the PTDH3-hemi-HRD1 allele, or both alleles. Lysates were prepared at the indicated times after cycloheximide addition and immunoblotted for the levels of 3HA-Hrd1p using an anti-HA mAb. (b) The degradation of full-length Hrd1p was programmed by the COOH-terminal RING-H2 domain. Degradation of Hrd1p, C399S-Hrd1p, RING-Hrd1p, C399S-RING-Hrd1p, or hemi-Hrd1p was assessed in otherwise isogenic strains with wild-type (wt), hrd3Δ, or ubc7Δ alleles by cycloheximide chase assay. The presence of the C399S mutation in the degradation substrate is indicated over the appropriate blot. Lysates were prepared at the indicated times after cycloheximide addition and immunoblotted for the levels of 3HA-Hrd1p and 3HA-RING-Hrd1p using an anti-HA mAb or for the levels of 1myc-hemi-hrd1p using the 9E10 anti-myc mAb.
The interaction between Hrd3p and hemi-Hrd1p was further tested with a native coimmunoprecipitation assay, in which membrane proteins were solubilized under nondenaturing conditions to preserve protein–protein associations (Stack et al. 1993). We used a myc epitope–tagged version of hemi-Hrd1p to test if the NH2-terminal domain could, by itself, interact with Hrd3p. 1myc-hemi-Hrd1p, containing a single, myc epitope sequence placed in frame after residue 231 of hemi-Hrd1p, was coexpressed in cells that also expressed 3HA-Hrd3p. When 3HA-Hrd3p was immunoprecipitated using anti-HA antibodies, 1myc-hemi-Hrd1p was coimmunoprecipitated (Fig. 1 e). In contrast, hemi-Hrd1p coimmunoprecipitation was not observed when untagged Hrd3p was coexpressed in the cells (Fig. 1 e). Coimmunoprecipitation was not the result of association between the two proteins through the action of the detergent lysis buffers, as 1myc-hemi-Hrd1p coimmunoprecipitation only occurred when coexpressed in the same cells as 3HA-Hrd3p (Fig. 1 f).
Together, these experiments demonstrated that Hrd1p physically interacted with Hrd3p through the Hrd1p NH2-terminal transmembrane domain. The COOH-terminal, RING-H2 domain was dispensable for the interaction and did not appear to be able to interact with Hrd3p when expressed as an isolated protein.
Phenotypes and Suppression of HRD1 Truncation Alleles
It has been shown that expression of a full-length, mutant version of Hrd1p, C399S-Hrd1p, results in a dominant negative block in ERAD (Bordallo and Wolf 1999), which is suppressed by increased Hrd3p expression (Plemper et al. 1999). We tested each half of Hrd1p for dominant negative ERAD phenotypes and suppression by Hrd3p overexpression.
Overproduction of hemi-Hrd1p in wild-type strains strongly stabilized the ERAD substrate Hmg2p-GFP, resulting in increased cellular fluorescence in hemi-HRD1 cells compared with control cells as measured by flow cytometry (Fig. 2 a). The hemi-HRD1 stabilizing effect was identical to the effect of a previously reported dominant negative stabilizing HRD1 allele encoding a protein with the RING-H2 motif deleted (Bordallo et al. 1998) (Fig. 2 a). Expression of hemi-Hrd1p also stabilized the regulated ERAD substrate 1myc-Hmg2p (Fig. 2 b) and the constitutive ERAD substrate 6myc-Hmg2p (data not shown), as measured by the direct assay of each protein's stability.
Figure 2.
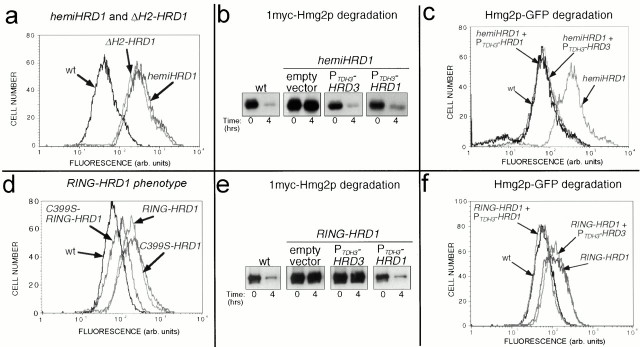
Dominant negative truncation mutants of Hrd1p. (a) hemi-Hrd1p expression stabilized Hmg2p-GFP similar to a RING-H2 deletion mutant of Hrd1p. Flow cytometry of log phase strains expressing Hmg2p-GFP (wt) or coexpressing hemi-Hrd1p or the RING-H2 motif deletion mutant of Hrd1p as indicated. The stabilizing effect of the hemi-HRD1 allele or the ΔH2-hrd1 allele was seen by the rightward shift in the fluorescence histogram compared with the wild-type strain. (b) Hrd3p overexpression reversed the hemi-HRD1 phenotype for 1myc-Hmg2p degradation. Isogenic strains expressing hemi-Hrd1p only (empty vector) or those coexpressing either the PTDH3-HRD3 or the PTDH3-HRD1 allele were assayed for 1myc-Hmg2p degradation by cycloheximide-chase assay, along with an isogenic wild-type strain (wt) without hemi-Hrd1p. Cell lysates were prepared at the indicated times after cycloheximide addition and immunoblotted using the 9E10 anti-myc mAb. (c) Hrd3p overexpression reversed the PTDH3-hemi-HRD1 phenotype for Hmg2p-GFP degradation. Flow cytometric analysis of strains expressing Hmg2p-GFP and the indicated alleles was performed as above in panel a. (d) RING-Hrd1p expression stabilized Hmg2p-GFP. Isogenic strains expressing Hmg2p-GFP and coexpressing either RING-Hrd1p (RING-HRD1), C399S-RING-Hrd1p (C399S-RING-HRD1), or full-length C399S-Hrd1p (C399S-hrd1) were analyzed by flow cytometry as in panel a. (e) Hrd3p overexpression did not suppress the RING-HRD1 phenotype for 1myc-Hmg2p degradation. Isogenic strains expressing RING-Hrd1p only (empty vector) or those coexpressing either the PTDH3-HRD3 allele or the PTDH3-HRD1 allele, were assayed for 1myc-Hmg2p degradation by cycloheximide chase assay along with a wild-type strain not expressing RING-Hrd1p. Cell lysates were prepared at the indicated times after cycloheximide addition and immunoblotted using the 9E10 anti-myc mAb. (f) Hrd3p overexpression did not reverse the PTDH3-RING-HRD1 phenotype for Hmg2p-GFP degradation. Flow cytometry of strains expressing Hmg2p-GFP and the indicated alleles was performed as in panel a.
Surprisingly, overexpression of the isolated Hrd1p COOH-terminal domain, RING-Hrd1p, also had a dominant negative stabilizing phenotype on Hmg2p-GFP degradation (Fig. 2 d). The stabilizing effect of RING-Hrd1p expression required an intact RING-H2 motif, as a point mutation in a critical cysteine within the motif, analogous to C399S in full-length Hrd1p, strongly suppressed the stabilizing effect of the RING-HRD1 allele (Fig. 2 d). This was in direct contrast to the stabilizing effect of the C399S point mutation in full-length Hrd1p (Fig. 2 d). The diminished stabilizing effect of C399S-RING-Hrd1p occurred despite its increased levels compared with RING-Hrd1p (see below). The dominant phenotype of the RING-HRD1 allele and its reversal by the C399S point mutation were also observed for degradation of 1myc-Hmg2p and 6myc-Hmg2p (data not shown).
The full-length C399S-Hrd1p dominant negative phenotype is suppressed by Hrd3p overexpression (Bordallo and Wolf 1999). Therefore, we evaluated the effect of Hrd3p overexpression on the dominant stabilizing phenotype of each Hrd1p truncation mutant. The dominant negative phenotype of the hemi-HRD1 allele was reversed by increased expression of Hrd3p from the TDH3 promoter, as measured by cycloheximide chase assay of 1myc-Hmg2p degradation (Fig. 2 b), or Hmg2p-GFP steady state fluorescence (Fig. 2 c). Suppression by HRD3 was complete, since the fluorescence histogram of the suppressed cells could be superimposed on that of the wild-type strain (Fig. 2 c). Importantly, neither the stability nor the steady state levels of hemi-Hrd1p were affected by increased Hrd3p expression (data not shown). Increased Hrd1p expression from the TDH3 promoter also reversed the hemi-HRD1 phenotype as expected (Fig. 2b and Fig. c).
Conversely, the dominant negative stabilizing phenotype of RING-Hrd1p was not suppressed by increased Hrd3p expression. The same expression levels of Hrd3p had no effect on the RING-HRD1–dependent stabilization of 1myc-Hmg2p or Hmg2p-GFP (Fig. 2e and Fig. f, respectively). The lack of suppression was not due to an inability of the RING-HRD1 effect to be suppressed, since increased Hrd1p expression did completely reverse the stabilizing effect of the RING-HRD1 allele (Fig. 2e and Fig. f). Thus, although RING-Hrd1p expression inhibited ERAD, the stabilizing effect was not through Hrd3p sequestration or inhibition, as would be expected by its lack of interaction with Hrd3p as shown above in the cross-linking studies.
The Hrd1p Transmembrane Region Mediated Hrd3p-dependent Stabilization of Hrd1p
One function of Hrd3p in ERAD is regulation of Hrd1p stability. In the absence of Hrd3p, the stability of Hrd1p is greatly reduced (Plemper et al. 1999). We evaluated the role of the Hrd1p NH2-terminal transmembrane domain in this action of Hrd3p. Expression of hemi-Hrd1p in a wild-type HRD3 strain mimicked the phenotype of the hrd3Δ allele, resulting in a similar decrease in the half-life and stability of Hrd1p (Fig. 3 a). Hrd1p was restabilized in this strain by increasing the expression of Hrd3p from the TDH3 promoter (Fig. 3 a). Thus, in this molecular readout of Hrd3p function, the isolated Hrd1p transmembrane domain sequestered Hrd3p and removed its stabilizing influence on endogenous Hrd1p.
The two domains of intact Hrd1p had distinct roles in Hrd3p-dependent regulation of Hrd1p stability. Hrd1p degradation was mediated by the COOH-terminal RING-H2 domain, whereas Hrd3p-dependent stabilization was mediated by the NH2-terminal transmembrane domain. In an hrd3Δ strain, Hrd1p degradation was dependent on a functional UBC7 gene and an intact RING-H2 motif. Loss of UBC7 function by introduction of a ubc7Δ allele completely stabilized Hrd1p (Fig. 3 b), as did introduction of the C399S mutation (Fig. 3 b). This was consistent with the previous analysis of Hrd1p degradation in the absence of HRD3 (Plemper et al. 1999). This was also consistent with the ubiquitin ligase activity of the Hrd1p RING-H2 domain (our unpublished results), and demonstrated the essential nature of the Hrd1p RING-H2 motif's activity in the degradation of Hrd1p. Degradation of isolated RING-Hrd1p, which lacks the NH2-terminal transmembrane region, was similarly dependent on UBC7 and its own functional RING-H2 motif (Fig. 3 b). However, RING-Hrd1p degradation was completely independent of Hrd3p, occurring with identical kinetics in the absence or presence of HRD3 (Fig. 3 b). This indicated that the transmembrane domain was critical for Hrd3p regulation of Hrd1p stability. In contrast, hemi-Hrd1p, which encompasses the isolated transmembrane domain, was exceedingly stable regardless of Hrd3p presence (Fig. 3 b).
The Hrd1p COOH-terminal RING-H2 Domain Was Located and Functioned in the Cytosol
It has been previously reported that the COOH-terminal RING-H2 domain is in the lumen of the ER (Bordallo et al. 1998). However, the homology of this domain with a class of ubiquitin ligases, and our concurrent studies of the Hrd1p biochemical function as a ubiquitin ligase (our unpublished results), led us to rigorously reevaluate the membrane orientation of the Hrd1p RING-H2 domain.
First, we tested the localization of intact Hrd1p and its truncation mutants by using the GFP fusions described in the interaction studies above. To aid visibility, all GFP fusions were expressed in strains with the ubc7Δ allele, since both the Hrd1p-GFP and the RING-Hrd1p-GFP were subject to degradation in a wild-type background (data not shown, and see Fig. 3). When expressed from the TDH3 promoter, both hemi-Hrd1p-GFP and RING-Hrd1p-GFP demonstrated identical cellular localization of GFP fluorescence (Fig. 4 a). The GFP fluorescence was primarily localized to a perinuclear band, as observed by 4,6-diamino-2-phenylindole (DAPI) staining of the nucleus (data not shown), with some GFP fluorescence localized to the periphery of the cell. This type of cellular localization is common for ER-localized membrane proteins (Parlati et al. 1995; Hampton et al. 1996b; Saito et al. 1999). The cellular localization of either hemi-Hrd1p-GFP or RING-Hrd1p-GFP was identical to that observed for full-length Hrd1p-GFP (Fig. 4 a), which has been previously determined to be ER localized (Bordallo et al. 1998). C399S-Hrd1p-GFP had identical ER localization as Hrd1p-GFP, as expected (Fig. 4 a). Furthermore, the cellular localization of each GFP construct was similar to Hmg2p-GFP (Fig. 4 a), a known ER-localized membrane protein (Hampton et al. 1996b).
Figure 4.

Orientation of Hrd1p proteins and complementation of a hrd1Δ allele by expression of both the RING-HRD1 and hemi-HRD1 alleles. (a) RING-Hrd1p-GFP was localized to the ER. Fluorescence microscopy was performed on otherwise isogenic ubc7Δ strains expressing RING-Hrd1p-GFP, Hrd1p-GFP, hemi-Hrd1p-GFP or C399S-Hrd1p-GFP. Cells were grown to log phase and observed by fluorescence microscopy. Position of the nucleus in each cell was visualized by DAPI staining (data not shown). Arrows indicate the perinuclear, ER localization of the cellular GFP fluorescence, which in all cases is similar to the ER localization of Hmg2p-GFP (right). (b) COOH-terminal RING-H2 domain of Hrd1p was exposed to the cytosol. Protease protection assay using isolated intact microsomes from strains expressing either 3HA-RING-Hrd1p, 3HA-Hrd1p, or 3HA-Hrd3p and 1myc-Hmg2p. Microsomes were treated with the indicated concentrations of trypsin in the absence or presence of Triton X-100 (X). Levels of each protein were assessed by immunoblotting using antibodies specific for the fused epitope tags or for the native protein in the case of Kar2p. For each protein observed, total amount in the cell was comparable to that found only in the crude microsomes (compare Lys lane with 0 lane). (c) Protease protection of untagged Hrd1p. Same experiment as in panel b, using both trypsin (left) or proteinase K (right). Hrd1p was immunoblotted with antisera against the 203 residue COOH-terminal region of Hrd1p. On the left, the black arrow indicates the trypsin-insensitive fragment of full-length Hrd1p, and the gray arrow indicates the trypsin-insensitive fragment for RING-Hrd1p. For the proteinase K experiment, the blot for 3HA-Hrd1p (second from left) was stripped and reprobed with an anti-HA mAb to detect both 3HA-Hrd1p and 3HA-Hrd3p (far right). (d) Coexpression of RING-Hrd1p and hemi-Hrd1p complemented the hrd1Δ allele for mevalonate pathway–regulated Hmg2p-GFP degradation. Cells containing the hrd1Δ allele and expressing Hmg2p-GFP and either RING-Hrd1p, hemi-Hrd1p, or both were grown to log phase and analyzed by flow cytometry (left). To assess proper regulation of Hmg2p-GFP degradation, zaragozic acid (10 μg/ml) or lovastatin (25 μg/ml) was added to cells early in log phase and cells were allowed to grow for 4 h before flow cytometric analysis (middle and right). (e) Coexpression of RING-Hrd1p and hemi-Hrd1p complemented the hrd1Δ allele for constitutive 6myc-Hmg2p degradation. Cells containing the hrd1Δ allele and expressing 6myc-Hmg2p and either RING-Hrd1p, hemi-Hrd1p, or both were subject to a cycloheximide chase assay. Cell lysates were prepared at the indicated times after cycloheximide addition and immunoblotted using the 9E10 anti-myc mAb.
ER membrane localization was expected for the fusions containing the NH2-terminal transmembrane domain. It was surprising that RING-Hrd1p-GFP behaved the same way since there are no predicted transmembrane spans or signal sequences within this protein, although such localization was consistent with its dominant inhibition of ERAD. From this, it seemed likely that RING-Hrd1p would be bound to the outside of the ER membrane. Therefore, we performed protease protection assays to test for cytosolic exposure of the RING-Hrd1p protein. When intact microsomes were isolated from cells expressing 3HA-RING-Hrd1p, nearly all of the immunoreactivity was associated with the pelleted microsomes (Fig. 4 b, 3HA-RING-Hrd1p; Lys). Brief trypsin treatment at several concentrations of protease completely removed the HA immunoreactivity without need for added detergent (.2 and .5). The loss of epitope immunoreactivity was identical to that of 1myc-Hmg2p (Fig. 4 b), which has a myc epitope sequence placed in the large COOH-terminal, cytosolic region (Hampton and Bhakta 1997). In contrast, the immunoreactivity of the completely lumenal Kar2p was unaffected in the absence of detergent (Fig. 4 b), whereas 3HA-Hrd3p showed a small shift in molecular weight (Fig. 4 b), consistent with removal of its small, cytoplasmic COOH-terminal region (Plemper et al. 1999; Saito et al. 1999). All protected proteins were rendered completely trypsin sensitive by pretreatment of the microsomes with detergent (Fig. 4 b, .5X).
The Hrd1p COOH-terminal RING-H2 domain was associated with the ER and accessible from the cytosolic face of the ER membrane when expressed as an isolated protein. However, it has been reported that this domain in full-length Hrd1p was exclusively on the lumenal side of the ER membrane (Bordallo et al. 1998). Therefore, we also evaluated the protease sensitivity of the same region when part of intact Hrd1p by using 3HA-Hrd1p, which contains the epitope tag at the extreme COOH terminus and completely complements an hrd1Δ allele for ER degradation. In an identical protease protection experiment, the HA immunoreactivity for the COOH-terminal tagged, full-length 3HA-Hrd1p was also completely destroyed by trypsin addition in the absence of any added detergent (Fig. 4 b), exactly like the cytosolic tag on Hmg2p and in striking contrast to Hrd3p or Kar2p. No accumulation of any protease-insensitive bands was seen for 3HA-Hrd1p (data not shown; see also Fig. 4 c, far right), indicating that the HA epitope tag was completely destroyed by protease treatment. Thus, the COOH terminus of full-length Hrd1p was also present on the cytosolic face of the ER membrane.
To ensure that these results were not caused by the added epitope sequences, we repeated our experiment using untagged Hrd1p and polyclonal antisera specific for residues 348–551 of Hrd1p. Using the polyclonal antisera, we found that COOH-terminal RING-H2 domain of normal, untagged, full-length Hrd1p was clearly trypsin sensitive in the absence of any added detergent and was no different from the HA-tagged version of full-length Hrd1p (Fig. 4 c, left panels). Control proteins Hrd3p, Kar2p and 1myc-Hmg2p showed identical responses to those determined in Fig. 4 b (data not shown). However, in contrast to the experiments using the HA mAb for detection, use of the polyclonal Hrd1p antisera did result in detection of a 32-kD trypsin-insensitive proteolytic fragment when microsomes containing either tagged or untagged full-length Hrd1p were subject to detergent-free trypsin digestion (Fig. 4 c, black arrow). The isolated, untagged COOH-terminal RING-Hrd1p protein was similarly trypsin sensitive (Fig. 4 c), also yielding a trypsin-protected proteolytic fragment that was 16 instead of 32 kD (gray arrow). To resolve whether the protease insensitivity of this fragment from the RING-H2 domains was specific for trypsin or was instead indicative of a protected lumenal localization, we repeated these studies using proteinase K. In fact, the entire COOH-terminal region of either full-length Hrd1p or the isolated RING-Hrd1p was completely proteinase K sensitive (>95% digestion) in the absence of detergent (Fig. 4 c, right panels), with no accumulation of a protected, immunoreactive fragment. In contrast, the mostly lumenal 3HA-Hrd3p was insensitive to proteinase K addition in the absence of detergent (Fig. 4 c, far right), indicating that the microsomes were intact. Thus, the partial trypsin insensitivity of the Hrd1p COOH-terminal domain appeared not to be a result of lumenal sequestration, but rather a structural feature of the COOH-terminal region.
Our protease protection data indicated that the Hrd1p COOH-terminal RING-H2 domain, whether expressed alone or as part of Hrd1p, was exposed to the cytosolic face of the ER membrane. As an independent and stringent test of this idea, we asked if the isolated Hrd1p COOH-terminal domain could function in ERAD when expressed with the isolated transmembrane domain. Indeed, coexpression of hemi-Hrd1p and RING-Hrd1p partially complemented the hrd1Δ allele for Hmg2p-GFP degradation, whereas expression of either hemi-Hrd1p or RING-Hrd1p alone did not (Fig. 4 d, left). Furthermore, the partially restored Hmg2p-GFP degradation was normally regulated by inhibitors of the mevalonate pathway, as seen by the expected effects on cellular Hmg2p-GFP fluorescence from addition of the degradation-stimulating zaragozic acid or degradation-slowing lovastatin (Fig. 4 d, middle and right, ZA and Lov, respectively). Coexpression of both hemi-Hrd1p and RING-Hrd1p also complemented the hrd1Δ allele for the constitutive degradation of misfolded 6myc-Hmg2p as measured directly by cycloheximide chase (Fig. 4 e).
Thus, the COOH-terminal RING-H2 domain of Hrd1p was localized to the cytosolic side of the ER membrane when part of full-length Hrd1p or when expressed in isolation as RING-Hrd1p.
Hrd3p Lumenal Determinants Mediated Stabilization of Hrd1p and a Separate Function in ERAD
The bulk of the Hrd3p sequence is found in the ER lumen with a small COOH-terminal region that is located in the cytosol (Plemper et al. 1999; Saito et al. 1999; and Fig. 4 b). The RING-H2 domain of Hrd1p, which is required for both ERAD and Hrd1p degradation, is located in the cytosol (Fig. 4b and Fig. c). Since Hrd3p regulates the activity of RING-H2 domain, we examined if this was mediated by the Hrd3p lumenal domain.
We prepared several truncation mutants of HRD3 in order to examine the role of the Hrd3p lumenal regions in both Hrd1p stability and ERAD. The domain organization of Hrd3p is shown in Fig. 5 a. The Hrd3p sequence includes an NH2-terminal lumenal domain (residues 1–767) containing a cleavable NH2-terminal signal sequence and a region (residues 391–767) with high homology to C. elegans SEL-1 and murine sel-1l (35% identity and 58% similarity) (Hampton et al. 1996a; Grant and Greenwald 1997; Donoviel et al. 1998), followed by a COOH-terminal transmembrane span and a poorly conserved COOH-terminal cytosolic domain. We engineered a truncated HRD3 allele that expressed only the lumenal domain of Hrd3p from the normal promoter (HRD31–767). When the resulting lumenal Hrd3p1–767 was expressed from single copy in hrd3Δ cells, both Hrd1p stability and ER degradation of Hmg2p were restored (Fig. 5b and Fig. c). Complementation of the hrd3Δ allele by the purely lumenal Hrd3p1–767 was complete, as observed by the superimposable Hmg2p-GFP cellular fluorescence histograms from the HRD31–767 cells and normal HRD3 cells (Fig. 5 d). Thus, the transmembrane and cytosolic regions of Hrd3p were dispensable for both Hrd3p-mediated stabilization of Hrd1p and ERAD. Since the critical Hrd1p RING-H2 domain is in the cytosol and Hrd3p1–767 is entirely lumenal (data not shown), the stable transmembrane domain of Hrd1p must transmit regulatory information across the ER membrane from lumenal determinants of Hrd3p to the cytosolic Hrd1p RING-H2 domain.
Figure 5.

Hrd3p was required for ER degradation, not just Hrd1p stability. (a) Topology of Hrd3p. The start and end of each putative domain are marked with the corresponding amino acid residue number in the Hrd3p coding sequence. (b) Hrd1p stability in the presence of Hrd3p truncation mutants. Stability of 3HA-Hrd1p was assessed by cycloheximide chase assay in isogenic strains coexpressing 3HA-Hrd1p and 1myc-Hmg2p with either the normal HRD3 allele, the hrd3Δ allele, the HRD3 1–767 allele, the hrd3 1–392 allele, or the hrd3 357–833 allele. Lysates were prepared at the indicated times after cycloheximide addition and immunoblotted for the levels of 3HA-Hrd1p using an anti-HA mAb (top) or levels of 1myc-Hmg2p using an anti-myc mAb (bottom). (c) Hmg2p-GFP degradation in the presence of various Hrd3p truncation mutants. Strains expressing Hmg2p-GFP with either the normal HRD3 allele, the hrd3Δ allele, the HRD3 1–767 allele, the hrd3 1–392 allele, or the hrd3 357–833 allele were analyzed by flow cytometry.
Although Hrd3p is clearly important for ERAD and Hrd1p stabilization, the effect of the hrd3Δ allele can be suppressed by overexpression of Hrd1p (Plemper et al. 1999; see Fig. 6 c). This could be interpreted to mean that the only function of Hrd3p is maintenance of Hrd1p steady state levels. To address this issue, we have looked for and discovered an allele of HRD3 that still allows Hrd1p stability, but does not allow normal ERAD. When codons 24–356 were excised from HRD3 (so that the signal sequence was still made), expression of the truncated Hrd3p, Hrd3p357–833, allowed stabilization of Hrd1p (Fig. 5 b), but did not allow ERAD of 1myc-Hmg2p (Fig. 5 c) or Hmg2p-GFP (Fig. 5 d). In fact, comparison of the cellular fluorescence histograms showed that cells expressing Hrd3p357–833 were equally deficient for ERAD as cells with the hrd3Δ allele, despite the restored stability and steady state levels of Hrd1p. Thus, the NH2-terminal region of Hrd3p (residues 1–356) appeared to be involved in aspects of ERAD distinct from Hrd1p stability. Expression of only the first 390 residues of Hrd3p did not allow Hrd1p stability or ERAD. Thus, Hrd1p stabilization required determinants in the COOH-terminal portion of Hrd3p lumenal domain (residues 357–783), whereas the NH2-terminal portion was dispensable for Hrd1p stability. Both regions were essential for ERAD when Hrd1p was expressed from its native locus.
Figure 6.
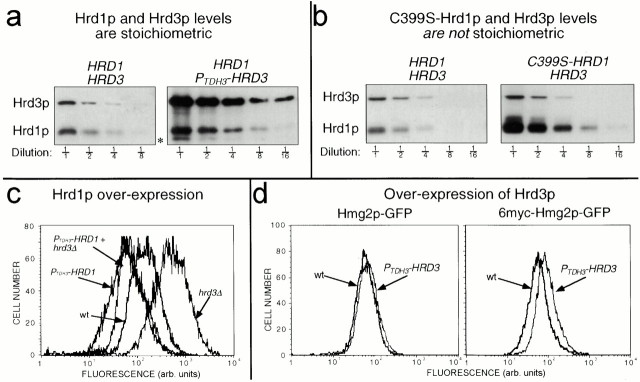
Regulation of Hrd1p by Hrd3p results in equal stoichiometry. (a) Hrd1p and Hrd3p are expressed at equivalent levels in cells. Log phase cells coexpressing 3HA-Hrd1p from its native promoter and either 3HA-Hrd3p from its native promoter or the TDH3 promoter (PTDH3HRD3) were lysed and immunoblotted with an anti-HA mAb to determine the levels of 3HA-Hrd1p and 3HA-Hrd3p. Fractions indicate dilutions of original lysates. Asterisk denotes a proteolytic product of Hrd3p as a result of cell lysis. (b) Effect of C399S mutation on Hrd1p levels. Strains that coexpressed 3HA-Hrd3p from its native promoter and either 3HA-Hrd1p or 3HA-C399S-Hrd1p from the native HRD1 promoter were lysed and immunoblotted with an anti-HA mAb as in panel a. (c) Hrd1p overexpression enhanced Hmg2p-GFP degradation and suppressed the stabilizing effect of the hrd3Δ allele. Strains expressing Hmg2p-GFP with either the HRD3 or hrd3Δ allele and carrying the PTDH3-HRD1 allele were analyzed by flow cytometry. (d) Hrd3p overexpression slightly stabilized both Hmg2p-GFP and 6myc-Hmg2p-GFP. Hmg2p-GFP or 6myc-Hmg2p-GFP steady state levels in otherwise isogenic strains were determined by flow cytometric analysis, with or without the PTDH3-HRD3 allele.
Stoichiometry of the Hrd1p/Hrd3p Complex
Hrd3p determines Hrd1p stability and, thus, Hrd1p steady state levels. The simplest model is that Hrd3p and Hrd1p form a complex and only Hrd1p molecules so engaged are stable. We compared the natural steady state levels of Hrd1p and Hrd3p to estimate the in vivo stoichiometry. To do this, we used strains coexpressing identically tagged (triple HA) versions of each protein from single coding regions under the control of their native promoters. In this way, each protein could be immunoblotted for identical epitopes using identical conditions and reagents. When identically tagged versions of Hrd1p and Hrd3p were expressed from their genomic loci, the resulting steady state levels of 3HA-Hrd1p and 3HA-Hrd3p were nearly equivalent, as determined by intensity of signal and diminution by dilution (Fig. 6 a, left). The similarity of Hrd1p and Hrd3p levels was consistent with the simple model that Hrd1p was stabilized by formation of a stoichiometric complex with Hrd3p.
We next examined if the amount of cellular Hrd3p determined the steady state level of Hrd1p through transstabilization. If Hrd1p were only stable when bound to Hrd3p, then increasing Hrd3p expression would be expected to result in increased Hrd1p levels by allowing increased stable complexes. Indeed, elevation of Hrd3p levels ∼20-fold by expression from the TDH3 promoter resulted in a 3-fold increase in Hrd1p steady state levels (Fig. 6 a, compare right with left panel). Clearly Hrd1p was not increased to the same extent as Hrd3p. However, by a costabilization model, the maximal possible effect of elevating Hrd3p would occur when every molecule of Hrd1p was stable. To evaluate this maximum effect, we compared the levels of 3HA-Hrd1p to the levels of the identically expressed, completely stable 3HA-C399S-Hrd1p (Fig. 3b). Cellular levels of the stable C399S-Hrd1p were approximately fourfold higher than wild-type Hrd1p levels (Fig. 6 b, compare right with left panel). Furthermore, elevation of Hrd3p levels had no effect on the steady state level of C399S-Hrd1p (data not shown). Thus, the effect of Hrd3p overexpression on Hrd1p levels was close to the maximal effect caused by the in cis stabilization of every Hrd1p molecule synthesized.
The cellular level of Hrd1p is demonstrably important in ERAD. Hrd1p is rate limiting for a variety of substrates, including Hmg2p and related proteins. Overexpression of Hrd1p resulted in enhanced degradation of Hmg2p-GFP (Fig. 6 c). Furthermore, Hrd1p overexpression suppressed the stabilizing effect of the hrd3Δ allele for Hmg2p-GFP degradation (Fig. 6 c), and CPY* degradation (Plemper et al. 1999). From these observations alone, it would seem that the sole function of Hrd3p is to ensure sufficient amounts of Hrd1p. If so, then Hrd3p overexpression, with the concomitant increase in the rate-limiting Hrd1p, would be expected to increase the degradation of HRD pathway substrates. However, the effect of Hrd3p overexpression on HRD-dependent degradation was more complex. The degradation of two separate substrates, mevalonate pathway–regulated Hmg2p-GFP and constitutive 6myc-Hmg2p-GFP, was slightly decreased (Fig. 6 d), despite the fact that Hrd1p levels were increased. These results further suggested that Hrd3p has separate functions in addition to simple maintenance of Hrd1p levels, and are consistent with our discovery of a Hrd3p allele that allowed Hrd1p stabilization, but did not allow ERAD in strains with normal levels of Hrd1p (Fig. 5).
Discussion
Both Hrd1p and Hrd3p are broadly involved in ER degradation. In this work, we have demonstrated that these two proteins interact via the NH2-terminal transmembrane region of Hrd1p. By rigorously evaluating the orientation of the Hrd1p, we have discovered that the RING-H2 domain is present and functions in the cytosol. The regions of Hrd3p required for control of Hrd1p stability and other aspects of ERAD reside solely in the ER lumen. Thus, the HRD complex, involving at least Hrd1p and Hrd3p, functions on both sides of the ER membrane and communicates across this barrier through the Hrd1p transmembrane domain to coordinate the cytosolic Hrd1p RING-H2 domain activity with events that occur on separate sides of the ER membrane.
Cytosolic Localization of the Hrd1p COOH-terminal RING-H2 Domain
To interpret our experiments and understand Hrd1p function, we evaluated the cellular location and orientation of the RING-H2 domain in relation to the ER membrane, both when part of the full-length, native protein and when expressed as an isolated protein. We have shown by numerous direct biochemical studies that, in either case, the Hrd1p COOH-terminal domain was exposed to the cytosol. Furthermore, the isolated Hrd1p COOH-terminal domain functioned normally in ERAD when expressed in the cytosol in trans with the isolated NH2-terminal transmembrane domain.
Thus, despite the report of a lumenal localization for the Hrd1p RING-H2 domain (Bordallo et al. 1998), we have clearly shown that the COOH-terminal RING-H2 domain of full-length Hrd1p was exposed to the cytosol. The reason why our results directly opposed those reported is not clear. The previous conclusions were reached by observation of Hrd1p's complete resistance to trypsin (Bordallo et al. 1998). Although we did not observe this, we did find that the Hrd1p COOH-terminal domain, whether expressed alone or as part of Hrd1p, was partially resistant to this protease. Perhaps the intrinsic trypsin insensitivity of the Hrd1p COOH terminus varies between different strains, allowing for the observation of complete protection in some circumstances. However, the Hrd1p COOH-terminal region was completely proteinase K sensitive in the same protocol, indicating that the trypsin resistance is a feature of Hrd1p structure and not lumenal sequestration.
A Functionally Important Stoichiometric Hrd1p–Hrd3p Complex Mediated by the Hrd1p Transmembrane Domain
Using different biochemical and genetic assays, we have shown that Hrd1p and Hrd3p physically interacted through the Hrd1p NH2-terminal transmembrane domain, which was both necessary and sufficient for complex formation. In the absence of Hrd3p, Hrd1p is subject to RING-H2 domain–dependent degradation mediated by the ER-localized Ubc7p (Plemper et al. 1999; and see above), whereas in the presence of Hrd3p, it is stable and abundant. The isolated Hrd1p RING-H2 domain similarly programs its own degradation, but in a manner independent of Hrd3p. Thus, the Hrd1p transmembrane domain allowed Hrd3p-dependent stabilization of the Hrd1p RING-H2 domain. Since the transmembrane domain itself was quite stable regardless of Hrd3p presence, it appeared that the transmembrane domain served as a transducer for lumenal Hrd3p regulation to control the cytosolic Hrd1p RING-H2 domain activity.
A model for the Hrd1p–Hrd3p complex required knowledge of the relative levels of the two proteins. It has been suggested that levels of Hrd1p and Hrd3p were highly disparate with Hrd1p in excess (Plemper et al. 1999), thus favoring a catalytic role for Hrd3p stabilization of Hrd1p. However, we directly evaluated the relative natural levels of Hrd1p and Hrd3p using identically tagged versions of each protein expressed in the same strain from the native promoters. By our analysis, the relative levels of Hrd1p and Hrd3p were actually quite similar. Thus, we propose that Hrd3p forms a stoichiometric complex with Hrd1p, via the Hrd1p transmembrane domain, allowing stabilization of Hrd1p and normal ERAD. By this model, the levels of Hrd1p are contingent on the levels of Hrd3p, with the maximal amount of Hrd1p occurring when all Hrd1p molecules are Hrd3p associated. Any residual, unbound Hrd1p would be degraded by the unbridled action of the RING-H2 domain. Indeed, the level of Hrd3p did influence the amount of Hrd1p in the cell, and this effect was similar to that observed when every Hrd1p molecule was stabilized by the in cis C399S mutation.
The Lumenal Functions of Hrd3p
Most of the Hrd3p sequence resides in the lumen of the ER (Plemper et al. 1999; Saito et al. 1999), yet one of its critical functions is to limit the degradation of Hrd1p, which depends on the ubiquitin ligase activity of the cytosolic Hrd1p RING-H2 domain. This suggested that Hrd3p regulation of Hrd1p stability was through lumen to cytosol signaling mediated by the Hrd1p transmembrane domain. We directly tested this idea by engineering an allele of HRD3 that expressed a version of Hrd3p containing only the signal sequence and the lumenal domain. This version of Hrd3p completely complemented the stabilizing effect of the hrd3Δ allele by restoration of both Hrd1p stability and ERAD, indicating that purely lumenal determinants of Hrd3p functioned in this regard.
This regulatory arrangement, in which the Hrd1p hydrophobic anchor functions in transmembrane communication between another molecule and the active RING-H2 domain, has interesting implications. Although the complete role of this signaling process in ERAD is not known, it could well be part of the communication that must occur in coupling lumenal substrate scanning to cytosolic substrate tagging and destruction. More generally, there are other examples of proteins with a RING-H2 domain connected to a transmembrane anchor (Gemmill et al. 1998, Okumoto et al. 1998; Lorick et al. 1999). If these and other unknown membrane-associated RING-H2 domains are regulated in a similar manner as Hrd1p, then the interactions and location of the corresponding anchors will play a key role in their function and biology.
In parallel studies, we have demonstrated that Hrd1p functions both in vivo and in vitro as a RING-H2 ubiquitin ligase (our unpublished results). Thus, Hrd1p and Hrd3p appear to be part of an ERAD-specific ubiquitin ligase complex that functions in the detection and targeting of ERAD substrates. One main function of Hrd3p is to effect the stabilization of Hrd1p, preventing the RING-H2 domain from programming Hrd1p degradation possibly through autoubiquitination. However, our studies with truncation alleles of Hrd3p and overexpression of Hrd3p indicated that Hrd1p stabilization, although clearly important for ERAD, was not the only function of Hrd3p. By our current model, Hrd3p acts to modulate Hrd1p RING-H2 ubiquitin ligase activity by communication through the Hrd1p NH2-terminal transmembrane domain. In the absence of substrate, Hrd3p stabilizes Hrd1p by binding the Hrd1p transmembrane domain and suppressing the cytosolic RING-H2 domain ubiquitin ligase activity. When substrate is present, Hrd3p senses the requirement for Hrd1p RING-H2 domain ubiquitin ligase activity, possibly by directly binding substrate, and signals through the Hrd1p transmembrane domain to activate Hrd1p function in a correct temporal and spatial manner, perhaps by coordinating retrotranslocation with Hrd1p RING-H2 ubiquitin ligase activity. From our truncation analysis, it may be that the sensing function is mediated by determinants in the NH2-terminal portion of the Hrd3p (1–390), whereas determinants that regulate the RING-H2 domain ubiquitin ligase activity and Hrd1p stability lie in the COOH-terminal portion of the lumenal domain, although clearly more analysis is required. It is important to note that Hrd1p overexpression did allow ERAD substrate degradation in the absence of Hrd3p, suggesting that Hrd3p was not required for ERAD. However, it is likely that the elevated levels of Hrd1p under these conditions, approximately eightfold higher than in wild-type cells (data not shown), are sufficient to allow lower efficiency interaction between Hrd1p and ERAD substrates, which may normally be aided by Hrd3p.
Implications of the Hrd1p–Hrd3p Complex
Hrd3p determines Hrd1p–Hrd3p stoichiometry by controlling Hrd1p degradation as a likely result of Hrd1p autoubiquitination. Stabilization as a tactic to ensure correct stoichiometry is a feature of other complexes such as T cell receptors, low density lipoproteins, and yeast transcription factors (Lippincott-Schwartz et al. 1988; Wileman et al. 1993; Fisher et al. 1997; Zhou et al. 1998). This mechanism may also be involved in self-regulation of ubiquitination machinery. In several cases, isolated RING-H2 proteins from known E3 complexes, including Hrd1p, catalyze their own ubiquitination in vitro (Joazeiro et al. 1999; Lorick et al. 1999; Skowyra et al. 1999; Fang et al. 2000; and our unpublished results). Similarly, the F box subunit of the SCF E3 ligase is susceptible to complex-catalyzed ubiquitination (Galan and Peter 1999). Furthermore, it has recently been demonstrated that the RING-H2 component of the SCF E3 complex, ROC1, is degraded in a proteasome-dependent manner, but is stabilized by association with cullins (Ohta et al. 1999b). Thus, it is likely that self-catalyzed ubiquitination and subsequent degradation of E3 components is a general mode of self-regulation to maintain complex stoichiometry. In any event, it is clear that the HRD gene–encoded ERAD E3 complex is a dynamic, self-regulating one, with a separate region of Hrd1p mediating ubiquitination of self and substrate and another region mediating Hrd3p-dependent communication across the ER membrane.
Acknowledgments
We thank Dr. Robert Rickert for use of the FACSCalibur™ flow microfluorimeter, and Dr. Suresh Subramani for the anti-GFP antiserum (Department of Biology, University of California at San Diego). R.G. Gardner thanks Pamela Kolinsky for her insight and participation in a complex interaction. R.Y. Hampton wishes to thank Vivek Malhotra for advice on biochemistry and for his complex philosophy.
This work was supported by National Institutes of Health grant DK5199601 (to R.Y. Hampton) and a Searle Scholarship (to R.Y. Hampton).
Footnotes
Abbreviations used in this paper: ERAD, ER-associated degradation; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GFP, green fluorescent protein; HA, hemagglutinin; SOEing, splicing by overlap extension.
References
- Biederer T., Volkwein C., Sommer T. Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin-proteasome pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:2069–2076. [PMC free article] [PubMed] [Google Scholar]
- Bitter G.A., Egan K.M. Expression of heterologous genes in Saccharomyces cerevisiae from vectors utilizing the glyceraldehyde-3-phosphate dehydrogenase gene promoter. Gene. 1984;32:263–274. doi: 10.1016/0378-1119(84)90002-7. [DOI] [PubMed] [Google Scholar]
- Bordallo J., Wolf D.H. A RING-H2 finger motif is essential for the function of Der3/Hrd1 in endoplasmic reticulum associated protein degradation in the yeast Saccharomyces cerevisiae . FEBS Lett. 1999;448:244–248. doi: 10.1016/s0014-5793(99)00362-2. [DOI] [PubMed] [Google Scholar]
- Bordallo J., Plemper R.K., Finger A., Wolf D.H. Der3p-Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell. 1998;9:209–222. doi: 10.1091/mbc.9.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun K.T., Bar-Nun S., Simoni R.D. The regulated degradation of 3-hydroxy-3-methylglutaryl-CoA reductase requires a short-lived protein and occurs in the endoplasmic reticulum. J. Biol. Chem. 1990;265:22004–22010. [PubMed] [Google Scholar]
- Ciccarelli E., Alonso M.A., Cresteil D., Bollen A., Jacobs P., Alvarez F. Intracellular retention and degradation of human mutant variant of a alpha 1-antitrypsin in stably transfected Chinese hamster ovary cell lines. Eur. J. Biochem. 1993;213:271–276. doi: 10.1111/j.1432-1033.1993.tb17759.x. [DOI] [PubMed] [Google Scholar]
- Donoviel D.B., Donoviel M.S., Fan E., Hadjantonakis A., Bernstein A. Cloning and characterization of Sel-1l, a murine homolog of the C. elegans sel-1 gene. Mech. Dev. 1998;78:203–207. doi: 10.1016/s0925-4773(98)00146-4. [DOI] [PubMed] [Google Scholar]
- Edwards P.A., Lan S.F., Tanaka R.D., Fogelman A.M. Mevalonolactone inhibits the rate of synthesis and enhances the rate of degradation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in rat hepatocytes. J. Biol. Chem. 1983;258:7272–7275. [PubMed] [Google Scholar]
- Fang S., Jensen J.P., Ludwig R.L., Vousden K.H., Weissman A.M. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 2000;275:8945–8951. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- Finger A., Knop M., Wolf D.H. Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur. J. Biochem. 1993;218:565–574. doi: 10.1111/j.1432-1033.1993.tb18410.x. [DOI] [PubMed] [Google Scholar]
- Fisher E.A., Zhou M., Mitchell D.M., Wu X., Omura S., Wang H., Goldberg A.L., Ginsberg H.N. The degradation of apolipoprotein B100 is mediated by the ubiquitin-proteasome pathway and involves heat shock protein 70. J. Biol. Chem. 1997;272:20427–20434. doi: 10.1074/jbc.272.33.20427. [DOI] [PubMed] [Google Scholar]
- Friedlander R., Jarosch E., Urban J., Volkwein C., Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2000;2:379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- Galan J.M., Peter M. Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc. Natl. Acad. Sci. USA. 1999;96:9124–9129. doi: 10.1073/pnas.96.16.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R., Cronin S., Leader B., Rine J., Hampton R. Sequence determinants for regulated degradation of yeast 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell. 1998;9:2611–2626. doi: 10.1091/mbc.9.9.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmill R.M., West J.D., Boldog F., Tanaka N., Robinson L.J., Smith D.I., Li F., Drabkin H.A. The hereditary renal cell carcinoma 3;8 translocation fuses FHIT to a patched-related gene, TRC8. Proc. Natl. Acad. Sci. USA. 1998;95:9572–9577. doi: 10.1073/pnas.95.16.9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B., Greenwald I. Structure, function, and expression of SEL-1, a negative regulator of LIN-12 and GLP-1 in C. elegans . Development. 1997;124:637–644. doi: 10.1242/dev.124.3.637. [DOI] [PubMed] [Google Scholar]
- Hampton R.Y., Rine J. Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J. Cell Biol. 1994;125:299–312. doi: 10.1083/jcb.125.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton R.Y., Bhakta H. Ubiquitin-mediated regulation of 3-hydroxy-3-methylglutaryl-CoA reductase. Proc. Natl. Acad. Sci. USA. 1997;94:12944–12948. doi: 10.1073/pnas.94.24.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton R.Y., Gardner R.G., Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein Mol. Biol. Cell. 7 1996. 2029 2044a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton R.Y., Koning A., Wright R., Rine J. In vivo examination of membrane protein localization and degradation with green fluorescent protein Proc. Natl. Acad. Sci. USA. 93 1996. 828 833b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M.M., Finger A., Schweiger M., Wolf D.H. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- Horton R.M., Hunt H.D., Ho S.N., Pullen J.K., Pease L.R. Engineering hybrid genes without the use of restriction enzymesgene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Joazeiro C.A.P., Wing S.S., Huang H., Leverson J.D., Hunter T., Liu Y. The tyrosine kinase negative regulator c-CBL as a RING-type E2-dependent ubiquitin-protein ligase. Science. 1999;286:309–312. doi: 10.1126/science.286.5438.309. [DOI] [PubMed] [Google Scholar]
- Knop M., Finger A., Braun T., Hellmuth K., Wolf D.H. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Bonifacino J.S., Yuan L.C., Klausner R.D. Degradation from the endoplasmic reticulumdisposing of newly synthesized proteins. Cell. 1988;54:209–220. doi: 10.1016/0092-8674(88)90553-3. [DOI] [PubMed] [Google Scholar]
- Lorick K.L., Jensen J.P., Fang S., Ong A.M., Hatakeyama S., Weissman A.M. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. USA. 1999;96:11364–11369. doi: 10.1073/pnas.96.20.11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcusson E.G., Horazdovsky B.F., Cereghino J.L., Gharakhanian E., Emr S.D. The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell. 1994;77:579–586. doi: 10.1016/0092-8674(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Nakanishi M., Goldstein J.L., Brown M.S. Multivalent control of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J. Biol. Chem. 1988;263:8929–8937. [PubMed] [Google Scholar]
- Ohta T., Michel J.J., Schottelius A.J., Xiong Y. ROC1, a homolog of APC11, represents a family of cullin partners with an associated ubiquitin ligase activity Mol. Cell. 3 1999. 535 541a [DOI] [PubMed] [Google Scholar]
- Ohta T., Michel J.J., Xiong Y. Association with cullin partners protects ROC proteins from proteasome-dependent degradation Oncogene. 18 1999. 6758 6766b [DOI] [PubMed] [Google Scholar]
- Okumoto K., Itoh R., Shimozawa N., Suzuki Y., Tamura S., Kondo N., Fujiki Y. Mutations in PEX10 is the cause of Zellweger peroxisome deficiency syndrome of complementation group B. Hum. Mol. Genet. 1998;7:1399–1405. doi: 10.1093/hmg/7.9.1399. [DOI] [PubMed] [Google Scholar]
- Parlati F., Dominguez M., Bergeron J.J., Thomas D.Y. Saccharomyces cerevisiae CNE1 encodes an endoplasmic reticulum (ER) membrane protein with sequence similarity to calnexin and calreticulin and functions as a constituent of the ER quality control apparatus. J. Biol. Chem. 1995;270:244–253. doi: 10.1074/jbc.270.1.244. [DOI] [PubMed] [Google Scholar]
- Peters J.M. Subunits and substrates of the anaphase-promoting complex. Exp. Cell Res. 1999;248:339–349. doi: 10.1006/excr.1999.4443. [DOI] [PubMed] [Google Scholar]
- Plemper R.K., Egner R., Kuchler K., Wolf D.H. Endoplasmic reticulum degradation of a mutated ATP-binding cassette transporter Pdr5 proceeds in a concerted action of Sec61 and the proteasome. J. Biol. Chem. 1998;273:32848–32856. doi: 10.1074/jbc.273.49.32848. [DOI] [PubMed] [Google Scholar]
- Plemper R.K., Bordallo J., Deak P.M., Taxis C., Hitt R., Wolf D.H. Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J. Cell Sci. 1999;112:4123–4134. doi: 10.1242/jcs.112.22.4123. [DOI] [PubMed] [Google Scholar]
- Saito Y., Yamanushi T., Oka T., Nakano A. Identification of SEC12, SED4, truncated SEC16, and EKS1/HRD3 as multicopy suppressors of ts mutants of Sar1 GTPase. J. Biochem. 1999;125:130–137. doi: 10.1093/oxfordjournals.jbchem.a022249. [DOI] [PubMed] [Google Scholar]
- Seol J.H., Feldman R.M., Zachariae W., Shevchenko A., Correll C.C., Lyapina S., Chi Y., Galova M., Claypool J., Sandmeyer S., Nasmyth K., Deshaies R.J. Cdc53/cullin and the essential Hrt1 RING-H2 subunit of SCF define a ubiquitin ligase module that activates the E2 enzyme Cdc34. Genes Dev. 1999;13:1614–1626. doi: 10.1101/gad.13.12.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra D., Koepp D.M., Kamura T., Conrad M.N., Conaway R.C., Conaway J.W., Elledge S.J., Harper J.W. Reconstitution of G1 cyclin ubiquitination with complexes containing SCFGrr1 and Rbx1. Science. 1999;284:662–665. doi: 10.1126/science.284.5414.662. [DOI] [PubMed] [Google Scholar]
- Stack J.H., Herman P.K., Schu P.V., Emr S.D. A membrane-associated complex containing the Vps15 protein kinase and the Vps34 PI 3-kinase is essential for protein sorting to the yeast lysosome-like vacuole. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:2195–2204. doi: 10.1002/j.1460-2075.1993.tb05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers K.J., Patil C.K., Wodicka D.J., Lockhart D.J., Weissman J.S., Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Ward C.L., Omura S., Kopito R.R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Wileman T., Kane L.P., Young J., Carson G.R., Terhost C. Associations between subunit ectodomains promote T cell antigen receptor assembly and protect against degradation in the ER. J. Cell Biol. 1993;122:67–78. doi: 10.1083/jcb.122.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhovsky S., Gardner R., Hampton R. HRD gene dependence of ER-associated degradation. Mol. Biol. Cell. 2000;11:1697–1708. doi: 10.1091/mbc.11.5.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Varshavsky A. The E2-E3 interaction in the N-end rule pathwaythe RING-H2 finger of E3 is required for the synthesis of multiubiquitin chain. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:6832–6844. doi: 10.1093/emboj/18.23.6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung S.J., Chen S.H., Chan L. Ubiquitin-proteasome pathway mediates intracellular degradation of apolipoprotein B. Biochemistry. 1996;35:13843–13848. doi: 10.1021/bi9618777. [DOI] [PubMed] [Google Scholar]
- Zhou M., Schekman R. The engagement of Sec61p in the ER dislocation process. Mol. Cell. 1999;4:925–934. doi: 10.1016/s1097-2765(00)80222-1. [DOI] [PubMed] [Google Scholar]
- Zhou M., Fisher E.A., Ginsberg H.N. Regulated co-translational ubiquitination of apolipoprotein B100. A new paradigm for proteasomal degradation of a secretory protein. J. Biol. Chem. 1998;273:24649–24653. doi: 10.1074/jbc.273.38.24649. [DOI] [PubMed] [Google Scholar]