

Abstract
Gap-junctional coupling among neurons is subject to regulation by a number of neurotransmitters including nitric oxide. We studied the mechanisms by which NO regulates coupling in cells expressing Cx35, a connexin expressed in neurons throughout the central nervous system. NO donors caused potent uncoupling of HeLa cells stably transfected with Cx35. This effect was mimicked by Bay 21-4272, an activator of guanylyl cyclase. A pharmacological analysis indicated that NO-induced uncoupling involved both PKG-dependent and PKG-independent pathways. PKA was involved in both pathways, suggesting that PKG-dependent uncoupling may be indirect. In vitro, PKG phosphorylated Cx35 at three sites: Ser110, Ser276 and Ser289. A mutational analysis indicated that phosphorylation on Ser110 and Ser276, sites previously shown also to be phosphorylated by PKA, had a significant influence on regulation. Ser289 phosphorylation had very limited effects. We conclude that NO can regulate coupling through Cx35 and that regulation is indirect in HeLa cells.
Keywords: Nitric oxide, Connexin35, Connexin36, Phosphorylation, HeLa cells
INTRODUCTION
Connexin35 (Cx35) and its highly conserved mammalian homologue Cx36 are widespread components of gap junctions throughout the central nervous system (Rash et al. 2000). This connexin is found abundantly in the retina (O’Brien et al. 1996; Guldenagel et al. 2000; O’Brien et al. 2004), and many regions of the brain including olfactory bulb, inferior olive and several brain stem nuclei, cerebellum, and the CA3/CA4 subfields of hippocampus (Condorelli et al. 1998; Belluardo et al. 2000). Neurons throughout these regions are known to be electrically and/or tracer coupled (Llinas and Yarom 1981; MacVicar and Dudek 1981; Yang and Hatton 1988) and Cx35 is thought to form many of these electrical synapses.
Electrical coupling among neurons is rarely static. The degree of coupling changes through development (Christie and Jelinek 1993; Penn et al. 1994; Rorig et al. 1995; Rorig and Sutor 1996), as a result of changes in the level of expression (Sohl et al. 1998; Al-Ubaidi et al. 2000; Gulisano et al. 2000). However, coupling also changes dynamically with activity or in response to transmitter secretion. This is most apparent in the retina, where light adaptation has profound effects on electrical coupling in many neural circuits. For example, in mammalian AII amacrine cells, which are homologously coupled to AII amacrine cells and heterologously coupled to cone ON bipolar cells (Famiglietti and Kolb 1975; Strettoi et al. 1994), adaptation to bright light reduces homologous coupling (Bloomfield et al. 1997). This process works in part through a dopaminergic system operating via cAMP and cAMP-dependent protein kinase (PKA) (Hampson et al. 1992). The heterologous gap junctions with cone ON bipolar cells are regulated in a different manner, showing greater responsiveness to nitric oxide (NO) and components of the cGMP pathway (Mills and Massey 1995; Xia and Mills 2004). Both homologous and heterologous gap junctions in the AII amacrine cells use Cx36 (Feigenspan et al. 2001; Mills et al. 2001), although some evidence suggests that the heterologous gap junctions may also be heterotypic (Famiglietti and Kolb 1975; Maxeiner et al. 2005). These observations suggest that electrical coupling between neurons can be controlled dynamically through compartmentalized signaling pathways.
Many studies have implicated protein kinase pathways in the short-term dynamic regulation of electrical coupling through gap junctions. Horizontal cell gap junctions have been found to be regulated by both dopamine/cAMP and NO/cGMP pathways (Lasater 1987; DeVries and Schwartz 1989), which use cyclic nucleotide-dependent protein kinases as effectors. Coupling through Cx35 gap junctions has been found to be regulated by cAMP-dependent protein kinase (PKA) activity (Mitropoulou and Bruzzone 2003; O’Brien et al. 2004), and we have shown that this regulation involves phosphorylation on several critical residues in the connexin (Ouyang et al. 2005). In this study, we examine the pathway through which NO and cGMP regulate coupling in Cx35 gap junctions.
MATERIALS AND METHODS
Expression of Connexins in HeLa Cells
The full coding sequence of perch Cx35 sub-cloned into pcDNA 3.1 Zeo (Invitrogen, Carlsbad, CA) was used to generate stably-transfected HeLa cell lines. HeLa cells (ATCC clone CCL2) were transfected with 2 μg of plasmid using GenePorterII reagent (Gene Therapy Systems, San Diego, CA) and grown under zeocin selection (100 μg/ml) for 3–6 weeks. Resistant colonies were isolated and amplified under selection. Clones were tested for connexin expression by immunostaining and RT-PCR. The Cx35 wild type cell line has been characterized previously (O’Brien et al. 2004; Ouyang et al. 2005).
For RT-PCR analysis, samples of each cell line were cultured in 75 mm2 flasks, rinsed twice with ice-cold PBS, and then scraped into 4 ml ice-cold PBS with a teflon cell scraper. The cells were centrifuged and total RNA was isolated from the cell pellets using an RNA miniprep kit (Qiagen, Valencia, CA) following the manufacturer’s protocol. RNA was stored as a 70% ethanolic solution at −80°C. For reverse transcription, 2 μg total RNA was precipitated from the ethanol solution by addition of sodium acetate. The dried pellets were dissolved in DEPC water and digested with RQ1 RNase-free DNaseI (Promega) for 1 hour at 37°C to destroy contaminating genomic DNA. The remaining RNA was reverse transcribed using a Superscript II first strand cDNA synthesis kit (Invitrogen) and random hexamers to prime synthesis. One microliter aliquots of the RT reactions were used as templates for subsequent PCR. Primers used for PCR amplification were GCACCAGTCGGCCAAGCAGAAG (forward) and TGCCCTCTTGTCGCCTCATTTTG (reverse). This primer pair is predicted to produce a product of 238 bp.
Immunostaining was performed on cultured HeLa cells on 12 mm glass coverslips. Cells were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer for 45 minutes at 4°C. Fixed cells were washed with PBST (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.7 mM KH2PO4, 0.5% Triton X-100 and 0.1% sodium azide) and then blocked in the same solution containing 3% normal donkey serum (Jackson ImmunoResearch, West Grove, PA.). Coverslips were incubated overnight at 4°C with an anti-perch Cx35 monoclonal antibody (Chemicon, Temecula, CA) at 1 μg/ml plus 1% normal donkey serum in PBST. Cy3-labeled donkey anti-mouse secondary antibodies (Jackson ImmunoResearch) were applied for 4 hours at 4°C. Samples were imaged on a Zeiss LSM 510 confocal microscope.
HeLa cell lines expressing mutant connexins were derived as above by transfection of mutated pcDNA constructs and selection of stably-transfected cell lines. Mutation of Ser289 to Ala was introduced into the wild-type Cx35 pcDNA clone using the same primers and cycling conditions used to make this mutation in the fusion protein construct. A Ser110Ala, Ser276Ala double mutant has been previously described (Ouyang et al. 2005). All mutants were confirmed by sequencing on both strands. Transfection was performed in the same manner. Stable cell lines were selected for each of the mutated connexin clones and used for tracer coupling assays.
Tracer Coupling Experiments
Connexin-transfected HeLa cells were maintained under zeocin selection until plating onto coverslips for tracer coupling experiments. One to two days prior to experiments, cells were fed with fresh medium, trypsinized, and plated at approximately 50% confluence onto glass cell culture coverslips (Fisher Scientific, Houston, TX). Cells were fed again 2 to 6 hours prior to experimentation. This was found to be important, as coupling declined if cultures had not been fed the same day.
Tracer coupling experiments were performed by scrape loading. HeLa cells were maintained at 25°C in oxygenated modified Ringer’s medium (containing 150 mM NaCl, 6.2 mM KCl, 1.2 mM NaH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, 10 mM Hepes, 10 mM glucose, pH 7.4). The medium was supplemented with 0.05 % Neurobiotin (Vector Laboratories, Burlingame, CA), and cells were scraped with a 25-guage needle. Incubation was continued for 10 minutes at 25°C to allow for loading and diffusion. Cells were washed three times for 2 minutes each with modified Ringer’s medium, fixed with 4% paraformaldehyde in 0.1 M phosphate buffer for 1 hour, then visualized with streptavidin-Cy3 (Jackson ImmunoResearch). Fluorescence signals were photographed on a Zeiss fluorescence microscope with a 12-bit Hamamatsu Orca-100 digital camera using Simple PCI software (Compix, Cranberry Township, PA).
Drugs were applied by exchanging the bathing solution with oxygenated incubation medium plus drug for 10 minutes prior to scrape-loading. Cells were exposed continuously to drugs thereafter without further supplementation or washout. Drugs used were the NO donors N-(2-Aminoethyl)-N-(2-hydroxy-2-nitrosohydrazino)-1,2-ethylenediamine (spermine NONOate; Calbiochem, La Jolla, CA; 1 to 100 μM) and sodium nitroprusside (Sigma; 50 μM), soluble guanylyl cyclase activator Bay41-2272 (Calbiochem; 5 μM), soluble guanylyl cyclase inhibitor 1H-[1,2,4]Oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; Calbiochem; 5 μM), cGMP-dependent protein kinase inhibitor KT5823 (Calbiochem; 5 μM), and PKA inhibitor Rp-8-cpt-cAMPS (Alexis, San Diego, CA; 20 μM). All tracer coupling experiments were performed on two to six separate days resulting from different platings of the HeLa cell lines. For all experiments, control and drug-treated conditions were examined on the same day with the same batch of cells. Due to some day-to-day variation in the level of tracer coupling, the diffusion data for some experiments were normalized to the mean diffusion coefficient for each cell line under control (no drug) conditions measured on the same day of experiments.
Data Analysis
Quantitative tracer coupling analyses were performed as previously described (O’Brien et al. 2004; Ouyang et al. 2005). Briefly, fluorescence intensities of coupled cells were used to calculate diffusion coefficients based upon a linear 25-compartment diffusion model of the type described by Zimmerman and Rose (Zimmerman and Rose 1985). In this model, the movement of tracer between adjacent compartments (cells) is described by a series of 25 differential equations that are solved for tracer flux given the total amount of diffusion time, the diffusion coefficient k, and a tracer loading coefficient bo. The diffusion coefficient, k, represents the proportion of tracer that diffuses from one compartment to another per second. Optimal fits of intensity data to the model were determined in MatLab (Mathworks, Natick, MA) by systematically varying k and bo. Data fits were determined by plotting cell intensities on a log intensity axis and determining the diffusion coefficient k that best fit the rate of decline with distance, and the rate of delivery, bo, that fit the overall tracer concentration.
cAMP assays
cAMP content of HeLa cell cultures was measured by competitive ELISA assays using a kit from Assay Designs (Ann Arbor, MI). For these assays, HeLa cells were plated in 12- or 24-well dishes and grown to confluency. Cells were rinsed with oxygenated modified Ringer’s medium and incubated with drugs as for tracer coupling assays. Drugs were not added to zero time point wells. At times from 1 to 20 minutes after beginning the incubation, the medium was removed and replaced with 0.1 M HCl to extract nucleotides. The extracts were assayed according to the manufacturer’s instructions. Extracted cells were subsequently dissolved in 2% SDS and assayed for protein using the BCA method (Pierce, Rockford, IL).
Fusion Protein Constructs
Portions of the perch Cx35 cDNA coding for intracellular domains were cloned separately into bacterial expression vectors with glutathione-S-transferase fusion partners. The intracellular loop domain, amino acids 102-178, was subcloned into pET42a (Novagen, Madison, WI) from a previously described clone in pET15b (O’Brien et al. 1998). The carboxyl-terminal domain, consisting of amino acids 254-304, was subcloned into pET42c from a pGexKG clone described previously (O’Brien et al. 2004). A pET42 control clone (Ouyang et al. 2005) was used as a phosphorylation control. This construct codes for a 31.7 kDa protein containing GST and the pET42 His-tag and multiple cloning site up to a point very close to the position into which the Cx35 cytoplasmic domains were cloned.
Mutations in the fusion protein constructs were made with a PCR-based site-directed mutagenesis protocol (Fisher and Pei 1997) using Pfu polymerase (Stratagene, La Jolla, CA). All constructs were sequenced on both strands to confirm mutations and screen for the presence of PCR-introduced mutations. The GST carrier protein was found to have a PKG phosphorylation site through in vitro phosphorylation assays (method below). This site was removed in all clones by mutation of Ser107 to alanine as described above. Primer sequences for this mutation were 5′GGTTTTGGATATTAGATACGGTGTTGCGAGAATTGC 3′ (forward) and 5′GCAATTCTCGCAACACCGTATCTAATATCCAAAACC 3′ (reverse).
Fusion proteins were expressed in E. coli strain BL21(DE3). Bacterial cells were lysed by sonication in PBS plus 0.5 mM PMSF and 10 μl/ml protease inhibitor cocktail (Sigma, St. Louis, MO), and cleared by centrifugation. GST fusion proteins were purified from extracts by binding to glutathione sepharose 4B (Amersham) and extensive washing with PBS plus 0.5% Igepal CA-630 and 5 mM dithiothreitol. Fusion proteins were eluted from the matrix with 250 mM glutathione in PBS. Fusion protein concentration was estimated by densitometry of Coomassie blue stained bands compared to a series of BSA standards run in the same minigels. In the case of the C-terminal fusion protein preparations that contained proteolytic cleavage products, only the intact protein band was used for the protein determination.
In Vitro Phosphorylation Assays
Fusion proteins (200 ng protein per reaction) were incubated with 1 unit PKG (Promega, Madison, WI) and 20 μCi γ33P-ATP (ICN, Irvine, CA) for 30 min. at 37°C. The final reaction solution contained 40 mM Tris-Cl (pH 7.4), 20 mM Mg-acetate, 200 μM ATP, 33 μM cGMP, 35 mM NaCl, 0.7 mM KCl, 2.5 mM Na2HPO4, 0.9 mM KH2PO4, 0.05 mM EDTA, and 25% glycerol. Reactions were stopped by addition of SDS sample buffer, heated to 70°C for 10 min., and electrophoresed on SDS polyacrylamide gels. Gels were blotted onto nitrocellulose membranes for imaging onto X-ray film (Kodak). For mass spectroscopic determination of phosphorylation sites, 200 μM ATP was used in place of γ33P-ATP, samples were electrophoresed on SDS polyacrylamide gels and stained with Coomassie blue.
Mass Spectroscopic Determination of Phosphorylated Residues
Posttranslational modifications of proteins were identified in the Proteomics Core Facility at the University of Texas Health Science Center at Houston. MS/MS analysis was performed on an Applied Biosystems QStar XL LC/MS/MS mass spectrometer equipped with an LC Packings HPLC for capillary chromatography. The HPLC is coupled to the mass spectrometer by a nanospray ESI head for direct analysis of the eluate. Protein bands were excised from the gels and subjected to in gel trypsin digestion essentially as described by Simpson (2003). Peptide extracts were separated by HPLC on a C18 reverse phase capillary column (Vydac 218MS3.07510) developed with a 2% to 50% acetonitrile gradient in 0.1% formic acid, 0.005% TFA over 30 minutes at a flow rate of 200 nl/min. The nanospray source was fitted with a 30 qm coated tapered fused silica tip (New Objective, Cambridge, MA). The QSTAR was operated in Information Dependent Acquisition mode using a 1 second survey scan followed by two consecutive 3 second product ion scans of 2+, 3+ and 4+ parent ions (m/z 350–1500). Identification was performed on MASCOT with an MS and MS/MS mass tolerance of 0.15 Da. Phosphopeptides were identified by neutral loss followed by MS/MS sequence analysis (Bennett et al. 2002). The neutral loss of phosphoric acid occurs via gas phase β-elimination of the phosphate ester. The reaction converts the phosphoserine to a dehydroalanine residue (69 amu) which is readily identified in the MS/MS spectrum and the neutral loss of 98 Da from the parent peptide mass.
RESULTS
NO Uncouples Cx35 Gap Junctions
NO is a second messenger used in many neurons throughout the retina. To study the effects of NO on Cx35 gap junctions, we examined the effects of NO donors on Neurobiotin tracer coupling in HeLa cells stably expressing Cx35. We have previously found that regulation of Cx35 gap junctional coupling by the cAMP/PKA pathway in this cell line followed closely the pattern of regulation of Cx35 gene homologues in retinal neurons (O’Brien et al. 2004; Ouyang et al. 2005). Preincubation of Cx35 HeLa cells for 10 minutes with the NO donor N-(2-Aminoethyl)-N-(2-hydroxy-2-nitrosohydrazino)-1,2-ethylenediamine (spermine NONOate; 100 μM) reduced the extent to which Neurobiotin tracer spread from damaged cells along a scrape to adjacent cells (figure 1A and C). Similar results were obtained with a different NO donor sodium nitroprusside (50 μM, data not shown). We measured the spread of tracer quantitatively by calculating the diffusion coefficient (k) for tracer spread using a linear diffusion model (see methods). Figure 1 (B and D) shows the fits of the model to fluorescence intensity data from these experiments. Under control conditions, the diffusion coefficient k was 0.0038 cells2/sec. The second free parameter, bo, represents the rate of tracer loading into the damaged cell. With spermine NONOate pretreatment, k was reduced to 0.0009 cells2/sec. A summary of the data for HeLa Cx35 cells and non-transfected HeLa cells (parental HeLa) under these conditions is shown in figure 1E. The reduction in diffusion coefficient caused by spermine NONOate treatment was significant (t = −12.5; n = 30 for spermine NONOate, n = 64 for control; p < 0.01).
Figure 1. NO donors cause uncoupling of Cx35-transfected HeLa cells.

Neurobiotin tracer coupling was examined in HeLa cell lines stably transfected with Cx35 (A). The tracer diffusion coefficients were measured quantitatively by fitting measurements of tracer fluorescence intensity to a linear diffusion model (B; see Methods for details). The free parameters required to fit the model to the data, the diffusion coefficient k and the cell loading rate bo, are shown in the inset to B. 10 minute pre-incubation with 100 μM spermine NONOate reduced tracer diffusion (C) and the measured diffusion coefficient (D). E. Summary of diffusion coefficients from 15 to 64 measurements under control conditions (cont) or in the presence of 100 μM spermine NONOate (SpNO) in Cx35-transfected HeLa cells (Cx35) or in non-transfected HeLa cells (parental HeLa). Error bars are 1 SEM. # indicates a significant difference from control at the p<0.01 level. See text for details of the statistical comparisons.
The parental HeLa cells always show a low level of Neurobiotin tracer coupling due to the presence of an endogenous connexin (O’Brien et al. 2004). Figure 1E shows that parental HeLa cells did not show any changes in coupling with spermine NONOate application under the conditions that caused uncoupling of Cx35 HeLa cells (t = 1.1; n = 15 for spermine NONOate, n = 16 for control; p > 0.05). Thus the changes in coupling caused by nitric oxide donors in Cx35 HeLa cells were due to modulation of Cx35 gap junctions, and not of the endogenous connexin.
Guanylyl Cyclase Activity Affects Tracer Coupling
One of the canonical pathways through which NO acts is the stimulation of a NO-sensitive soluble guanylyl cyclase (sGC). We examined the role of this pathway in NO-induced uncoupling in Cx35 HeLa cells by applying Bay41-2272 (Bay), a NO-independent activator of sGC. Figure 2 shows that 5 μM Bay significantly uncoupled Cx35 HeLa cells (t = −6.9; n = 38 for Bay, n = 64 for control; p < 0.01), while it had no effect on parental HeLa cells (t = 0.7; n = 14 for Bay, n = 16 for control; p > 0.05). This suggests that sGC activity and cGMP production led to uncoupling of Cx35 gap junctions. Paradoxically, the sGC inhibitor 1H-[1,2,4]Oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; 5 μM) also reduced coupling in Cx35 HeLa cells (t = −7.5; n = 35 for ODQ, n = 64 for control; p < 0.01), while it had no effect on parental HeLa cells (t = −0.2; n = 17 for ODQ, n = 16 for control; p > 0.05). This suggests that there are at least two signaling mechanisms driven by cGMP in HeLa cells and that these mechanisms have opposite effects on coupling through Cx35 gap junctions.
Figure 2. Guanylyl cyclase activity affects tracer coupling.

Summary of scrape-loading data for Cx35-transfected HeLa and parental HeLa cells pre-incubated 10 minutes with an activator of soluble guanylyl cyclase (5 μM Bay41-2272) and an inhibitor of soluble guanylyl cyclase (5 μM ODQ). Both activator (Bay) and inhibitor (ODQ) of sGC caused significant uncoupling of gap junctions in Cx35 HeLa cells, but did not affect gap junctions in parental HeLa cells. Data are means derived from 14 to 64 measurements of each condition + 1 SEM.
Protein Kinases Mediate NO and cGMP-induced Uncoupling
Protein kinase signaling has been widely shown to regulate gap junctional coupling, including that mediated by Cx35 (Mitropoulou and Bruzzone 2003; Ouyang et al. 2005). One of the downstream effectors of guanylyl cyclase activity is cGMP-dependent protein kinase (PKG), so we examined whether this kinase was involved in cGMP-mediated uncoupling of Cx35. KT5823 (KT; 10μM), a selective inhibitor of PKG, blocked uncoupling caused by Bay41-2272, and even increased the diffusion coefficient slightly above the control level (figure 3; Bay vs. control − same data as in figure 2; Bay+KT vs. control − t = 2.1, n = 30 for Bay+KT, n = 64 for control, p < 0.05). Thus PKG is involved in the uncoupling caused by activation of sGC.
Figure 3. NO and cGMP-induced uncoupling is mediated by protein kinases.
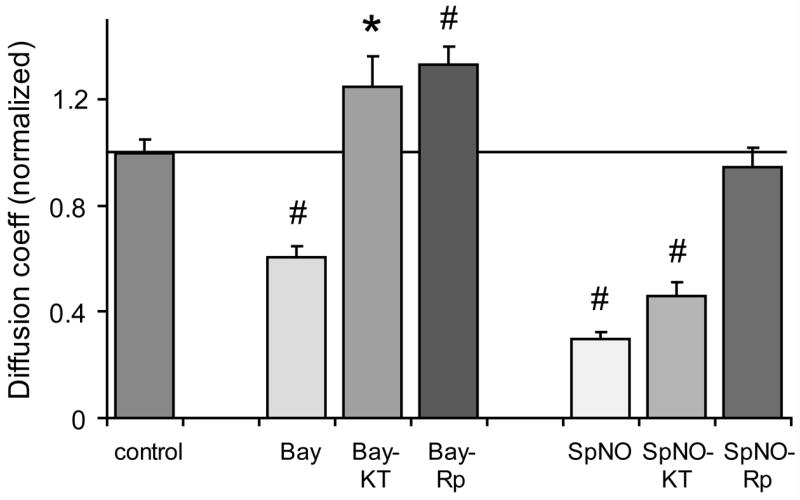
Summary of scrape-loading data for Cx35-transfected HeLa cells pre-incubated 10 minutes with either 5 μM Bay41-2272 or 100 μM spermine NONOate. Co-application of either the selective PKG inhibitor KT5823 (10 μM; Bay-KT) or the selective PKA inhibitor Rp-8-cpt-cAMPS (20 μM; Bay-Rp) blocked Bay-induced uncoupling. However, KT5823 only partially blocked uncoupling caused by spermine NONOate (SpNO-KT) while Rp-8-cpt-cAMPS (SpNO-Rp) completely blocked uncoupling. Data are means derived from 17 to 64 measurements of each condition + 1 SEM. * indicates statistical significance at the p<0.05 level.
In our previous work (Ouyang et al. 2005), we have shown that cAMP-dependent protein kinase (PKA) phosphorylation regulates Cx35 coupling. We examined whether there might be crosstalk between cGMP and cAMP signaling pathways. Figure 3 shows that the selective PKA inhibitor Rp-8-cpt-cAMPS (Rp) had the same effect as the PKG inhibitor, blocking uncoupling caused by Bay41-2272 and increasing coupling above the control level (Bay+Rp vs. control − t = 4.1; n = 19 for Bay+Rp, n = 64 for control, p < 0.01). This finding suggests that there is crosstalk between cGMP and cAMP signaling pathways. PKA is also involved in the pathway leading from sGC activation to Cx35 gap junction uncoupling. Since PKG is directly stimulated by cGMP and both PKG and PKA inhibitors prevent uncoupling, it is likely that PKA is downstream of PKG in this pathway.
We subsequently examined whether PKG or PKA inhibitors could block the uncoupling caused by NO. Figure 3 shows that inhibiting PKG with KT5823 only partially reversed the uncoupling caused by 100 μM spermine NONOate. The normalized diffusion coefficient for tracer coupling in HeLa Cx35 cells treated with KT5823 plus spermine NONOate was significantly larger than that for cells treated with spermine NONOate alone (t = 2.6; n = 17 for SN+KT, n = 28 for SN alone; p < 0.05), but was significantly smaller than for no drug controls (t = −7.4; n = 17 for SN+KT, n = 42 for control; p < 0.01). On the other hand, inhibiting PKA with Rp-8-cpt-cAMPS completely blocked uncoupling (SN+Rp vs. control: t = −0.6; n = 22 for SN+Rp, n = 42 for control; p > 0.05; SN+Rp vs. SN alone: t = 8.3, p < 0.01). These data suggest that the cGMP/PKG-dependent pathway accounts for only a portion of the uncoupling caused by NO. A portion of the pathway is not dependent on sGC. However, all of the measurable uncoupling caused by NO depended upon PKA activity.
Crosstalk Between Signaling Pathways
Several results of this study have suggested the existence of crosstalk between signaling pathways in HeLa cells. These include inhibition of uncoupling caused by Bay41-2272 by a selective PKA inhibitor, and uncoupling of Cx35 gap junctions by ODQ, a selective sGC inhibitor. Since regulation of Cx35 gap junctional coupling in this cell line seems to rely substantially on PKA activity, we examined whether any of the drug treatments targeted at changing cGMP concentration affect the cellular cAMP content. Figure 4A shows the time course of variation in cAMP content in Cx35 HeLa cells treated with 100 μM spermine NONOate, 5 μM Bay41-2272, or 5 μM ODQ. There were no consistent changes in cAMP content with any of the treatments (t-tests performed at each data point vs. time 0: n=4, p>0.05 for all). This suggests that if there were any changes in overall cAMP content, they were small and were within the experimental error of the assays. This does not rule out localized changes in cAMP concentration.
Figure 4. Analysis of signaling pathway crosstalk.

A. Variation in cAMP content with time of Cx35- transfected HeLa cells treated with 100 μM spermine NONOate (SpNO), 5 μM Bay41-2272 (Bay), 5 μM ODQ, or no drug (control). For each time course, time 0 represents cells without any drug treatment. T-tests of each time point vs. time 0 showed no significant changes in cAMP content for any of the treatment conditions. Data are means of 4 experiments ± SEM.
B. Test of the involvement of PKA activity in the anomalous uncoupling caused by ODQ. 5 μM ODQ significantly reduced coupling in Cx35-transfected HeLa cells both in the absence and the presence of 20 μM Rp-8-cpt-cAMPS. Data are means of 14 to 16 measurements + 1 SEM.
We further analyzed whether the uncoupling caused by ODQ was mediated by PKA activity. Figure 4B shows that the PKA inhibitor Rp-8-cpt-cAMPS (20 μM) did not block the uncoupling caused by ODQ (ODQ vs. control: t = −3.1, n = 16 for ODQ, n = 14 for control, p < 0.01; ODQ+Rp vs control: t = −2.3, n = 14 for ODQ+Rp, n = 14 for control, p < 0.05). Thus PKA is not involved in this pathway, which is consistent with the observation that overall cAMP content was not changed by ODQ. This suggests either that an off-target effect of ODQ uncouples Cx35 gap junctions, or that some minimal level of cGMP is required to maintain Cx35 in a coupled state.
PKG Phosphorylates Cx35 Directly
The complex nature of the tracer coupling data lead one to question whether PKG may phosphorylate Cx35 at all. To examine this question, we performed in vitro phosphorylation experiments with purified PKG and Cx35 cytoplasmic domain fusion proteins purified from a bacterial expression system. Figure 5A shows that PKG did indeed phosphorylate Cx35 in both its intracellular loop and carboxyl terminal tail domains. We identified the sites of phosphorylation by in-gel trypsin digestion and tandem mass spectrometry of the phosphorylated peptides. Figure 5B shows the reconstructed TOF spectrum of the phosphorylated C-terminal fusion protein. The data showed that PKG phosphorylated the same two sites, Ser110 in the intracellular loop and Ser276 in the C-terminal tail, that were phosphorylated by PKA (Ouyang et al. 2005). PKG also phosphorylated a second site in the C-terminal tail, Ser289, which was not phosphorylated by PKA in previous in vitro assays. Thus, the protein kinases phosphorylate a partially overlapping set of sites.
Figure 5. PKG can phosphorylate Cx35 directly.

A. Autoradiogram of in vitro phosphorylation experiments with Cx35 intracellular loop domain (Cx35 IL) and carboxyl-terminal domain (Cx35 CT) GST fusion proteins. Both domains were phosphorylated.
B. Tandem mass spectroscopic identification of phosphorylated peptides in the C-terminal domain. The spectrum shown is a reconstructed MS TOF scan of singly charged parent ions from the LC/MS/MS analysis. The labeled peptides represent phosphorylated Cx35 peptides (phosphate represented by (P)) and several non-phosphorylated peptides that were sequenced by MS/MS. Serines 276 and 289 were found to be phosphorylated in this domain. Similar analysis of the IL domain found Serine 110 to be phosphorylated.
Phosphorylation-deficient Mutants Reveal Complex Regulation of Coupling
The involvement of protein kinases in the signaling pathways that cause uncoupling of Cx35 gap junctions does not necessarily mean that those actions are due to phosphorylation of the connexin itself. Regulation may be accomplished indirectly via other signaling molecules that have influence on gap junctional coupling. To evaluate whether phosphorylation of the connexin was involved in the uncoupling caused by NO and cGMP, we studied the effects of these agents on a mélange of Cx35 mutants that lacked phosphorylation sites. Our earlier study showed that PKA regulation of Cx35 coupling did depend on phosphorylation at two sites within the connexin: Ser110 and Ser276 (Ouyang et al. 2005). This regulation was complex and required phosphorylation at both sites. Since PKG also phosphorylated these same sites, we chose to study these as a pair, using a Cx35 Ser110Ala, Ser276Ala double mutant cell line. In addition, we prepared a Ser289Ala single mutant cell line, targeting the site that was phosphorylated by PKG but not by PKA in our in vitro assays. Figure 6 shows summary data for diffusion coefficients measured in tracer coupling experiments with wild-type and the two mutant cell lines. In the double mutant, Cx35-S110A, S276A, neither spermine NONOate nor Bay41-2272 (the sGC activator) caused any significant change in coupling, whereas both significantly uncoupled wild-type Cx35 HeLa cells (SpNO: t = 0.6; n = 20 for SpNO, n = 20 for control; p > 0.05; Bay: t = 1.4; n = 21 for Bay, n = 20 for control; p > 0.05). ODQ had the reverse effect on S110A, S276A cells as it had on wild-type Cx35 cells. That is, ODQ caused a small but significant increase in coupling compared to no drug controls (t = 2.5; n = 21 for ODQ, n = 20 for control; p < 0.05). This argues against the action of ODQ being a direct inhibition of gap junctional coupling in wild-type Cx35 HeLa cells. It also implies that coupling can be regulated apart from the action of phosphorylation on Ser110 and Ser276. The latter is consistent with the observation that ODQ’s uncoupling effect is mediated by an effector other than PKA.
Figure 6. Mutation of phosphorylation sites changes regulation.

Pharmacological analysis of tracer coupling in Cx35 wild-type transfected HeLa cells (Cx35) or in two mutant cell lines lacking either the two major phosphorylation sites shared by PKG and PKA (S110A, S276A) or the phosphorylation site so far found only to be phosphorylated by PKG (S289A). Elimination of phosphorylation at Ser110 and Ser276 blocked most of the significant changes in coupling seen in wild-type Cx35, and weakly reversed the uncoupling action of ODQ. Eliminating Ser289 produced minimal changes from the wild-type behavior. Cx35 data are the same as in figures 2 and 3; remaining data are means of 16 to 41 measurements + SEM.
Mutation of Ser289, a site phosphorylated by PKG in vitro but which we have not previously found to be phosphorylated by PKA, resulted in few changes in regulation. In the Cx35-S289A cell line, spermine NONOate, Bay41-2272, and ODQ each caused significant uncoupling (SpNO: t = −5.4, n = 23 for SpNO, n = 22 for control, p < 0.01; Bay: t = −5.4, n = 13 for Bay, n = 13 for control, p < 0.01; ODQ: t = −5.3; n = 31 for ODQ, n = 36 for control; p < 0.01), similar to their effects in wild-type Cx35 HeLa cells. As in wild-type Cx35 HeLa cells, the uncoupling caused by Bay41-2272 was blocked by both KT5823 and Rp-8-cpt-cAMPS (Bay+KT vs. control: t = 0.1, n = 10 for Bay+KT, n = 13 for control, p > 0.05; Bay+Rp vs. control: t = 1.0, n = 11 for Bay+Rp, n = 13 for control, p > 0.05). However, the small increase in coupling above control levels that was seen in wild-type Cx35 HeLa cells with these drugs was not observed in the Cx35-S289A mutant cell line.
DISCUSSION
Electrical synapses play an important role in neural circuitry of the central nervous system, and Cx35 is the most prevalent connexin protein forming these synapses. Substantial evidence suggests that coupling through electrical synapses is dynamically regulated, both in an activity-dependent manner (Yang et al. 1990; Pereda and Faber 1996; Pereda et al. 2003) and in response to certain neurotransmitters (Hampson et al. 1992; Mills and Massey 1995; O’Donnell and Grace 1997). In this study, we have found that NO, one transmitter known to modulate gap junction coupling, can uncouple gap junctions formed by Cx35.
NO can initiate cellular signaling through at least two major pathways. The most widely studied pathway involves the direct activation of soluble guanylyl cyclase (sGC) through binding to a heme iron (Russwurm and Koesling 2002). This increases intracellular cGMP concentration, resulting in the activation of cGMP-dependent protein kinase (PKG), certain cGMP-dependent ion channels, and either activation or inhibition of several phosphodiesterases (Hofmann et al. 2000). In our study of Cx35 coupling in HeLa cells, we found that both application of NO donors and activation of sGC in the absence of exogenous NO caused uncoupling. Thus a cGMP-dependent pathway in these cells led to uncoupling.
The second major route for NO signaling is through redox signaling involving direct S-nitrosylation of thiols, including cysteine thiols in many proteins. This reaction affects such diverse proteins as caspases, tyrosine phosphatases, JNK and Src kinases, NSF, NMDA receptors, ryanodine receptors, and transcription factors such as NF-KB (Hess et al. 2005). Similar oxidative changes to cysteine residues have been proposed to reduce gap junction hemichannel activity in Cx43 (Saez et al. 2005). In our HeLa cell expression system, Cx35 uncoupling due to NO included a distinct cGMP-independent component.
The mechanisms through which NO leads to uncoupling of Cx35 gap junctions in HeLa cells are clearly complex. Our evidence suggests that the final step in regulation of coupling involved phosphorylation of the connexin. However, it was not at all clear that this was accomplished by PKG phosphorylation of Cx35, even though PKG can phosphorylate this connexin. The cGMP-dependent component of uncoupling depended on PKG activity, but this component could also be blocked by a specific PKA inhibitor. Thus there is crosstalk in this signaling pathway and PKA is an important component. Figure 7A outlines a hypothetical model that describes this regulatory scheme and cellular constituents that might influence regulation. Central to this model is a significant amount of PKA activity that phosphorylates Cx35 at two conserved sites, causing uncoupling at some steady-state level. PKG activity and cytoplasmic cGMP may influence this equilibrium at a number of points. cGMP can inhibit one phosphodiesterase, PDE3 (Manganiello et al. 1995), resulting in an increase in cytoplasmic cAMP and concomitant increase in PKA activity. This is considered unlikely in the HeLa cell system, since the pathway we observed depended on PKG activity. PKG also phosphorylates small polypeptides such as G-substrate and DARPP-32 that act as inhibitors of protein phosphatases (Wang and Robinson 1997). Such PKG phosphorylation can lead to inhibition of protein phosphatase I and potentially shift the equilibrium toward more phosphorylated Cx35. Finally, it is possible that PKG has unknown effects directly on PKA or on a phosphodiesterase that may lead to increased phosphorylation in this system.
Figure 7. Hypothetical model for Cx35 regulation.

A. Schematic summary of the nitric oxide signaling system in HeLa cells. NO acts through two distinct pathways, one depending on the activity of guanylyl cyclase and the other independent of this activity. The pathways converge to uncouple Cx35 gap junctions through a mechanism that requires PKA activity. cGMP can either activate or inhibit different isoforms of phosphodiesterase (PDEs), thereby regulating local cAMP concentration. The cGMP-inhibited PDE3 could account for uncoupling (but see discussion). Activation of PKG by cGMP could result in phosphorylation of small polypeptide inhibitors of protein phosphatases (PPase) such as G-substrate (GS) or DARPP-32, and subsequent inhibition of phosphatase activity. PKG could also activate PKA through an unknown mechanism. It is unclear if PKG phosphorylates Cx35 directly. In the cGMP-independent pathway, NO may activate PKA directly, may inhibit a protein phosphatase activity, or may sensitize Cx35 to the action of PKA.
B. Summary of the phosphorylation sites on Cx35 that regulate its coupling. Ser110 in the intracellular loop and Ser276 in the C-terminal domain are key targets of phosphorylation that uncouple the gap junctions. Both PKA and PKG can phosphorylate these sites. Other sites in the outer half of the C-terminus such as Ser289 have weaker effects, but may modulate the effects of phosphorylation on the two major sites.
The cGMP-independent component of regulation did not depend on PKG activity, but did depend on PKA activity. Thus this pathway also relies on connexin phosphorylation. Our experiments did not examine the mechanisms by which NO might activate PKA. Redox signaling mechanisms involving adenylyl cyclases and PKA have both been reported. A number of reports suggest that NO inhibits adenylyl cyclase isoforms 5 and 6 (Klein 2002). This would not produce an increase in PKA activity. The catalytic subunit of PKA has been found to be inhibited by glutathionylation on a cysteine residue near the active site (Humphries et al. 2002). It is unclear whether nitrosylation would have a similar effect on activity, and neither of these phenomena directly fit with our observations in HeLa cells. Some potential alternative mechanisms are that NO nitrosylates and inhibits a phosphatase, or that NO nitrosylates Cx35 itself and either sensitizes the connexin to the effects of phosphorylation or increases the affinity of PKA phosphorylation sites for PKA. Cx35 has several free cysteine residues that could be subject to nitrosylation (O’Brien et al. 1998). Such direct effects on the connexin are similar to those proposed to mediate the oxidation-dependent reduction in Cx43 hemichannel activity that can be alleviated by reducing agents (Saez et al. 2005). This cGMP-independent mechanism will require further studies to elucidate.
One anomalous finding of this study was that a guanylyl cyclase inhibitor, ODQ, caused uncoupling of wild-type Cx35 HeLa cells. This conflicts with the observation that activating sGC also uncoupled these gap junctions, suggesting that ODQ may have an off-target effect in this system. We consider it unlikely that this is a result of direct inhibition of the Cx35 gap junctions because coupling through the S110A, S276A double mutant was increased in the presence of ODQ. This reversal of regulatory behavior is qualitatively similar to the effect of a small C-terminal truncation of Cx35 on PKA regulation of coupling (Ouyang et al. 2005). Both effects suggest that signaling mechanisms separate from phosphorylation at the major PKA sites have an important influence on the state of coupling. Although it is not clear what these mechanisms are in either case, it appears that some types of signaling control a “state switch” that interacts with the major phosphorylation sites to affect coupling. In the case of the ODQ effect, it is possible that this depends on the maintenance of a normal resting level of cGMP. However, it is also possible that the ODQ effect is entirely unrelated to its nominal target, guanylyl cyclase.
With the identification of a number of functionally relevant phosphorylation sites on Cx35, a picture of the regulatory scheme for this connexin is emerging. An outline of this scheme is shown in figure 7B. First, there are two sites, Ser110 in the intracellular loop and Ser276 in the C-terminal tail, that appear to be the major sites responsible for changes in coupling. Phosphorylation on one or both of these residues is essential for most changes in coupling that we have observed. These two sites are targets for PKA and PKG. Although PKG may not phosphorylate these sites directly in HeLa cells, it is reasonable to predict that it may do so in other cells. In addition, sequence analyses indicate that these sites may also serve as targets for Calmodulin-dependent Kinase, S6 Kinase, Casein Kinase II (Ser276) and perhaps other kinases (authors’ unpublished analyses). Second, a “state switch” serves to modulate the response to phosphorylation at the two primary sites. The behavior of the C-terminal truncation mutant (Ouyang et al. 2005) suggests that this switch may be located on the outer half of the C-terminal tail. However, it is still unclear whether this is a property of Cx35 itself or is the result of signaling complexes associated with it.
Signaling pathways that regulate the function of Cx35 may be highly localized. For example, cAMP concentration is controlled in spatially and temporally separate microdomains even in simple cells (Rich et al. 2001), and these microdomains may strongly influence the phosphorylation state of Cx35. Since the residues critical for regulation may be substrates for phosphorylation by several different protein kinases, it is apparent that coupling may be controlled locally by many signaling pathways. It is likely that the functional state of the gap junction relies on the combinatorial effects of several pathways.
In neurons, regulation of coupling through Cx35 gap junctions depends substantially on the components of signaling pathways present in the near vicinity of the gap junctions. For example, in retinal AII amacrine cells, gap junctions are made with other AII amacrine cells and with-cone ON bipolar cells (Famiglietti and Kolb 1975; Strettoi et al. 1994). Mills and Massey (1995) found that the AII-AII gap junctions were regulated by dopamine and cAMP analogs while the AII-bipolar gap junctions were regulated by NO and not by dopamine. However, Xia and Mills (2004) found that the AII-bipolar gap junction could be closed by membrane-permeant cAMP analogs that activate PKA. In this case, signaling is compartmentalized by the presence of appropriate receptors at the cell surface and signaling pathways within the cell. It is possible that such compartments are very limited in size, providing for individual control of gap junctions. By relying upon signals from several different residues, Cx35 gap junctions are capable of responding to a variety of environmental cues. This is likely to happen dynamically, allowing electrical synapses between neurons that use this connexin to vary their resistance. Such processes can account for the wide range of adaptive changes in electrical coupling that have been observed in retinal and other central nervous system neurons.
Acknowledgments
The authors wish to thank Ms. Gigi Mayorga-Wark for expert technical assistance, and Mr. Larry Saltzman for insightful conversations. This research was supported by grants from the National Institutes of Health (EY12857, EY10608) and Research to Prevent Blindness.
References
- Al-Ubaidi MR, White TW, Ripps H, Poras I, Avner P, Gomes D, Bruzzone R. Functional properties, developmental regulation, and chromosomal localization of murine connexin36, a gap-junctional protein expressed preferentially in retina and brain. J Neurosci Res. 2000;59:813–26. doi: 10.1002/(SICI)1097-4547(20000315)59:6<813::AID-JNR14>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Belluardo N, Mudo G, Trovato-Salinaro A, Le Gurun S, Charollais A, Serre-Beinier V, Amato G, Haefliger JA, Meda P, Condorelli DF. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000;865:121–38. doi: 10.1016/s0006-8993(00)02300-3. [DOI] [PubMed] [Google Scholar]
- Bennett KL, Stensballe A, Podtelejnikov AV, Moniatte M, Jensen ON. Phosphopeptide detection and sequencing by matrix-assisted laser desorption/ionization quadrupole time-of-flight tandem mass spectrometry. J Mass Spectrom. 2002;37:179–90. doi: 10.1002/jms.271. [DOI] [PubMed] [Google Scholar]
- Bloomfield SA, Xin D, Osborne T. Light-induced modulation of coupling between AII amacrine cells in the rabbit retina. Vis Neurosci. 1997;14:565–76. doi: 10.1017/s0952523800012220. [DOI] [PubMed] [Google Scholar]
- Christie MJ, Jelinek HF. Dye-coupling among neurons of the rat locus coeruleus during postnatal development. Neuroscience. 1993;56:129–37. doi: 10.1016/0306-4522(93)90568-z. [DOI] [PubMed] [Google Scholar]
- Condorelli DF, Parenti R, Spinella F, Salinaro AT, Belluardo N, Cardile V, Cicirata F. Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. Eur J Neurosci. 1998;10:1202–8. doi: 10.1046/j.1460-9568.1998.00163.x. [DOI] [PubMed] [Google Scholar]
- DeVries SH, Schwartz EA. Modulation of an electrical synapse between solitary pairs of catfish horizontal cells by dopamine and second messengers. J Physiol (Lond) 1989;414:351–75. doi: 10.1113/jphysiol.1989.sp017692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famiglietti EV, Jr, Kolb H. A bistratified amacrine cell and synaptic cirucitry in the inner plexiform layer of the retina. Brain Res. 1975;84:293–300. doi: 10.1016/0006-8993(75)90983-x. [DOI] [PubMed] [Google Scholar]
- Feigenspan A, Teubner B, Willecke K, Weiler R. Expression of Neuronal Connexin36 in AII Amacrine Cells of the Mammalian Retina. J Neurosci. 2001;21:230–239. doi: 10.1523/JNEUROSCI.21-01-00230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher CL, Pei GK. Modification of a PCR-based site-directed mutagenesis method. Biotechniques. 1997;23:570–1. 574. doi: 10.2144/97234bm01. [DOI] [PubMed] [Google Scholar]
- Guldenagel M, Sohl G, Plum A, Traub O, Teubner B, Weiler R, Willecke K. Expression patterns of connexin genes in mouse retina. J Comp Neurol. 2000;425:193–201. [PubMed] [Google Scholar]
- Gulisano M, Parenti R, Spinella F, Cicirata F. Cx36 is dynamically expressed during early development of mouse brain and nervous system. Neuroreport. 2000;11:3823–8. doi: 10.1097/00001756-200011270-00045. [DOI] [PubMed] [Google Scholar]
- Hampson EC, Vaney DI, Weiler R. Dopaminergic modulation of gap junction permeability between amacrine cells in mammalian retina. Journal of Neuroscience. 1992;12:4911–22. doi: 10.1523/JNEUROSCI.12-12-04911.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Ammendola A, Schlossmann J. Rising behind NO: cGMP-dependent protein kinases. J Cell Sci. 2000;113 ( Pt 10):1671–6. doi: 10.1242/jcs.113.10.1671. [DOI] [PubMed] [Google Scholar]
- Humphries KM, Juliano C, Taylor SS. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J Biol Chem. 2002;277:43505–11. doi: 10.1074/jbc.M207088200. [DOI] [PubMed] [Google Scholar]
- Klein C. Nitric oxide and the other cyclic nucleotide. Cell Signal. 2002;14:493–8. doi: 10.1016/s0898-6568(01)00283-2. [DOI] [PubMed] [Google Scholar]
- Lasater EM. Retinal horizontal cell gap junctional conductance is modulated by dopamine through a cyclic AMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1987;84:7319–23. doi: 10.1073/pnas.84.20.7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Yarom Y. Electrophysiology of mammalian inferior olivary neurones in vitro. Different types of voltage-dependent ionic conductances. J Physiol. 1981;315:549–67. doi: 10.1113/jphysiol.1981.sp013763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar BA, Dudek FE. Electrotonic coupling between pyramidal cells: a direct demonstration in rat hippocampal slices. Science. 1981;213:782–5. doi: 10.1126/science.6266013. [DOI] [PubMed] [Google Scholar]
- Manganiello VC, Taira M, Degerman E, Belfrage P. Type III cGMP-inhibited cyclic nucleotide phosphodiesterases (PDE3 gene family) Cell Signal. 1995;7:445–55. doi: 10.1016/0898-6568(95)00017-j. [DOI] [PubMed] [Google Scholar]
- Maxeiner S, Dedek K, Janssen-Bienhold U, Ammermuller J, Brune H, Kirsch T, Pieper M, Degen J, Kruger O, Willecke K, Weiler R. Deletion of connexin45 in mouse retinal neurons disrupts the rod/cone signaling pathway between AII amacrine and ON cone bipolar cells and leads to impaired visual transmission. J Neurosci. 2005;25:566–76. doi: 10.1523/JNEUROSCI.3232-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills SL, Massey SC. Differential properties of two gap junctional pathways made by AII amacrine cells. Nature. 1995;377:734–7. doi: 10.1038/377734a0. [DOI] [PubMed] [Google Scholar]
- Mills SL, O’Brien JJ, Li W, O’Brien J, Massey SC. Rod pathways in the mammalian retina use connexin36. J Comp Neurol. 2001;436:336–350. [PMC free article] [PubMed] [Google Scholar]
- Mitropoulou G, Bruzzone R. Modulation of perch connexin35 hemi-channels by cyclic AMP requires a protein kinase A phosphorylation site. J Neurosci Res. 2003;72:147–57. doi: 10.1002/jnr.10572. [DOI] [PubMed] [Google Scholar]
- O’Brien J, Al-Ubaidi MR, Ripps H. Connexin 35: a gap-junctional protein expressed preferentially in the skate retina. Molecular Biology of the Cell. 1996;7:233–43. doi: 10.1091/mbc.7.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J, Bruzzone R, White TW, Al-Ubaidi MR, Ripps H. Cloning and expression of two related connexins from the perch retina define a distinct subgroup of the connexin family. J Neurosci. 1998;18:7625–37. doi: 10.1523/JNEUROSCI.18-19-07625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J, Nguyen HB, Mills SL. Cone photoreceptors in bass retina use two connexins to mediate electrical coupling. J Neurosci. 2004;24:5632–5642. doi: 10.1523/JNEUROSCI.1248-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell P, Grace AA. Cortical afferents modulate striatal gap junction permeability via nitric oxide. Neuroscience. 1997;76:1–5. doi: 10.1016/s0306-4522(96)00433-2. [DOI] [PubMed] [Google Scholar]
- Ouyang X, Winbow VM, Patel LS, Burr GS, Mitchell CK, O’Brien J. Protein kinase A mediates regulation of gap junctions containing connexin35 through a complex pathway. Brain Res Mol Brain Res. 2005;135:1–11. doi: 10.1016/j.molbrainres.2004.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn AA, Wong RO, Shatz CJ. Neuronal coupling in the developing mammalian retina. J Neurosci. 1994;14:3805–15. doi: 10.1523/JNEUROSCI.14-06-03805.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda A, O’Brien J, Nagy JI, Smith M, Bukauskas F, Davidson KG, Kamasawa N, Yasumura T, Rash JE. Short-range functional interaction between connexin35 and neighboring chemical synapses. Cell Commun Adhes. 2003;10:419–23. doi: 10.1080/15419060390263254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda AE, Faber DS. Activity-dependent short-term enhancement of intercellular coupling. J Neurosci. 1996;16:983–92. doi: 10.1523/JNEUROSCI.16-03-00983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash JE, Staines WA, Yasumura T, Patel D, Furman CS, Stelmack GL, Nagy JI. Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc Natl Acad Sci U S A. 2000;97:7573–8. doi: 10.1073/pnas.97.13.7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DM, Karpen JW. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc Natl Acad Sci U S A. 2001;98:13049–54. doi: 10.1073/pnas.221381398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorig B, Klausa G, Sutor B. Dye coupling between pyramidal neurons in developing rat prefrontal and frontal cortex is reduced by protein kinase A activation and dopamine. J Neurosci. 1995;15:7386–400. doi: 10.1523/JNEUROSCI.15-11-07386.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorig B, Sutor B. Regulation of gap junction coupling in the developing neocortex. Mol Neurobiol. 1996;12:225–49. doi: 10.1007/BF02755590. [DOI] [PubMed] [Google Scholar]
- Russwurm M, Koesling D. Isoforms of NO-sensitive guanylyl cyclase. Mol Cell Biochem. 2002;230:159–64. [PubMed] [Google Scholar]
- Saez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV. Connexin-based gap junction hemichannels: gating mechanisms. Biochim Biophys Acta. 2005;1711:215–24. doi: 10.1016/j.bbamem.2005.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RJ. Proteins and proteomics : a laboratory manual. Cold Spring Harbor, NY; Cold Spring Harbor Laboratory Press: 2003. [Google Scholar]
- Sohl G, Degen J, Teubner B, Willecke K. The murine gap junction gene connexin36 is highly expressed in mouse retina and regulated during brain development. FEBS Lett. 1998;428:27–31. doi: 10.1016/s0014-5793(98)00479-7. [DOI] [PubMed] [Google Scholar]
- Strettoi E, Dacheux RF, Raviola E. Cone bipolar cells as interneurons in the rod pathway of the rabbit retina. J Comp Neurol. 1994;347:139–49. doi: 10.1002/cne.903470111. [DOI] [PubMed] [Google Scholar]
- Wang X, Robinson PJ. Cyclic GMP-dependent protein kinase and cellular signaling in the nervous system. J Neurochem. 1997;68:443–56. doi: 10.1046/j.1471-4159.1997.68020443.x. [DOI] [PubMed] [Google Scholar]
- Xia X, Mills S. Gap junctional regulatory mechanisms in the AII amacrine cell of the rabbit retina. Vis Neurosci. 2004;21:791–805. doi: 10.1017/S0952523804045122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang QZ, Hatton GI. Direct evidence for electrical coupling among rat supraoptic nucleus neurons. Brain Res. 1988;463:47–56. doi: 10.1016/0006-8993(88)90525-2. [DOI] [PubMed] [Google Scholar]
- Yang XD, Korn H, Faber DS. Long-term potentiation of electrotonic coupling at mixed synapses. Nature. 1990;348:542–5. doi: 10.1038/348542a0. [DOI] [PubMed] [Google Scholar]
- Zimmerman AL, Rose B. Permeability properties of cell-to-cell channels: kinetics of fluorescent tracer diffusion through a cell junction. J Membr Biol. 1985;84:269–83. doi: 10.1007/BF01871390. [DOI] [PubMed] [Google Scholar]