

Abstract
Background and purpose:
Non-steroidal anti-inflammatory drugs (NSAIDs) are analgesic and anti-inflammatory by virtue of inhibition of the cyclooxygenase (COX) reaction that initiates biosynthesis of prostaglandins. Findings in a pulmonary pharmacology project gave rise to the hypothesis that certain members of the NSAID class might also be antagonists of the thromboxane (TP) receptor.
Experimental approach:
Functional responses due to activation of the TP receptor were studied in isolated airway and vascular smooth muscle preparations from guinea pigs and rats as well as in human platelets. Receptor binding and activation of the TP receptor was studied in HEK293 cells.
Key results:
Diclofenac concentration-dependently and selectively inhibited the contraction responses to TP receptor agonists such as prostaglandin D2 and U-46619 in the tested smooth muscle preparations and the aggregation of human platelets. The competitive antagonism of the TP receptor was confirmed by binding studies and at the level of signal transduction. The selective COX-2 inhibitor lumiracoxib shared this activity profile, whereas a number of standard NSAIDs and other selective COX-2 inhibitors did not.
Conclusions and implications:
Diclofenac and lumiracoxib, in addition to being COX unselective and highly COX-2 selective inhibitors, respectively, displayed a previously unknown pharmacological activity, namely TP receptor antagonism. Development of COX-2 selective inhibitors with dual activity as potent TP antagonists may lead to coxibs with improved cardiovascular safety, as the TP receptor mediates cardiovascular effects of thromboxane A2 and isoprostanes.
Keywords: human platelet aggregation, airway smooth muscle, vascular smooth muscle, guinea pig lung, cyclooxygenase inhibitors
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) comprise one of the major classes of pharmaceuticals, widely used to alleviate occasional pain and fever, and to manage chronic inflammatory musculo-skeletal and joint diseases (Paulose-Ram et al., 2005).
The mode of action of NSAIDs has been debated since the introduction of aspirin in 1897. Following the observation that prostaglandin (PG) formation was inhibited by aspirin and other NSAIDs (Smith and Willis, 1971; Vane, 1971), it has been established that NSAIDs inhibit the cyclooxygenase (COX) reaction that is an early key step in the biosynthesis of prostaglandins and thromboxane (TX) from arachidonic acid. Hence, NSAIDs are generally named COX inhibitors.
Because of their widespread use, NSAIDs are also a major quantitative cause of drug adverse reactions. In particular, gastrointestinal bleedings account for a large number of deaths every year (Wolfe et al., 1999). There are at least two COX-related actions of NSAIDs contributing to the adverse gastrointestinal effects, inhibition of local production of cytoprotective prostaglandins in the gastric mucosa, and inhibition of haemostasis as a consequence of suppressed formation of the platelet aggregating thromboxane A2 (TXA2) (Whittle, 2003).
The identification of a second COX isoform (COX-2) that was inducible by inflammatory cytokines (Fu et al., 1990) seemed to open the avenue for a new class of NSAIDs with improved gastrointestinal safety. Accordingly, evidence was obtained supporting the notion that the cytoprotective and pro-aggregatory actions of prostaglandins and TXA2 were due to COX products formed in reactions catalysed by the constitutively expressed COX-1, whereas proinflammatory effects of prostaglandin E2 were due to increased biosynthesis along the COX-2 pathway (FitzGerald and Patrono, 2001). Selective COX-2 inhibitors, referred to as coxibs, were therefore introduced as a second generation of safer anti-inflammatory drugs.
Initial data were promising, documenting that the coxibs had the same efficacy as COX non-selective, that is, traditional, NSAIDs, but were associated with less gastrointestinal bleeding (Bombardier et al., 2000; Silverstein et al., 2000). The simple hypothesis that COX-2 inhibition only affected proinflammatory prostaglandins was however soon questioned (Hawkey et al., 1998; Wallace, 2001). Moreover, the discovery that COX-2 inhibition in humans suppressed the systemic biosynthesis of prostacyclin (prostaglandin I2) (McAdam et al., 1999) represented a turning point in the risk-benefit assessment of coxibs. Regrettably, the importance of this observation was not initially fully appreciated by the scientific community nor the pharmaceutical industry. This finding suggested that the physiological balance between the pro-thrombotic and vasoconstrictory actions of COX-1-derived TXA2 in platelets and the anti-aggregatory and vasorelaxant actions of COX-2-derived prostaglandin I2 in endothelium would be tilted by COX-2 inhibition in favour of aggregation, implicating cardiovascular hazard. In fact, with some delay, the adverse cardiovascular effects of coxibs emerged most clearly from several long term studies with primary endpoints other than cardiovascular safety, where an increased number of serious adverse cardiovascular events associated with the use of rofecoxib (Bresalier et al., 2005), celecoxib (Solomon et al., 2005) and valdecoxib (Nussmeier et al., 2005) was observed. In addition, long-term follow-up of gastrointestinal outcomes associated with the use of some coxibs suggested similar incidence of upper gastrointestinal ulcer complications as those associated with NSAIDs (Juni et al., 2002). Thus, it has been difficult to develop safer NSAIDs, and this illustrates the complexity of the biologically active products formed along the COX pathway.
In this communication, we report how we discovered a previously unknown mode of action of one of the most frequently used NSAIDs, diclofenac. The observation was made in a project where we investigated the mode of action of the mast cell mediator, prostaglandin D2 (PGD2) in airways. It was found that diclofenac had a profile of activity that differed from other NSAIDs. As reported here, pharmacological characterization showed that diclofenac was a competitive antagonist of the thromboxane receptor (TP). Moreover, we also found that this additional mode of action was shared by the highly COX-2 selective derivative of diclofenac, lumiracoxib (Esser et al., 2005). The data presented in this report highlight that, in spite of the established action of NSAIDs and coxibs as COX inhibitors, individual compounds may possess additional and compound-related mechanisms of action that could have an impact on their efficacy and safety.
Methods
Animals
The study was approved by the local Ethical Review Board (127/04 for KI and 124/2003-A for UNIMI).
Isolated perfused and ventilated guinea pig lungs
Male Dunkin Hartley guinea pigs (n=30) weighing 300–500 g were used. Whole lungs were prepared as described previously (Sundstrom et al., 2003). Briefly, the lungs were perfused with Krebs–Ringer bicarbonate buffer (composition in mM: NaCl, 118.0; KCl, 4.7; CaCl2, 2.5; MgSO4, 1.2; NaHCO3, 24.9; KH2PO4, 1.2) with the addition of glucose (5.5 mM) and HEPES (12.6 mM).
The lungs were allowed first to equilibrate for 10 min before bolus administration of PGD2 dissolved in 0.1 ml ethanol and 0.9% NaCl, 1:10, v:v and injected into the pulmonary artery. This concentration of solvent had no effect on preparation. The challenges with PGD2 were performed twice in each lung preparation, in control and after pretreatment with one of the substances under investigation: the COX non-selective NSAIDs diclofenac and flurbiprofen (10 μM each, 20 min), the TP receptor antagonist SQ 29548 (1 μM, 5 min) and the TX synthase inhibitor ozagrel (30 μM, 15 min). Drug stock solutions were made in ethanol or dimethylsulphoxide (DMSO) and added to perfusion buffer in 1000-fold dilutions or more, to avoid solvent effects on the preparation.
Tracheal airflow was measured with a heated pneumotachograph (Hans Rudolph, Inc., Kansas City, Missouri, USA) connected to the transducer in the EMKA system (EMKA Technologies, Paris, France) that also measured pulmonary pressure. Lung-function parameters, airway conductance and dynamic compliance, were calculated and recorded by a computerized data acquisition system with software IOX (EMKA Technologies). The peak effects on airway conductance and dynamic compliance were evaluated as the percent of baseline values. Bronchoconstriction in the preparation is expressed as percent decreases in these parameters. Since changes in dynamic compliance followed similar pattern as the changes in airway conductance throughout all experiments, we show only the values of airway conductance.
Guinea pig trachea, guinea pig aorta and rat aorta preparations
Male Dunkin Hartley guinea pigs (n=30) weighing 500–900 g and male Sprague–Dawley rats (n=3) weighing 180–220 g were used. The animals were killed by inhalation of high concentrations of CO2 in air. The heart–lung–trachea package and the middle part of aorta were quickly removed and placed in ice-cold Tyrode's solution. The trachea and the aorta were dissected free from surrounding tissue and prepared as rings. The tracheal and aortic rings were placed in 5 ml organ baths filled with Tyrode's solution (composition in mM: NaCl, 142.9; KCl, 2.7; NaHCO3, 11.9; glucose, 5.5; CaCl2, 1.8; MgCl2 6H2O, 0.5; NaH2PO4, 0.4). The pH was kept at 7.4 by gassing with 6.5% CO2 in O2 and the temperature was kept constant at 37 °C. The tracheal and aortic rings were mounted on lower and upper organ hooks, connected to the isometric force-displacement transducers (EMKA Technologies). Changes in smooth-muscle tension in the preparations, that is, airway and vascular smooth muscle contractions and relaxations, were recorded and displayed by a computerized data acquisition system with software IOX (EMKA Technologies). Calculations of tension changes were made with help of data analysis software Datanalyst (EMKA Technologies).
The guinea pig tracheal rings were allowed to equilibrate for 60 min with a resting tension set at 30 mN with a load of 3 g. The capacity of the tracheal rings to contract was checked by the challenge with histamine in final molar concentrations of 0.3–30 μM. Preparations that showed less than 10-mN increases above the resting tension in response to the highest concentration of histamine were excluded. After histamine wash-out, another equilibration period of 60 min and pretreatment period of 60 min with 10 μM flurbiprofen followed, at the end of which the baseline tension was always adjusted to 30 mN. Thereafter, cumulative concentration–response relations for the following agonists: PGD2, the stable TX analogue U-46619 or leukotriene D4 were established in control or in the presence of substances under investigation: diclofenac, flurbiprofen and the selective COX-2 inhibitors lumiracoxib, celecoxib or rofecoxib (only U-46619), added 20 min before the cumulative concentration-response curves for agonists were performed. Drug stock solutions were made in ethanol or DMSO and added to tissue baths in 1000-fold dilutions or more, to avoid solvent effects on the preparation. All responses were expressed as percent of the maximum contractions induced by KCl (40 mM). Control preparations received the respective solvent only.
The guinea pig and rat aortic rings were allowed to equilibrate for 60 min; the baseline resting tension was set at 10 mN with a load of 1 g and the preparations were treated for 20 min with 10 μM indomethacin. The capacity of the aortic rings to contract and to relax was checked by challenges with 10 μM noradrenaline and 0.1–10 μM acetylcholine, respectively. After another equilibration period of 60 min and the pretreatment period of 20 min with 10 μM indomethacin, cumulative concentration–response relations for U-46619 were established in control or in the presence of diclofenac or lumiracoxib, added to the organ bath fluid 20 min before the concentration–response curves for U-46619 were performed. All responses were expressed as percent of the maximum contractions induced by addition of KCl (40 mM).
Isolation of human platelets and analysis of platelet aggregation
Human blood was taken from the antecubital vein of healthy volunteers of both genders who had not taken medications for at least 72 h and had no history of cardiovascular diseases; age range spanned from 25 to 60 years (mean 35 years). Blood was anticoagulated with anticoagulant citrate dextrose solution (ACD) (84 mM sodium citrate, 41 mM citric acid and 136 mM glucose; 1:7, v:v) and treated with 1 mM acetylsalicylic acid. Platelet-rich plasma was obtained by centrifugation at 180 g for 15 min at room temperature, and further centrifugation at 650 g for 10 min at room temperature, to obtain a platelet pellet that was resuspended in HEPES-buffered Tyrode's solution (2.5 mM KCl, 120 mM NaCl, 1 mM MgCl2, 25 mM NaHCO3, 5 mM glucose and 4.2 mM HEPES, pH 7.4). Washed platelet suspension was adjusted to 2 × 108 cell ml−1. CaCl2 (0.9 mM) was added immediately before drug or vehicle incubation.
Agonist-induced platelet aggregation was determined using the Born turbidimetric assay (Born and Cross, 1963) in a 0.5-ml sample of washed platelets at 37 °C, using a Chrono-Log aggregometer (Mascia Brunelli, Milano, Italy). The baseline was set using HEPES-buffered Tyrode's solution as blank (100% light transmission vs platelet suspension). The platelet samples were incubated with drug or vehicle (DMSO, maximum 0.2%, v:v) for 5 min at 37 °C, challenged with the TP agonist U-46619 (0.5–1 μM) with stirring and the aggregation followed for 6 min. In a few selected experiments, platelet aggregation was induced by thrombin (1 U ml−1) or by the calcium ionophore A-23187 (3 μM). The use of DMSO did not affect either thrombin or U-46619-induced aggregation. Experiments were repeated in triplicate using platelets from different subjects (n=3–5). Given the significant inter-subject variability of the platelet response to agonist challenge, the anti-aggregating activity of different compounds was compared with the appropriate control aggregation, recorded immediately before and after drug testing.
Culture and transfection of HEK293 cells
Human embryonic kidney cell line (HEK293) cells (ATCC, Rockville, MD, USA) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% foetal bovine serum, 2 mM glutamine, 50 U ml−1 penicillin, 100 μg ml−1 streptomycin and 20 mM HEPES buffer pH 7.4, at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Transfection with human TPα receptor construct was performed as previously described (Capra et al., 2004). Briefly, cells were plated into 12-well (total inositol phosphate (IP) formation assay) or 24-well (binding assay) tissue culture dishes previously coated with 5 μg ml−1 poly-D-lysine gelatin, and transfected at 50–60% confluence with Lipofectamine 2000, in Dulbecco's modified Eagle's medium plus 10% foetal bovine serum with an optimized 2:1 Lipofectamine 2000/DNA ratio. Equal protein content was assured at the end of each assay by Lowry dye binding procedure. Transfection reagent Lipofectamine 2000, cell culture media, serum, and supplements were purchased from Invitrogen-Life Technologies (Carlsbad, CA, USA).
Ligand binding assays in HEK293 cells
Receptor expression was monitored 48 h after transfection. Equilibrium mixed-type binding curve of [3H]SQ 29548 (Perkin-Elmer, Boston, MA, USA) together with heterologous competition curves of the specified ligands were generated as previously described (Capra et al., 2003, 2004). Briefly, confluent adherent cells in 250 μl of serum-free Dulbecco's modified Eagle's medium, containing 0.2% (w:v) bovine serum albumin, were assayed in the presence of 0.1–1 nM of the specific receptor antagonist [3H]SQ 29548 (48 Ci mmol−1), 3 nM–10 μM of the homologous cold ligand or 1–300 μM of the heterologous cold ligands. All samples contained 0.2% ethanol (v:v) as vehicle for SQ 29548, and 0.3% DMSO (v:v) as the drug vehicle. After 30 min incubation at 25 °C, cells were lysed in 0.5 N NaOH. Radioactivity was measured by liquid scintillation counting (Ultima Gold; Packard Instruments, Meriden, CT, USA).
Measurement of total IP in HEK293 cells
Functional activity of the receptor was assessed 48 h after transfection by measuring accumulation of total IPs as previously described (Capra et al., 2004). Briefly, cells labelled with 1 μCi of myo-[2–3H]inositol (18 Ci mmol−1; Perkin-Elmer) for 24 h in serum-free, inositol-free Dulbecco's modified Eagle's medium (ICN Pharmaceuticals Inc., Costa Mesa, CA, USA), containing 20 mM HEPES buffer pH 7.4 and 0.5% (w:v) Albumax I, were incubated with 25 mM LiCl for 10 min. Following pretreatment with inhibitors or antagonists (SQ 29548, diclofenac, lumiracoxib), cells were stimulated for 30 min with either vehicle or U-46619 (1 μM), lysed with 10 mM formic acid for 30 min and total IPs were extracted by means of anion exchange AG 1X-8 columns, formate form, 200–400 mesh (BioRad Laboratories, Hercules, CA, USA).
Data analysis
Statistical analysis of the data obtained in isolated lung model (Figure 1) was performed using Student's t-test for paired observations; P<0.05 was considered significant.
Figure 1.

Antagonism by diclofenac of PGD2-induced bronchoconstriction in isolated perfused and ventilated guinea pig lungs. Bronchoconstriction induced by a single dose of PGD2, administered intravascularly to the isolated, perfused and ventilated guinea pig lungs; effect of pretreatment with COX inhibitors, (a) diclofenac (Diclo) or (b) flurbiprofen (Flur) with the selective TP receptor antagonist SQ 29548 (c) or with the TX synthase inhibitor ozagrel (d). Bronchoconstriction (mean±s.e.) is expressed as percent decrease in airway conductance related to baseline. **P<0.01; ***P<0.001. COX, cyclooxygenase; PGD2, prostaglandin D2; s.e., standard error.
For the antagonist assays, agonist concentration–response curves, in the absence and presence of antagonist (Figures 2, 3 and 4), were globally fitted to the modified Gaddum/Schild model using Prism 4 (GraphPad Software Inc., San Diego, CA, USA):
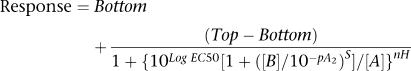 |
where Top represents the maximal asymptote of the curves, Bottom represents the lowest asymptote (basal response) of the curves, Log EC50 represents the logarithm of the agonist EC50 in the absence of antagonist, [A] represents the concentration of the agonist, [B] represents the concentration of the antagonist, nH represents the Hill slope of the agonist curve, s represents the Schild slope for the antagonist and pA2 represents the negative logarithm of the concentration of antagonist that shifts the agonist EC50 by a factor of 2. All curves are computer generated. The concentration–response curves of platelet aggregation (Figure 5) were analysed by Prism-4 software utilizing the four-parameter logistic model.
Figure 2.

Antagonism by diclofenac of guinea pig airway smooth-muscle contractions induced by PGD2 and the TX mimetic U-46619. Contractions induced by challenges with cumulatively increasing concentrations of PGD2 in guinea pig tracheal rings, pretreated with 10 μM flurbiprofen, (a) effect of diclofenac (pA2=6.7±0.28 s.e.) and (b) lack of effect of flurbiprofen; (c) lack of the effect of diclofenac on airway smooth-muscle contractions induced by challenges with cumulatively increasing concentrations of leukotriene D4 in guinea pig tracheal rings pretreated with 10 μM flurbiprofen. (d) Effect of diclofenac (pA2=5.83±0.16 s.e.) on airway smooth-muscle contractions evoked by challenges with cumulatively increasing the concentrations of the selective TP receptor agonist U-46619 in guinea pig tracheal rings pretreated with 10 μM flurbiprofen. Airway smooth-muscle contractions (mean±s.e.) are expressed as percent of a maximal contraction induced with 40 mM KCl at the end of the experiments. LTD4, leukotriene D4; PGD2, prostaglandin D2; s.e., standard error.
Figure 3.

Antagonism by lumiracoxib of guinea pig airway smooth-muscle contractions induced by the TX mimetic U-46619. (a–c) Contractions induced by challenges with cumulatively increasing concentrations of U-46619 in guinea pig tracheal rings pretreated with 10 μM flurbiprofen; (a) effect of lumiracoxib (pA2=5.05±0.11 s.e.). Lack of inhibitory effect of two other selective COX-2 inhibitors, celecoxib(b) and rofecoxib (c). (d) Contractions induced by challenges with cumulatively increasing concentrations of U-46619 in na¿ve (untreated) guinea pig tracheal rings; effect of lumiracoxib (pA2=4.8±0.13 s.e.). Airway smooth-muscle contractions (mean±s.e.) are expressed as percent of a maximal contraction induced with 40 mM KCl at the end of the experiments. COX-2, cyclooxygenase 2; s.e., standard error.
Figure 4.

Antagonism by diclofenac and lumiracoxib of guinea pig and rat vascular smooth-muscle contractions induced by the TX mimetic U-46619. Contractions of vascular smooth muscle induced by challenges with cumulatively increasing concentrations of U-46619 in guinea pig (a–b) and rat (c) aortic rings pretreated with 10 μM indomethacin; (a) effect of diclofenac (pA2=6.33±0.11 s.e.) and (b) lumiracoxib (pA2=4.4±0.10 s.e.); (c) contractions of rat aortic rings challenged with cumulatively increasing concentrations of U-46619 in the absence (EC50=25 nM±8 % CV) or the presence (EC50=285 nM±8 % CV) of 60 μM diclofenac. Partial reversibility of the effect of diclofenac after drug removal by change of media (U-46619, EC50=57 nM±9 % CV). Vascular smooth-muscle contractions (mean±s.e.) are expressed as percent of a maximal contraction induced with 40 mM KCl at the end of the experiments. CV, coefficient of variation; s.e., standard error.
Figure 5.

Representative experiments of aggregation of human aspirin-treated platelets (see Methods). Challenge with thrombin in the presence of solvent (DMSO) or lumiracoxib (60 μM) (left panel). Challenge with U-46619 in the presence of DMSO or lumiracoxib (60 μM). After response to U-46619 was blocked by lumiracoxib, the preparation responded to ionophore (3 μM) (right panel). DMSO, dimethylsulphoxide.
Statistical analysis of ligand binding data (Figure 6) was performed with the LIGAND programme (Munson and Rodbard, 1980). Nonspecific binding was calculated as an unknown parameter of the model. Parameter errors are always expressed in percentage coefficient of variation (% CV) and calculated by simultaneous analysis of at least two different independent experiments performed in duplicates or triplicates. Data are presented as means±s.e. of multiple independent experiments each performed at least in duplicate. A statistical level of significance of P<0.05 was accepted. All curves are computer generated.
Figure 6.

Antagonism by diclofenac and lumiracoxib of platelet aggregation induced by the TX mimetic U-46619. Aggregation of washed human platelets induced by challenges with increasing concentrations of the TX analogue U-46619; (a) effect of diclofenac (pA2=4.97±0.09 s.e.) and (b) lumiracoxib (pA2=4.60±0.06 s.e.). Gaddum–Schild analysis indicated a Schild slope of 1.6 for diclofenac and 2.3 for lumiracoxib, statistically different from 1, indicating that the effect of diclofenac and lumiracoxib cannot be defined as a pure competitive receptor antagonism. Blood was collected in the presence of 1 mM acetylsalicylic acid. Platelet aggregation (mean) is expressed as percent of a maximal aggregation induced with U-46619 (0.5–1 μM). s.e., standard error.
Chemicals and drugs
All chemicals were fine grade and purchased from Sigma (St Louis, MI, USA) and other commercial suppliers. Agonists and NSAIDs were purchased from Cayman Chemicals (Ann Arbor, MI, USA), SynphaBase AG (Basel, Switzerland) or Sigma.
Results
Experiments in perfused guinea pig lungs and isolated airway and vascular smooth-muscle preparations of guinea pig and rat
In a study of bronchoconstriction evoked by PGD2 in the isolated, perfused and ventilated guinea pig lung, it was observed that diclofenac (10 and 100 μM) inhibited the response to PGD2 in a concentration-dependent manner (Figure 1a). In contrast, another non-selective COX inhibitor, flurbiprofen (10 and 100 μM), had no significant effect on bronchoconstriction induced by PGD2 (Figure 1b). The response to PGD2 in this model is mediated by the activation of TP receptors, as demonstrated by total blockade of the response to PGD2 by the selective TP receptor antagonist SQ 29548 (Ogletree et al., 1985) (1 μM) (Figure 1c). In contrast, the selective TX synthase inhibitor ozagrel (30 μM) had no effect on the bronchoconstriction evoked by PGD2 (Figure 1d). The latter observation excluded the possibility that the response to PGD2 involved release of secondarily formed TXA2, a mechanism that at least theoretically might have contributed to the activation of TP receptors by PGD2.
The findings in the isolated lung model therefore gave rise to the hypothesis that diclofenac might exert TP receptor antagonism. This was further tested in the guinea pig trachea, a standard airway pharmacology in vitro model, where the contraction response to PGD2 is mediated by TP receptors (Featherstone et al., 1990). In order to optimize the detection of a pharmacological action of diclofenac distinct from COX inhibition, we performed the experiments in a protocol where the preparation's endogenous COX was inhibited by pretreatment with flurbiprofen (10 μM). Under these conditions, diclofenac (1–100 μM) concentration dependently inhibited the smooth-muscle contractions evoked by PGD2 (Figure 2a). As in the isolated lungs (Figure 1b), flurbiprofen (1–100 μM) failed to inhibit the response to PGD2 (Figure 2b). Nor did diclofenac (1–100 μM) inhibit airway smooth-muscle contractions induced by another agonist, leukotriene D4 (1 pM–1 μM) (Figure 2c). Together, the findings supported that the inhibitory effect of diclofenac was selective for a contractile agonist acting on TP receptors.
In the same protocol, it was further examined whether the inhibitory and COX-independent effect of diclofenac could be confirmed when airway smooth-muscle contractions were evoked by the selective TP receptor agonist U-46619. This was indeed the case; diclofenac concentration dependently inhibited the airway smooth-muscle contractions evoked by U-46619 (Figure 2d). Furthermore, also in a model of peripheral lung reactivity, the guinea pig lung parenchyma (Drazen and Schneider, 1978), diclofenac and lumiracoxib concentration dependently inhibited the contraction response to U-46619 in an indomethacin pretreated protocol similar to that used in the trachea (data not shown).
On the basis of our findings with diclofenac (Figures 1, 2), we next examined the actions of the potent selective COX-2 inhibitor lumiracoxib, which is a structural analogue of diclofenac. We hypothesized that lumiracoxib might share the TP receptor antagonism observed for diclofenac. This was first tested in the guinea pig trachea protocol. Lumiracoxib (10–100 μM) was indeed found to inhibit the airway smooth-muscle contraction induced by U-46619 in a concentration-dependent manner (Figure 3a). In contrast, two other COX-2 inhibitors, celecoxib (Figure 3b) and rofecoxib (Figure 3c), failed to inhibit airway smooth-muscle contractions evoked by U-46619.
At this stage, we concluded that both diclofenac and lumiracoxib in addition to being COX inhibitors had the action to inhibit TP receptor-dependent contractions of airway smooth muscle. In order to assess the relative contribution of the two different modes of action at different drug concentrations, we next studied the influence of lumiracoxib on U-46619-induced contractions in naïve guinea pig trachea preparations, that is, without the flurbiprofen pretreatment that had been used in the experiments that provided evidence for TP antagonism. Also in the naïve preparation, lumiracoxib (10–100 μM) concentration dependently inhibited the response to U-46619 (Figure 3d). In the low-concentration range (0.1–1 μM), lumiracoxib, however, only enhanced the contraction response to U-46619 (Figure 3d).
Furthermore, the TP antagonist effects of diclofenac and lumiracoxib were confirmed also in preparations of vascular smooth muscle. Thus, in the guinea pig aorta, in a protocol where the preparation was pretreated with indomethacin (10 μM), diclofenac (Figure 4a) or lumiracoxib (Figure 4b) concentration dependently inhibited the response to U-46619. In the aorta from another species, the rat, diclofenac (60 μM) also caused significant inhibition of vascular smooth-muscle contractions evoked by U-46619 (Figure 4c). The antagonistic effect of diclofenac appeared readily reversible, as the responsiveness to U-46619 in the rat model returned to control level 15 min after drug removal by changing of media (Figure 4c).
Human platelet aggregation experiments
Blood was collected in 1 mM acetylsalicylic acid; this pretreatment made the platelets unresponsive to the challenge with arachidonic acid (1–3 μM), but they were fully responsive to the calcium ionophore A-23187 (3 μM) or thrombin (0.2 U ml−1) (Figure 5, left panel). When platelets were challenged with U-46619, a concentration-dependent platelet aggregation occurred, with an EC50 value of 64 nM±17 % CV. This response was thus truly independent of endogenous TXA2 formation.
As further evidence that diclofenac exerted TP antagonism, pretreatment with increasing concentrations (20–100 μM) of diclofenac (Figure 6a) or lumiracoxib (Figure 6b) inhibited the aggregation of human platelets. Both drugs caused a rightward shift of the concentration–response curve for U-46619. However, pretreatment with lumiracoxib (60 μM) left thrombin- or A-23187-induced aggregation of human platelets unaffected (Figure 5, right panel). In contrast, neither the selective COX-2 inhibitor, celecoxib, nor the non-selective COX inhibitor, flurbiprofen, inhibited the platelet aggregation evoked by U-46619 (n=3, data not shown).
Human TPα receptor transiently expressed in HEK293 cells: whole-cell binding and total IP determination
The TP antagonistic effects of diclofenac and lumiracoxib were confirmed in radioligand binding studies in HEK293 cells labelled with [3H]-SQ 29548 (Figure 7). Mixed-type curves of [3H]-SQ 29548 and heterologous competition curves of diclofenac or lumiracoxib were monophasic, fitting a single-site model. The data indicated typical binding parameters for the interaction of SQ 29548 with the TPα receptor, as previously reported (Capra et al., 2004). In agreement with the results obtained in airway and vascular preparations, as well as in the human platelets, both diclofenac and lumiracoxib were able to compete for the labelled antagonist, albeit with lower affinity than SQ 29548 (Figure 7). No detectable binding in mixed-type curve of [3H]-SQ 29548 was observed when cells were transfected with the empty vector (data not shown).
Figure 7.

Equilibrium binding of [3H]SQ 29548 in HEK293 transiently expressing human TPα receptor. Mixed-type binding curve of SQ 29548 was generated using 0.1–1 nM [3H]SQ 29548 (saturation part of the curve) and 3 nM–10 μM of the homologous ligand (Kd=4.47 nM±36 % CV) (competition part of the curve). Heterologous competition curves were performed using 1 nM [3H]SQ 29548 and 1–300 μM of diclofenac (Ki=26.5 μM±29 % CV) or lumiracoxib (Ki=122 μM±33 % CV). Binding is expressed as the ratio of bound ligand concentration over total ligand concentration, (B/T, dimensionless), vs the logarithm of total ligand concentration (log T). B (in M) is the sum of ‘hot', ‘cold' and nonspecific binding; T (in M) is the sum of ‘hot' and ‘cold' ligand incubated. Experiments, performed in duplicate, and were analysed simultaneously with LIGAND. CV, coefficient of variation; HEK293, human embryonic kidney cell line 293.
Signalling of TPα receptor was also investigated by measuring the capacity of diclofenac and lumiracoxib to inhibit agonist-induced total IP production. As expected, HEK293 cells expressing the human TPα receptor responded to agonist stimulation (1 μM U-46619), with approximately a threefold increase of the total IP production. Pretreatment with diclofenac or lumiracoxib inhibited U-46619-induced IP production, whereas ATP-stimulated IP production was unaffected (n=3, data not shown), supporting the proposal that the effect of diclofenac and lumiracoxib was linked selectively to antagonism of the TP receptor.
Discussion
During an investigation of the mechanism of action of the mast-cell mediator PGD2 in airways, it was observed that diclofenac had a profile of activity in the perfused guinea pig lung that was distinct from that of several other NSAIDs (Selg et al., manuscript in preparation). This new action of diclofenac was displayed at a concentration of drug that also was required to inhibit COX activity. Further analysis in airway and vascular smooth-muscle preparations, as well as in human platelets, disclosed that diclofenac was a competitive TP receptor antagonist. This mode of action was confirmed by performing receptor binding experiments in human recombinant TP receptors, as well as by the assessment in the same system of the effect of diclofenac on the TP receptor signalling.
Moreover, the selective COX-2 inhibitor, lumiracoxib, being structurally related to diclofenac, was found to share the TP receptor antagonist activity established for diclofenac, although at about half a log order of magnitude higher concentrations than diclofenac. The TP antagonistic effect of lumiracoxib was also documented in functional studies of airway and vascular smooth muscle and in human platelets. This novel pharmacodynamic activity of diclofenac and lumiracoxib was not shared by the collection of comparative NSAIDs and coxibs that were tested. Although the antagonism exerted by diclofenac and lumiracoxib was classically surmountable, strict pharmacological analysis indicated a complex interaction in some of the test systems (Schild plot slopes sometimes being different from 1) (Figure 5). This can presumably be explained by the dual activity of these drugs.
Diclofenac and lumiracoxib exhibited TP antagonism, that is, inhibition of U-46619-induced bronchoconstriction, vasoconstriction as well as aggregation of human platelets at micromolar concentrations (Figures 2, 3, 4 and 5) that in fact can be reached following administration of therapeutic doses of either drug (Todd and Sorkin, 1988; Lyseng-Williamson and Curran, 2004). Furthermore, in our experiments in naïve guinea pig airways (Figure 3d), we also showed that lumiracoxib displayed TP antagonism in the micromolar concentration range. Interestingly, in submicromolar concentrations, lumiracoxib potentiated the response to U-46619 in that particular assay, exactly as other NSAIDs do (Selg et al., manuscript in preparation). However, we are not claiming that lumiracoxib at therapeutic doses predominantly acts as a TP antagonist, and the TP antagonistic property may not add significantly to the pharmacodynamic profile of diclofenac, as it primarily should inhibit formation of all COX products.
What is of great interest in our opinion is the observation that lumiracoxib acts as a dual drug with a new and previously unknown pharmacodynamic profile, that is, COX-2 inhibition and TP antagonism. The implication of this observation is that new compounds can be developed with dual activity as selective COX-2 inhibitors and TP antagonists. Such compounds may have, besides a better gastrointestinal tolerability than NSAIDs, also a broader and more favourable pharmacodynamic profile than coxibs.
First, a TP antagonistic property might be an advantage with respect to anti-inflammatory and analgesic effect of these compounds. The dual activity has a great potential to add anti-inflammatory mode of action beyond COX-2 inhibition, as biosynthesis of TXA2 and PGD2 along the COX-1 pathway is unaffected by coxibs. The actions of TXA2 on TP receptors have thus been reported to augment cellular immune responses and inflammatory tissue injury (Thomas et al., 2003) and cause inflammatory tachycardia (Takayama et al., 2005). Moreover, there is in fact a COX-independent pathway for isoprostane formation during oxidative stress and tissue injury (Morrow et al., 1990), and the involvement of TP receptors is suggested in the nociceptor sensitization caused by isoprostanes (Evans et al., 2000). It is thus possible that selective COX-2 inhibitors with the TP antagonistic property may have a favourable anti-inflammatory and analgesic effect. As mentioned above, the enhanced anti-inflammatory activity due to TP antagonism may seem less likely to occur for the COX non-selective diclofenac. However, diclofenac may have a favourable anti-inflammatory and analgesic effect, at least in clinical settings with a substantial role for isoprostanes.
Second, the TP antagonism may have relevance with respect to the cardiovascular hazard of anti-inflammatory drugs other than aspirin. It is now generally accepted that COX inhibition and suppression of the biosynthesis of prostaglandin I2 is the major cause of adverse cardiovascular events to coxibs in particular and to NSAIDs in general (Grosser et al., 2006; Mitchell et al., 2006). There are a number of reports of TP receptor-mediated detrimental cardiovascular actions exerted by COX-1-derived TXA2 and by non-enzymatically formed isoprostanes. Thus, the unopposed effect of endogenous TP agonists on platelets increases the risk for thrombosis (Hennan et al., 2001). Moreover, TP receptors mediate the effects of isoprostanes on platelet function and vascular tone, with relevance for atherogenesis and injury after ischaemia and reperfusion (Audoly et al., 2000). The TP-mediated effects of TXA2 contribute to the development of cardiac hypertrophy and fibrosis in hypertension (Francois et al., 2005) and to atherogenesis (Worth et al., 2005). There is in fact data supporting the view that TP antagonism decreases the proliferative response to carotid vascular injury (Cheng et al., 2002). It is thus possible that a COX-2 inhibitor that also is a TP antagonist might have a favourable cardiovascular effect profile. However, the events that link the TP receptor to platelets, the vessel wall and finally to the thrombotic event are complex and the proof-of-principle in vivo experiments should ideally be done later on, with newly developed dual compounds with improved TP antagonist potency compared to that of lumiracoxib.
The limited TP antagonistic potency displayed by lumiracoxib and diclofenac in the present study may make the TP antagonism of dubious clinical relevance with respect to the cardiovascular effect profile of these particular drugs. The full understanding of the clinical impact of our results for the cardiovascular effects of diclofenac and lumiracoxib is far beyond the purpose of this work, and will require a number of different sub-studies, with many endpoints to consider. Nevertheless, the only long-term study assessing the cardiovascular safety of lumiracoxib in 18 325 patients failed to detect a significant cardiovascular hazard (Farkouh et al., 2004). The latter study has however been criticized as the number of subjects at risk for cardiovascular events was low in the trial that primarily was designed to study gastrointestinal bleeding during treatment of osteoarthritis. Certainly, new studies in risk groups are warranted.
In summary, the specific implication of our current findings is that diclofenac and lumiracoxib could represent prototypes of novel chemical entities that may be designed with selectivity to COX isoenzymes, combined with improved TP antagonist potency. This may give raise to a new generation of coxibs with increased efficacy and, potentially, decreased cardiovascular adverse effects. A general implication of our findings is that it may be worthwhile to develop new and optimized NSAIDs that have several synergistic pharmacologic activities. Finally, as a historical note, before the discovery that NSAIDs were COX inhibitors, certain results obtained in vitro with this class of drugs had been interpreted as if NSAIDs were receptor antagonists (Collier et al., 1963). It appears that the concepts of the early investigators were not altogether incorrect.
Acknowledgments
This work was supported by grants to Sven-Erik Dahlén by the Swedish Medical Research Council, the Swedish Heart and Lung Foundation, the Stockholm County Council Research Funds and Karolinska Institutet, and by EU Grant LSHM-CT-2004-005033 EICOSANOX to Giancarlo Folco.
Abbreviations
- HEK293
human embryonic kidney cell line
- NSAIDs
non-steroidal anti-inflammatory drugs
- PGD2
prostaglandin D2
- PGI2
prostaglandin I2
- TP
thromboxane receptor
- TXA2
thromboxane A2
Conflict of interest
The authors state no conflict of interest.
References
- Audoly LP, Rocca B, Fabre JE, Koller BH, Thomas D, Loeb AL, et al. Cardiovascular responses to the isoprostanes iPF(2alpha)-III and iPE(2)-III are mediated via the thromboxane A(2) receptor in vivo. Circulation. 2000;101:2833–2840. doi: 10.1161/01.cir.101.24.2833. [DOI] [PubMed] [Google Scholar]
- Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- Born GV, Cross MJ. The aggregation of blood platelets. J Physiol. 1963;168:178–195. doi: 10.1113/jphysiol.1963.sp007185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- Capra V, Habib A, Accomazzo MR, Ravasi S, Citro S, Levy-Toledano S, et al. Thromboxane prostanoid receptor in human airway smooth muscle cells: a relevant role in proliferation. Eur J Pharmacol. 2003;474:149–159. doi: 10.1016/s0014-2999(03)02014-4. [DOI] [PubMed] [Google Scholar]
- Capra V, Veltri A, Foglia C, Crimaldi L, Habib A, Parenti M, et al. Mutational analysis of the highly conserved ERY motif of the thromboxane A2 receptor: alternative role in G protein-coupled receptor signaling. Mol Pharmacol. 2004;66:880–889. doi: 10.1124/mol.104.001487. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, et al. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–541. doi: 10.1126/science.1068711. [DOI] [PubMed] [Google Scholar]
- Collier HOJ, Hammond AR, Whiteley B. Anti-anaphylactic action of acetylsalicylate in guinea pig lung. Nature. 1963;200:176–178. doi: 10.1038/200176b0. [DOI] [PubMed] [Google Scholar]
- Drazen JM, Schneider MW. Comparative responses of tracheal spirals and parenchymal strips to histamine and carbachol in vitro. J Clin Invest. 1978;61:1441–1447. doi: 10.1172/JCI109063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser R, Berry C, Du Z, Dawson J, Fox A, Fujimoto RA, et al. Preclinical pharmacology of lumiracoxib: a novel selective inhibitor of cyclooxygenase-2. Br J Pharmacol. 2005;144:538–550. doi: 10.1038/sj.bjp.0706078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AR, Junger H, Southall MD, Nicol GD, Sorkin LS, Broome JT, et al. Isoprostanes, novel eicosanoids that produce nociception and sensitize rat sensory neurons. J Pharmacol Exp Ther. 2000;293:912–920. [PubMed] [Google Scholar]
- Farkouh ME, Kirshner H, Harrington RA, Ruland S, Verheugt FW, Schnitzer TJ, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), cardiovascular outcomes: randomised controlled trial. Lancet. 2004;364:675–684. doi: 10.1016/S0140-6736(04)16894-3. [DOI] [PubMed] [Google Scholar]
- Featherstone RL, Robinson C, Holgate ST, Church MK. Evidence for thromboxane receptor mediated contraction of guinea-pig and human airways in vitro by prostaglandin (PG) D2, 9 alpha,11 beta-PGF2 and PGF2 alpha. Naunyn Schmiedebergs Arch Pharmacol. 1990;341:439–443. doi: 10.1007/BF00176337. [DOI] [PubMed] [Google Scholar]
- FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345:433–442. doi: 10.1056/NEJM200108093450607. [DOI] [PubMed] [Google Scholar]
- Francois H, Athirakul K, Howell D, Dash R, Mao L, Kim HS, et al. Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metab. 2005;2:201–207. doi: 10.1016/j.cmet.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Fu JY, Masferrer JL, Seibert K, Raz A, Needleman P. The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J Biol Chem. 1990;265:16737–16740. [PubMed] [Google Scholar]
- Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkey CJ, Tulassay Z, Szczepanski L, van Rensburg CJ, Filipowicz-Sosnowska A, Lanas A, et al. Randomised controlled trial of Helicobacter pylori eradication in patients on non-steroidal anti-inflammatory drugs: HELP NSAIDs study. Helicobacter eradication for lesion prevention. Lancet. 1998;352:1016–1021. doi: 10.1016/s0140-6736(98)04206-8. [DOI] [PubMed] [Google Scholar]
- Hennan JK, Huang J, Barrett TD, Driscoll EM, Willens DE, Park AM, et al. Effects of selective cyclooxygenase-2 inhibition on vascular responses and thrombosis in canine coronary arteries. Circulation. 2001;104:820–825. doi: 10.1161/hc3301.092790. [DOI] [PubMed] [Google Scholar]
- Juni P, Rutjes AW, Dieppe PA. Are selective COX 2 inhibitors superior to traditional non-steroidal anti-inflammatory drugs. BMJ. 2002;324:1287–1288. doi: 10.1136/bmj.324.7349.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyseng-Williamson KA, Curran MP.Lumiracoxib Drugs 2004642237–2246.discussion 2247–2248 [DOI] [PubMed] [Google Scholar]
- McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci USA. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Lucas R, Vojnovic I, Hasan K, Pepper JR, Warner TD. Stronger inhibition by nonsteroid anti-inflammatory drugs of cyclooxygenase-1 in endothelial cells than platelets offers an explanation for increased risk of thrombotic events. FASEB J. 2006;20:2468–2475. doi: 10.1096/fj.06-6615com. [DOI] [PubMed] [Google Scholar]
- Morrow J, Hill K, Burk R, Nammour T, Badr K, Roberts LJI. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci USA. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson PJ, Rodbard D. Ligand: a versatile computerized approach for characterization of ligand-binding systems. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- Nussmeier NA, Whelton AA, Brown MT, Langford RM, Hoeft A, Parlow JL, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med. 2005;352:1081–1091. doi: 10.1056/NEJMoa050330. [DOI] [PubMed] [Google Scholar]
- Ogletree ML, Harris DN, Greenberg R, Haslanger MF, Nakane M. Pharmacological actions of SQ 29548, a novel selective thromboxane antagonist. J Pharmacol Exp Ther. 1985;234:435–441. [PubMed] [Google Scholar]
- Paulose-Ram R, Hirsch R, Dillon C, Gu Q. Frequent monthly use of selected non-prescription and prescription non-narcotic analgesics among US adults. Pharmacoepidemiol Drug Saf. 2005;14:257–266. doi: 10.1002/pds.983. [DOI] [PubMed] [Google Scholar]
- Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: a randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA. 2000;284:1247–1255. doi: 10.1001/jama.284.10.1247. [DOI] [PubMed] [Google Scholar]
- Smith JB, Willis AL. Aspirin selectively inhibits prostaglandin production in human platelets. Nat New Biol. 1971;231:235–237. doi: 10.1038/newbio231235a0. [DOI] [PubMed] [Google Scholar]
- Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- Sundstrom E, Lastbom L, Ryrfeldt A, Dahlen S-E. Interactions among three classes of mediators explain antigen-induced bronchoconstriction in the isolated perfused and ventilated guinea pig lung. J Pharmacol Exp Ther. 2003;307:408–418. doi: 10.1124/jpet.103.053546. [DOI] [PubMed] [Google Scholar]
- Takayama K, Yuhki K, Ono K, Fujino T, Hara A, Yamada T, et al. Thromboxane A2 and prostaglandin F2alpha mediate inflammatory tachycardia. Nat Med. 2005;11:562–566. doi: 10.1038/nm1231. [DOI] [PubMed] [Google Scholar]
- Thomas DW, Rocha PN, Nataraj C, Robinson LA, Spurney RF, Koller BH, et al. Proinflammatory actions of thromboxane receptors to enhance cellular immune responses. J Immunol. 2003;171:6389–6395. doi: 10.4049/jimmunol.171.12.6389. [DOI] [PubMed] [Google Scholar]
- Todd PA, Sorkin EM. Diclofenac sodium. A reappraisal of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs. 1988;35:244–285. doi: 10.2165/00003495-198835030-00004. [DOI] [PubMed] [Google Scholar]
- Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Pathogenesis of NSAID-induced gastroduodenal mucosal injury. Best Pract Res Clin Gastroenterol. 2001;15:691–703. doi: 10.1053/bega.2001.0229. [DOI] [PubMed] [Google Scholar]
- Whittle BJ. Gastrointestinal effects of nonsteroidal anti-inflammatory drugs. Fundam Clin Pharmacol. 2003;17:301–313. doi: 10.1046/j.1472-8206.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340:1888–1899. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- Worth NF, Berry CL, Thomas AC, Campbell JH. S18886, a selective TP receptor antagonist, inhibits development of atherosclerosis in rabbits. Atherosclerosis. 2005;183:65–73. doi: 10.1016/j.atherosclerosis.2005.02.034. [DOI] [PubMed] [Google Scholar]