

Abstract
Background and Aims
Inherited syndromes of intrahepatic cholestasis commonly result from mutations in the genes SERPINA1 (α1-antitrypsin deficiency), JAG1 (Alagille syndrome), ATP8B1 (Progressive Familial Intrahepatic Cholestasis type 1/PFIC1), ABCB11 (PFIC2), and ABCB4 (PFIC3). However, the large gene sizes and lack of mutational hotspots make it difficult to survey for disease-causing mutations in clinical practice. Here, we aimed to develop a technological tool that reads out the nucleotide sequence of these genes rapidly and accurately.
Methods
25-mer nucleotide probes were designed to identify each base for all exons, 10 bases of intronic sequence bordering exons, 280–500 bases upstream from the first exon for each gene, and 350 bases of the second intron of the JAG1 gene, and tiled using the Affymetrix resequencing platform. We then developed high-fidelity PCRs to produce amplicons using 1 ml of blood from each subject; amplicons were hybridized to the chip, and nucleotide calls were validated by standard capillary sequencing methods.
Results
Hybridization of amplicons with the chip produced a high nucleotide sequence readout for all five genes in a single assay, with an automated call rate of 93.5% (range: 90.3–95.7%). The accuracy of nucleotide calls was 99.99% when compared with capillary sequencing. Testing the chip on subjects with cholestatic syndromes identified disease-causing mutations in SERPINA1, JAG1, ATP8B1, ABCB11 or ABCB4.
Conclusion
The resequencing chip efficiently reads SERPINA1, JAG1, ATP8B1, ABCB11 and ABCB4 with a high call rate and accuracy in one assay, and identifies disease-causing mutations.
INTRODUCTION
The discovery of the genetic basis of syndromes of intrahepatic cholestasis allows for the potential for diagnostic and treatment plans tailored to the patient’s genetic makeup. Inherited syndromes of intrahepatic cholestasis represent a heterogeneous group of disorders that begin during childhood, most commonly manifesting as neonatal cholestasis, and lead to ongoing liver dysfunction in children and adults.1 These disorders share many clinical features, yet discrete clinical, biochemical and histological patterns suggest specific entities. Identification of specific diseases as causes of intrahepatic cholestasis began with the report that α1-antitrypsin (α1AT) deficiency can present as neonatal cholestasis.2, 3 Subsequent studies led to the recognition of the inherited nature of syndromic and non-syndromic forms of cholestasis.4 For example, patients with the Alagille syndrome have chronic cholestasis and variable syndromic features that may be shared by family members.5, 6 Application of genetic mapping approaches to study these patients discovered disease-causing mutations in the JAG1 gene.7, 8 A significant portion of the remaining subjects with non-syndromic forms of intrahepatic cholestasis comprise a group commonly referred to as Progressive Familial Intrahepatic Cholestasis (PFIC).9–14 Patients with the most common types of PFIC are now known to harbor mutations in genes encoding proteins involved in bile acid transport: 1) ATP8B1 gene, encoding FIC1 (patients with PFIC1);15, 16 ABCB11 gene, encoding the bile salt export pump (BSEP, patients with PFIC2);17 and ABCB4 gene, encoding the multi-drug resistance protein-3 (MDR3, patients with PFIC3).18, 19 Despite the remarkable progress in identifying the genes that cause the most common inherited syndromes of intrahepatic cholestasis, current diagnostic algorithms do not incorporate mutation analysis in the clinical evaluation of affected patients. In addition, the large gene size and the lack of highly predominant mutational hotspots form obstacles to studies exploring gene-gene interactions and genotype-phenotype relationships. To fulfill this technological gap, we developed a customized resequencing gene chip that reads SERPINA1 (gene encoding α1AT), JAG1, ATP8B1, ABCB11, and ABCB4 genes simultaneously in a single assay, with high call rate and accuracy.
METHODS
Length of genes and selection of target sequences
A sequence analysis of all exon-intron elements for the SERPINA1, JAG1, ATP8B1, ABCB11, and ABCB4 genes showed that sequencing by standard methods would require the identification of 313,288 bases (data from GenBank at www.ncbi.nlm.nih.gov/entrez). An alternative high-throughput method is resequencing technology using the Affymetrix GeneChip® CustomSeq™ Resequencing Array platform that can identify 30,000 nucleotides of double-stranded contiguous or non-contiguous sequence (60,000 total) (www.affymetrix.com/products/arrays/specific/custom_seq.affx). This resequencing platform identifies individual nucleotides by a comparative, high-fidelity hybridization using four 25-mer probes designed per strand of each individual nucleotide (base), where the central position of the probes varies to incorporate each of the four possible bases (A, T, C, G). In this system, the probe that matches 100% of the target sequence (which occurs in 1 of 4 probes according to the precise match of the central position base) generates a specific signal and identifies the individual base. To select the target sequences of interest, we developed a database for all exons, 10 bases of the intronic sequence bordering exons (potential splice sites), and 280–500 bases upstream from the first exon for each gene. We also selected 350 bases of the second intron of the JAG1 gene based on its identification as a putative regulatory region as defined by Trafac, an application that surveys for potential regulatory regions by the direct comparison of conserved non-coding DNA sequences between human and mouse orthologous genes20; no similar region was identified for any of the other four genes. We included the potential regulatory sequences to facilitate studies addressing whether they may be sites of novel mutations that segregate with disease phenotypes or that modify the level of gene expression. Combined, these fragments formed the target sequences of interest and totaled 26,725 bases. The target sequences were converted to FASTA format and analyzed for repetitive elements, internal duplications and homologous sequences. The entire sequence was then submitted to Affymetrix, Inc (Santa Clara, CA) for primer design and chip assembly.
Amplification of target sequences
DNA was isolated from 1 ml of peripheral blood obtained from subjects with intrahepatic cholestasis evaluated at the Cincinnati Children’s Hospital Pediatric Liver Care Center. Informed consent was obtained from the patient’s legal guardians as approved by the Institutional Review Board of Cincinnati Children’s Hospital Medical Center. DNA was isolated using the Puregene® Purification Kit (Gentra Systems, Minneapolis, MN), according to the manufacturer’s protocol. To produce DNA samples of sufficient amounts for proper labeling and hybridization against the chip, we designed PCR primers to amplify the target sequences from each patient’s DNA (herein referred to as “amplicons”). In order to develop a simpler amplification assay with a small number of reactions that can be run simultaneously, we designed short- and long-range, high fidelity PCR protocols. They included an initial incubation of DNA with RNase A solution, followed by a total of 4 μg of purified DNA divided into small aliquots to serve as template in three amplification programs containing 35 individual reactions (Supplementary Figure 1). A 7.5 kb control fragment was amplified using primers and template included in the CustomSeq™ Control kit provided by Affymetrix.
Chip hybridization and analysis
Individual amplicons were identified in 1% agarose electrophoreses and quantified by PicoGreen dsDNA Quantitation Kit (Invitrogen, Carlsbad, CA); equimolar amounts of amplicons were pooled and subjected to fragmentation and labeling according to protocols provided by Affymetrix. In brief, fragmentation was performed with the GeneChip Fragmentation Reaction Kit, which contains an enzyme to fragment amplicons at 37 °C × 15 minutes, followed by labeling with TdT buffer, Labeling Reaction Mix and TdT enzyme at 37 °C × 2 hours, and a denaturing step at 95 °C × 15 min. Prior to hybridization, the chip was incubated with 200 μl of pre-hybridization buffer (Tris, pH7.8 and 1% Tween-20) at 45 °C in a hybridization oven for 15 min. Then, the buffer was removed and a 200 μl of hybridization solution (Tris, pH7.8, Tween-20, acetylated BSA, herring sperm DNA, labeled oligonucleotide control reagent, and labeled fragments of amplicons) was added to the resequencing chip. Following incubation at 45 °C for 16 hours, the chip was washed with decreasing concentrations of SSPE and Tween-20, stained with anti-biotin antibody, and scanned with the Affymetrix GeneChip® 3000 Scanner. Specific signals were captured by the Affymetrix GeneChip® Operating Software (GCOS) and analyzed with the Affymetrix GeneChip® DNA Analysis Software (GDAS). To examine the accuracy of the chip readout, unused fractions of the original amplicons were subjected to automated capillary sequencing using ABI Prism® 3730 DNA Analyzer at the Gene Sequencing Core at Cincinnati Children’s Research Foundation.
Statistical analysis
Base call accuracy was expressed as a percentage of the number of base calls correctly classified when compared to automated capillary sequencing. We estimated the need to sequence at least 32,431 bases to enable the detection of an error rate of 0.0005 and a no more than 0.0002 error (95% confidence interval) with 80% power and 5% significance level.21 The agreement between resequencing and capillary sequencing was measured using intraclass kappa statistics implemented in SAS, Version 9.1 (SAS Institute, Cary, NC).
RESULTS
Assembly of the Jaundice Chip
A key feature of CustomSeq™ Arrays is that the target gene sequences do not have to be contiguous, allowing for an efficient method to sequence gene domains that are separated by long intervening DNA sequences that are not the focus of interrogation. This feature allowed us to select 26,725 bases encompassing nucleotide elements that represent the best candidates for mutations that alter the structure/function of the proteins and are likely to cause a clinical phenotype. Analysis of the entire target sequences did not find repetitive elements, internal duplications or highly homologous sequences, which could decrease reading accuracy due to potential cross-hybridization of amplicons with similar gene segments. Therefore, the dataset was submitted to Affymetrix, where 25-mer probes were designed to identify individual bases in the forward and reverse strands. The primers were tiled onto 25×20 μm features in a standard array format, and the array was assembled into a hybridization cartridge; the protocols for production of 25-mer probes, tiling, and assembling constitute proprietary information of Affymetrix.
Efficient amplification of target sequences
In order to develop a convenient assay that efficiently produces amplicons for all target sequences in a short period of time using a small volume of blood from patients, we designed 35 PCR primer pairs that produced single amplicons in reactions containing optimal amplification buffer and three cycling programs (Supplementary Table 1). The relationship between target sequences, amplicons, fragmentation, labeling, and hybridization with the resequencing chip are summarized in Supplementary Figure 2. Sufficient amounts of DNA for all amplicons were produced in the first run of PCR reactions in ~55% of the samples from subjects included in this report; the remainder required 1-3 additional PCR reactions to produce enough quantities of individual amplicons with low copy number in the first run. The low yield was not related to the same amplicons, but varied among the reactions. Altogether, amplicons were successfully produced at desired quantities within 48 hours.
Highly accurate readout by the resequencing chip
To determine whether hybridization of the chips with fragmented and labeled amplicons identified the target gene sequences, the reference nucleotide sequences for SERPINA1, JAG1, ATP8B1, ABCB11, and ABCB4 were entered in the GDAS software. Then, using the software to make automated base calling according to hybridization intensities, we found high sequence readout from the first chip, with a base call rate of 91.4%. Base call rate was defined as the fraction of individual bases that can be clearly detected and identified (or “called”) by the method.22 Because the GDAS call algorithm computes a background correction that requires multiple samples for an effective estimate, we hybridized amplicons from 15 additional subjects. The average call rate for all 18 samples was 93.5% (range: 90.3–95.7%); the percent of “no calls” was not influenced by the lengths of the amplicons (Supplementary Table 2). The “no call” positions were not sequence dependent (average GC percentage of 49.63%), did not reside near the ends of amplicons, and were dispersed throughout the target sequences of all five genes (Supplementary Figure 3). We were able to manually analyze “no call” regions and identify individual nucleotides; however, because the manual calls could not be uniformly validated by automated capillary sequencing, we decided not to use manual analysis and to accept the standard filter conditions, algorithm parameters, and reliability rules of GDAS.
To determine the accuracy of the automated nucleotide calls, we compared the chip readout with the readout produced by standard automated capillary sequencing (Figure 1). Capillary sequencing was performed for the first and last exons of all five genes and 1–2 other exons randomly chosen for each gene using the amplicons from 10 of the 18 subjects. In the comparison, we included all bases that were identified/called by both methods. Direct comparison of 40,216 bases identified by the chip and capillary sequencing revealed 4 discordant calls in the chip readout, representing an automated call accuracy of 99.99%. Notably, all 4 discordant calls were heterozygous calls, with one of the alleles containing a base that did not match the automated sequence call (Table 1). Both methods were in overall agreement, as supported by a kappa value of 0.7498 (95% CI: 0.644–0.856, P<0.0005). Lastly, the reproducibility by capillary sequencing of 52 single nucleotide mutations (minus the 4 discordant calls) present among the 40,216 bases translates into a positive predictive value of 92.31%.
Figure 1. Sequence readout by the resequencing “Jaundice Chip”.
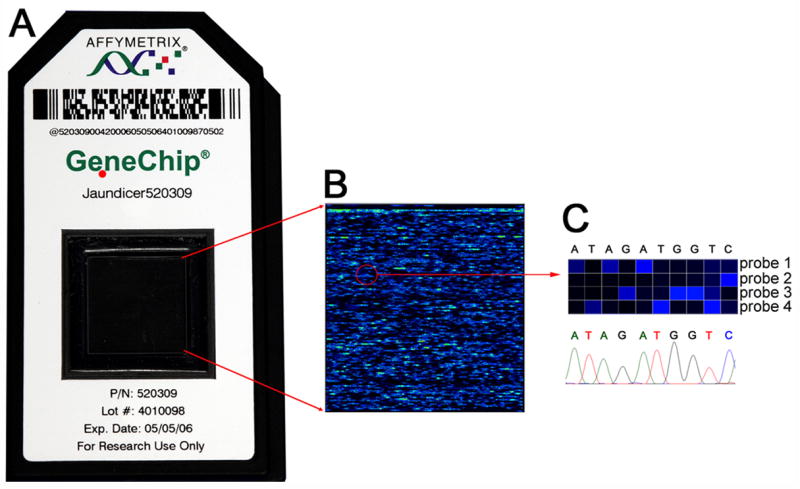
Panel A displays the hybridization cartridge assembled by Affymetrix, Inc., which contains a solid support featuring 25-mer probes designed to sequence 26,725 nucleotides (bases) of SERPINA1, JAG1, ATP8B1, ABCB11, and ABCB4. Panel B shows signals obtained from a high-resolution scanning after hybridization of a chip with amplicons from a subject. In the top portion of panel C, magnification of a region of the chip shows that the brightest signal for each set of four 25-mer probes for each column identifies a base. In the lower panel, identical base calls were made by standard capillary sequencing methodology.
Table 1.
Performance data for the Jaundice Chip with accuracy examined by direct comparison with automated capillary sequencing.
| Automated call rate | Mean: 93.5% |
| Range: 90.3–95.7% | |
| Number of bases called by both methods | 40,216 |
| Number of errors by the resequencing chip | 4* |
| Call accuracy | 99.99% |
All four errors were heterozygous calls
Detection of disease-causing mutations by the Jaundice Chip
To investigate if the chip identifies disease-causing mutations, we analyzed the sequence readout of subjects known to have intrahepatic cholestasis secondary to α1AT deficiency, the Alagille syndrome, or PFIC1-3 based on clinical, histological, and biochemical criteria (reviewed in reference #1). We also analyzed the readout from five subjects with biliary atresia, a non-inherited cholestatic disease due to a fibroinflammatory obstruction of extrahepatic bile ducts (as “disease controls”). Each subject with inherited forms of intrahepatic cholestasis was found to harbor at least one disease-causing mutation previously reported or newly identified mutations containing a new stop codon (Table 2 and Figure 2). Interestingly, one of the subjects with PFIC1 (subject #10) presented with neonatal cholestasis and has had progressive liver disease, with severe pruritus that was unresponsive to external biliary diversion. Analysis of the gene readout revealed a homozygous deletion of the 10 intronic bases continuing with the first 17 bases of exon 23 of ATP8B1. This finding was validated by capillary sequencing, which further showed that the extent of deletion included 569 bases 5′ of exon 23 (552 bases of intron 22 + 17 bases of exon 23). In subjects with cholestasis due to biliary atresia, non-synonymous polymorphisms were heterozygous and infrequent (Table 2).
Table 2.
Non-synonymous changes identified by the Jaundice Chip in subjects with inherited forms of intrahepatic cholestasis (subjects #1–13). Disease-causing mutations for subjects #1, 4, 8,11 and 13 were reproduced by standard capillary sequencing as shown in Figure 2. Also included are the changes found after the sequence analysis for all five genes in patients with biliary atresia (#14–18).
| Patient # | Diagnosis* | Mutation |
|---|---|---|
| 1 | α1AT deficiency | SERPINA1 T638C (Val213Ala, homozygous) and G1024A (Glu342Lys, homozygous)45, 46 |
| 2 | α1AT deficiency | SERPINA1T638C (Val213Ala, homozygous) and G1024A (Glu342Lys, homozygous)45, 46 |
| 3 | α1AT deficiency | SERPINA1 T638C (Val213Ala, homozygous) and G1024A (Glu342Lys, homozygous)45, 46 |
| 4 | Alagille syndrome | JAG1 C2230T (Arg744stop, heterozygous)47 |
| 5 | Alagille syndrome | JAG1 IVS19 +1 G to A, heterozygous 48 |
| 6 | Alagille syndrome | JAG1 C2650T (Glu884Stop, heterozygous)** |
| 7 | Alagille syndrome | JAG1 C2650T (Glu884Stop, heterozygous)** |
| 8 | PFIC1 | ATP8B1 C2788T (Arg930stop, heterozygous)28 |
| 9 | PFIC1 | ATP8B1 T1982C (Ile661Thr, heterozygous) 15 |
| 10 | PFIC1 | ATP8B1 569 bp deletion (including first 17 bp in exon 23, homozygous)** |
| 11 | PFIC2 | ABCB11 C3457T (Arg1153Cys, heterozygous)17 |
| 12 | PFIC2 | ABCB11 C2782T (Arg928Stop, heterozygous)** |
| 13 | PFIC3 | ABCB4 A1954G (Arg652Gly, homozygous)49 |
| 14 | Biliary atresia | ATP8B1 IVS 26 +8 G to T, heterozygous** |
| 15 | Biliary atresia | No non-synonymous polymorphism |
| 16 | Biliary atresia | No non-synonymous polymorphism |
| 17 | Biliary atresia | JAG1 C2612G (Pro871Arg, heterozygous) 50 |
| 18 | Biliary atresia | SERPINA1 G302A (Arg101His, heterozygous) 51 |
Figure 2. Comparison of nucleotide calls made by the resequencing chip and by standard capillary sequencing.
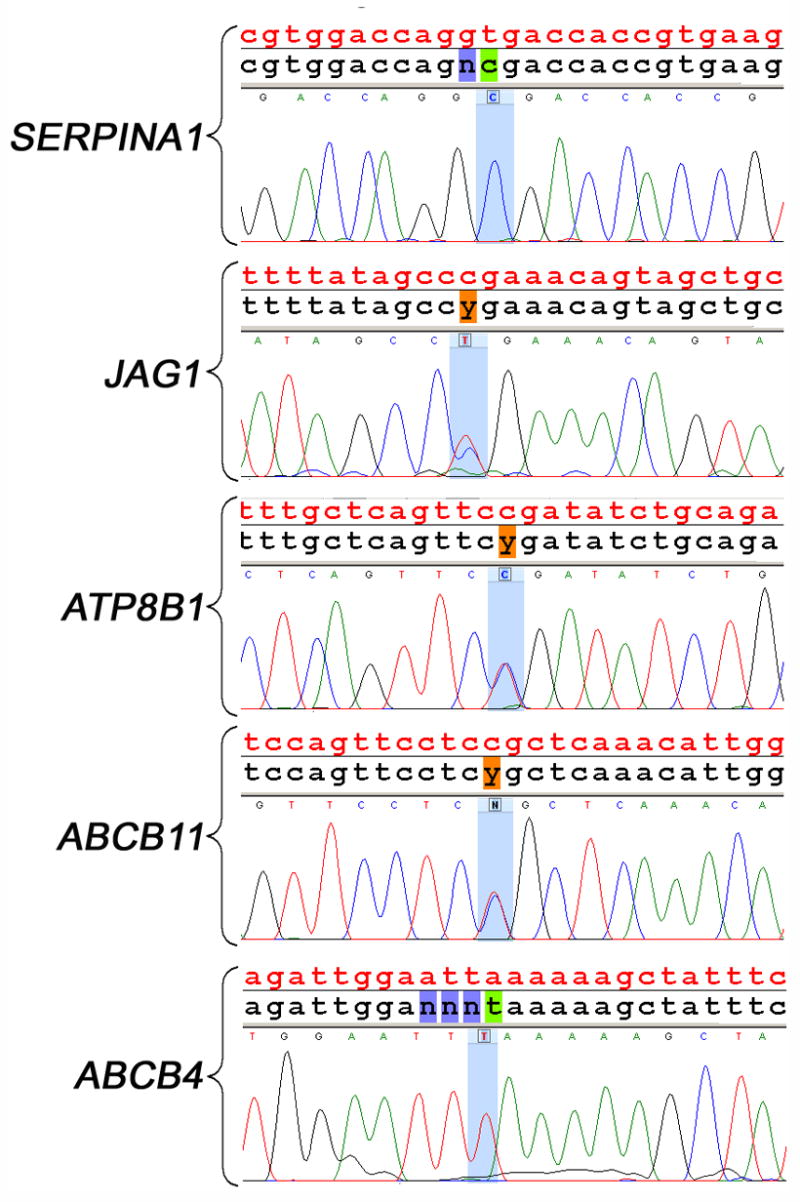
The top panel depicts the reference nucleotide sequence for SERPINA1 in red, the chip readout in black, and nucleotides identified by capillary sequencing. The T>C homozygous mutation identified by the chip (shaded in green) is reproduced by capillary sequencing. The same sequencing organization is presented for the other genes in the four lower panels. In JAG1, ATP8B1, and ABCB11, the chip produced heterozygous C/T calls (shown as a typical “y” symbol shaded in orange) that could be identified by red and blue peaks produced by capillary sequencing. In the lower panel, a homozygous A>T mutation in ABCB4 was identified by both methods. The mutations presented in this figure reflect the mutations described in patients #1,4,8,11 and 13 of Table 2.
Finally, to examine the ability of the chip to detect a broader spectrum of genetic mutations reported for the clinical phenotypes, we developed a dataset of mutations for all 5 genes based on data from the published literature (www.ncbi.nlm.nih.gov/entrez/, December 2005) and from the Human Gene Mutation Database (www.hgmd.cf.ac.uk/ac/index.php). Collectively, the most common types of mutations were base changes and comprised a dataset of 360 positions reported as missense/nonsense mutations (N=200), single nucleotide polymorphisms (N=119), and mutations in splice sites (N=41). Among these positions, only 11 (or 3.06%) resided in regions of the chip that consistently generated failed signals (or “no calls;” Table 3). Thus, the nucleotide sequences identified by the Jaundice Chip efficiently screened for >95% of the disease-causing mutations due to single base changes reported to date. A second group of disease-causing mutations consisted of insertions, deletions or duplications, which may be as high as 55% in the JAG1 gene in patients with the Alagille disease,23 or cause a more severe phenotype as reported for ATP8B1 (as illustrated by patient #10 of Table 2).24 Although most of these insertions, deletions or duplications fall within the reading ability of the chip (Supplementary Table 3), our current dataset does not allow a precise analysis of how reliable the chip is in detecting these types of mutation.
Table 3.
Relationship between the number and type of mutations reported for each gene and the number of regions that consistently failed to generate signals (“no calls”).
| Type of mutation | Number of mutations | Number of mutations in “no call” regions |
|---|---|---|
| Single nucleotide changes | ||
| Missense/nonsense | 200 | 6 |
| Single nucleotide polymorphism | 119 | 4 |
| Splicing | 41 | 1 |
| Total number of mutations | 360 | 11* |
| Insertion and deletion** | ||
| Insertion | 50 | 1 (?) |
| Insertion/deletion | 5 | 0 |
| Small deletion | 86 | 4 (?) |
| Large deletion (>20 nucleotides) | 6 | 0 |
| Duplication | 2 | 0 |
6 mutations in SERPINA1, 4 mutations in JAG1, and 1 mutation in ABCB11
The resequencing technology may not reliably identify insertion, deletion, or duplication. Although most of these mutations reside in regions of the gene fully sequenced by the chip, the ability to identify these mutations requires further validation.
DISCUSSION
We used the resequencing technology to develop a chip that reads the nucleotide signature for five genes known to cause the most common inherited syndromes of intrahepatic cholestasis: SERPINA1, JAG1, ATP8B1, ABCB11, and ABCB4. The chip readout is highly accurate and has an error rate estimated at 0.001 due primarily to heterozygous base calls when compared to automated capillary sequencing. The readout encompasses all coding sequences, intron-exon boundaries, and >200 bases upstream of the first exon, thus enabling the detection of nucleotide changes that may alter structure/function and/or level of expression of encoded proteins. Detection of individual bases for all five genes occurs simultaneously during a single hybridization assay and without manual editing; automated identification has an average call rate of 93.5%. In addition to the accurate sequence readout with minimal need for data curation, the amplification of all target sequences from as little as 4 μg of whole blood DNA in PCR reactions run in three cycling programs simultaneously form a robust system that yields sequence for mutational analysis in 3–4 days. These features will greatly facilitate the use of the chip as a research tool to study the role of individual or combined genes in determination of clinical phenotypes, and as a potential diagnostic tool for patients with inherited syndromes of intrahepatic cholestasis.
The high-throughput features of resequencing microarrays offer the potential to meet investigative and clinical needs to identify nucleotide variations along different gene segments rapidly and efficiently. This approach is particularly relevant to genomic regions that are not contiguous and/or have high rates of nucleotide polymorphisms. The Jaundice Chip reported herein can screen for >95% of disease-causing mutations due to single nucleotide changes and appears to also detect short homozygous deletions. While we expect that the chip will identify other homozygous deletions of short and long DNA segments, it is possible that heterozygous deletions (regardless of the length) or insertions may not be detected by the chip because the strand with normal sequences will produce a normal readout. If this occurs, complementary technologies such as single strand conformational polymorphism, conformation sensitive gel electrophoresis and capillary sequencing of selected regions may be required for the most comprehensive detection of all types of mutations. More accurate evaluation of these scenarios will require formal bench-testing of the chip against DNA from subjects known to carry insertions, deletions or duplications. This notwithstanding, the Jaundice Chip allows for the accurate screening for the vast majority of reported mutations due to single nucleotide changes accurately and in a predictable fashion.
The ability to rapidly survey for genetic mutations in children and adults with inherited syndromes of intrahepatic cholestasis generates unique opportunities to investigate the molecular basis of the clinical heterogeneity observed in these patients. For example, although mutations in ATP8B1 and ABCB11 have been reported for subjects with severe forms of PFIC with low serum levels of gamma-glutamyltranspeptide (γGTP), specific mutations within each gene have also been associated with milder clinical phenotypes.15, 17, 24–30 Equally notable is the phenotypic pleomorphism of mutations in ABCB4, which ranges from high γGTP-PFIC (or PFIC3), to intrahepatic cholestasis of pregnancy, and gallstone formation,19, 25, 31–36 and an array of mutations in JAG1 in subjects with liver and/or non-hepatic malformations (e.g.: cardiovascular and renal defects).4, 7, 8, 23, 37–39 In view of such variability in clinical presentation, the ability to analyze the nucleotide sequence facilitates studies of phenotype-genotype relationship for individual genes. In addition, we chose to add SERPINA1 to the chip as an investigative tool for studies examining how α1AT polymorphisms may serve as disease modifiers in subjects harboring mutations in one of the other cholestatic genes, or in subjects with other forms of liver disease in a fashion described for patients with chronic pulmonary diseases.40–42
In summary, we customized a state-of-the-art resequencing technology to read a comprehensive nucleotide sequence of the five genes responsible for the most common forms of inherited cholestasis. The combined large size of these genes and the lack of hot spots for disease-causing mutations make it time-consuming and costly to use genetic testing by standard sequencing methodologies in the evaluation of patients with cholestasis. These limitations may be overcome by the resequencing tool here reported, and named Jaundice Chip. The immediate patient population that may benefit from the chip consists of children with chronic cholestasis, in whom current diagnostic algorithms are aimed at excluding known diseases and frequently include: 1) extensive biochemical evaluation of blood and urine, 2) radiological studies of the liver and extra-hepatic systems, and 3) liver biopsy for histological examination and electron microscopy. An additional application of the chip is as an investigative tool to study the role of synonymous and non-synonymous polymorphisms of individual genes, or of heterozygous polymorphisms in functionally related genes, in phenotype determination of children and adults with chronic cholestasis, intrahepatic cholestasis of pregnancy, and gallstone formation associated with mutations in ABCB4.43, 44 One specific disease that may benefit from such investigative approach is biliary atresia, in which the systematic analysis of all five genes may provide insight into the potential role of genetic variations in long-term survival with the native liver. For all of these settings, the current version of the Jaundice Chip offers a real opportunity to translate laboratory discoveries into a tool that may simplify the diagnostic algorithm at the bedside, and facilitate the design of treatment protocols that take into account the genetic makeup of the patient.
Supplementary Material
Supplementary Figure 1 Panels A–E shows the relationship between amplicon lengths and regions of the target gene (table on the left side) and the appearance of individual amplicons as resolved in 1% agarose gel (picture on the right; MW = molecular weight marker).
Supplementary Figure 2 Relationship between exons/introns, target gene sequences and amplicons in the Jaundice Chip using SERPINA1 as a prototype. The resequencing technology reads out the nucleotide sequence for the SERPINA1 gene (red vertical bars representing exons; red interrupted line representing introns). Using a reference nucleotide sequence, a database is produced containing 300 nucleotides upstream from exon 1, all exons, and 22 nucleotides forming intron/exon boundaries. Collectively, they form non-contiguous “target sequences” (shown in green). Based on the target sequences, 25-mer probes are produced, tiled onto a solid support and inserted into a hybridization cartridge (bottom of the panel and depicted in Figure 1). To produce DNA templates from a patient to hybridize with the chip, short/long range PCR primers, buffer, and thermocycling protocols are designed to amplify a fragment of DNA containing the target sequences (“Amplicons,” shown in blue). Then, amplicons undergo labeling and fragmentation, followed by hybridization with the chip. Scanning of the chip allows for the identification of individual nucleotides based on the high hybridization signal produced by 25-mer probes that fully match complementary sequences.
Supplementary Figure 3 Distribution of consistent “no call” regions along the target sequences read by the Jaundice Chip in the SERPINA1 gene. The “no calls” are depicted by black vertical bars. Target sequences are defined as bases upstream from the first exon, intronic sequence bordering exons, and exons.
Acknowledgments
Grant support: This work was supported by the NIH grant DK075162 and by the Bioinformatics and Microarray Cores of the Digestive Disease Research Development Center (DK032512). Dr. Miethke is recipient of a Fellowship Award from the NIH-funded Rare Liver Disease Network (DK 078377). The authors acknowledge the support of Susan Krug and staff of the Cincinnati Children’s Hospital Pediatric Liver Care Center with patient recruitment, and the critical review of the manuscript by Dr. William Balistreri and Dr. Ursula Matte.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Balistreri WF, Bezerra JA. Whatever happened to "neonatal hepatitis"? Clin Liver Dis. 2006;10:27–53. v. doi: 10.1016/j.cld.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Sharp HL, Bridges RA, Krivit W, Freier EF. Cirrhosis associated with alpha-1-antitrypsin deficiency: a previously unrecognized inherited disorder. J Lab Clin Med. 1969;73:934–9. [PubMed] [Google Scholar]
- 3.Odievre M, Martin JP, Hadchouel M, Alagille D. Alpha1-antitrypsin deficiency and liver disease in children: phenotypes, manifestations, and prognosis. Pediatrics. 1976;57:226–31. [PubMed] [Google Scholar]
- 4.Balistreri WF, Bezerra JA, Jansen P, Karpen SJ, Shneider BL, Suchy FJ. Intrahepatic cholestasis: summary of an American Association for the Study of Liver Diseases single-topic conference. Hepatology. 2005;42:222–35. doi: 10.1002/hep.20729. [DOI] [PubMed] [Google Scholar]
- 5.Alagille D, Estrada A, Hadchouel M, Gautier M, Odievre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr. 1987;110:195–200. doi: 10.1016/s0022-3476(87)80153-1. [DOI] [PubMed] [Google Scholar]
- 6.Alagille D, Odievre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J Pediatr. 1975;86:63–71. doi: 10.1016/s0022-3476(75)80706-2. [DOI] [PubMed] [Google Scholar]
- 7.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–51. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 8.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–42. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 9.Henriksen NT, Drablos PA, Aagenaes O. Cholestatic jaundice in infancy. The importance of familial and genetic factors in aetiology and prognosis. Arch Dis Child. 1981;56:622–7. doi: 10.1136/adc.56.8.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whitington PF, Freese DK, Alonso EM, Schwarzenberg SJ, Sharp HL. Clinical and biochemical findings in progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 1994;18:134–41. doi: 10.1097/00005176-199402000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Alonso EM, Snover DC, Montag A, Freese DK, Whitington PF. Histologic pathology of the liver in progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 1994;18:128–33. doi: 10.1097/00005176-199402000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Clayton RJ, Iber FL, Ruebner BH, McKusick VA. Byler disease. Fatal familial intrahepatic cholestasis in an Amish kindred. Am J Dis Child. 1969;117:112–24. [PubMed] [Google Scholar]
- 13.Jacquemin E, Setchell KD, O’Connell NC, Estrada A, Maggiore G, Schmitz J, Hadchouel M, Bernard O. A new cause of progressive intrahepatic cholestasis: 3 beta-hydroxy-C27- steroid dehydrogenase/isomerase deficiency. J Pediatr. 1994;125:379–84. doi: 10.1016/s0022-3476(05)83280-9. [DOI] [PubMed] [Google Scholar]
- 14.Balistreri WF. Inborn errors of bile acid biosynthesis and transport. Novel forms of metabolic liver disease. Gastroenterol Clin North Am. 1999;28:145–72. vii. doi: 10.1016/s0889-8553(05)70048-0. [DOI] [PubMed] [Google Scholar]
- 15.Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, Klomp LW, Lomri N, Berger R, Scharschmidt BF, Knisely AS, Houwen RH, Freimer NB. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–24. doi: 10.1038/ng0398-219. [DOI] [PubMed] [Google Scholar]
- 16.Bull LN, Carlton VE, Stricker NL, Baharloo S, DeYoung JA, Freimer NB, Magid MS, Kahn E, Markowitz J, DiCarlo FJ, McLoughlin L, Boyle JT, Dahms BB, Faught PR, Fitzgerald JF, Piccoli DA, Witzleben CL, O’Connell NC, Setchell KD, Agostini RM, Jr, Kocoshis SA, Reyes J, Knisely AS. Genetic and morphological findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC-1] and Byler syndrome): evidence for heterogeneity. Hepatology. 1997;26:155–64. doi: 10.1002/hep.510260121. [DOI] [PubMed] [Google Scholar]
- 17.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, Dahan K, Childs S, Ling V, Tanner MS, Kagalwalla AF, Nemeth A, Pawlowska J, Baker A, Mieli-Vergani G, Freimer NB, Gardiner RM, Thompson RJ. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–8. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 18.Deleuze JF, Jacquemin E, Dubuisson C, Cresteil D, Dumont M, Erlinger S, Bernard O, Hadchouel M. Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology. 1996;23:904–8. doi: 10.1002/hep.510230435. [DOI] [PubMed] [Google Scholar]
- 19.de Vree JM, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, Deleuze JF, Desrochers M, Burdelski M, Bernard O, Oude Elferink RP, Hadchouel M. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A. 1998;95:282–7. doi: 10.1073/pnas.95.1.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jegga AG, Sherwood SP, Carman JW, Pinski AT, Phillips JL, Pestian JP, Aronow BJ. Detection and visualization of compositionally similar cis-regulatory element clusters in orthologous and coordinately controlled genes. Genome Res. 2002;12:1408–17. doi: 10.1101/gr.255002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rubinson L, Neutons JJ. Research Techniques for the Health Sciences. MacMillan; 1987. [Google Scholar]
- 22.Affymetrix. Technical note: GeneChip CustomSeq Resequencing Array: Performance data for base calling algorithm in GeneChip DNA Analysis Software (GDAS) 2003 www.affymetrix.com.
- 23.Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in alagille syndrome. Hum Mutat. 2001;17:18–33. doi: 10.1002/1098-1004(2001)17:1<18::AID-HUMU3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 24.Klomp LWJ, Bull LN, Knisely AS, Doelen MAMvd, Juijn JA, Berger R, Forget S, Nielsen I-M, Eiberg H, Houwen RHJ. A missense mutation in FIC1 is associated with Greenland familial cholestasis. Hepatology. 2000;32:1337–1341. doi: 10.1053/jhep.2000.20520. [DOI] [PubMed] [Google Scholar]
- 25.Chen HL, Chang PS, Hsu HC, Ni YH, Hsu HY, Lee JH, Jeng YM, Shau WY, Chang MH. FIC1 and BSEP defects in Taiwanese patients with chronic intrahepatic cholestasis with low gamma-glutamyltranspeptidase levels. J Pediatr. 2002;140:119–24. doi: 10.1067/mpd.2002.119993. [DOI] [PubMed] [Google Scholar]
- 26.Egawa H, Yorifuji T, Sumazaki R, Kimura A, Hasegawa M, Tanaka K. Intractable diarrhea after liver transplantation for Byler’s disease: successful treatment with bile adsorptive resin. Liver Transpl. 2002;8:714–6. doi: 10.1053/jlts.2002.34384. [DOI] [PubMed] [Google Scholar]
- 27.Lykavieris P, van Mil S, Cresteil D, Fabre M, Hadchouel M, Klomp L, Bernard O, Jacquemin E. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: no catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol. 2003;39:447–52. doi: 10.1016/s0168-8278(03)00286-1. [DOI] [PubMed] [Google Scholar]
- 28.Klomp LW, Vargas JC, van Mil SW, Pawlikowska L, Strautnieks SS, van Eijk MJ, Juijn JA, Pabon-Pena C, Smith LB, DeYoung JA, Byrne JA, Gombert J, van der Brugge G, Berger R, Jankowska I, Pawlowska J, Villa E, Knisely AS, Thompson RJ, Freimer NB, Houwen RH, Bull LN. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004;40:27–38. doi: 10.1002/hep.20285. [DOI] [PubMed] [Google Scholar]
- 29.Wang R, Salem M, Yousef IM, et al. Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci USA. 2001;98:2011–2016. doi: 10.1073/pnas.031465498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Mil SW, van der Woerd WL, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11 . Gastroenterology. 2004;127:379–384. doi: 10.1053/j.gastro.2004.04.065. [DOI] [PubMed] [Google Scholar]
- 31.Jacquemin E. Role of multidrug resistance 3 deficiency in pediatric and adult liver disease: one gene for three diseases. Semin Liver Dis. 2001;21:551–62. doi: 10.1055/s-2001-19033. [DOI] [PubMed] [Google Scholar]
- 32.Jacquemin E, De Vree JM, Cresteil D, Sokal EM, Sturm E, Dumont M, Scheffer GL, Paul M, Burdelski M, Bosma PJ, Bernard O, Hadchouel M, Elferink RP. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001;120:1448–58. doi: 10.1053/gast.2001.23984. [DOI] [PubMed] [Google Scholar]
- 33.Jacquemin E, Cresteil D, Manouvrier S, Boute O, Hadchouel M. Heterozygous non-sense mutation of the MDR3 gene in familial intrahepatic cholestasis of pregnancy. Lancet. 1999;353:210–1. doi: 10.1016/S0140-6736(05)77221-4. [DOI] [PubMed] [Google Scholar]
- 34.Dixon PH, Weerasekera N, Linton KJ, Donaldson O, Chambers J, Egginton E, Weaver J, Nelson-Piercy C, de Swiet M, Warnes G, Elias E, Higgins CF, Johnston DG, McCarthy MI, Williamson C. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genet. 2000;9:1209–17. doi: 10.1093/hmg/9.8.1209. [DOI] [PubMed] [Google Scholar]
- 35.Lucena JF, Herrero JI, Quiroga J, et al. A multidrug resistance 3 gene mutation causing cholelithiasis, cholestasis of pregnancy, and adulthood biliary cirrhosis. Gastroenterology. 2003;124:1037–1042. doi: 10.1053/gast.2003.50144. [DOI] [PubMed] [Google Scholar]
- 36.Pauli-Magnus C, Lang T, Meier Y, Zodan-Marin T, Jung D, Breymann C, Zimmermann R, Kenngott S, Beuers U, Reichel C, Kerb R, Penger A, Meier PJ, Kullak-Ublick GA. Sequence analysis of bile salt export pump (ABCB11) and multidrug resistance p-glycoprotein 3 (ABCB4, MDR3) in patients with intrahepatic cholestasis of pregnancy. Pharmacogenetics. 2004;14:91–102. doi: 10.1097/00008571-200402000-00003. [DOI] [PubMed] [Google Scholar]
- 37.Warthen DM, Moore EC, Kamath BM, Morrissette JJ, Sanchez P, Piccoli DA, Krantz ID, Spinner NB. Jagged1 (JAG1) mutations in Alagille syndrome: increasing the mutation detection rate. Hum Mutat. 2006;27:436–43. doi: 10.1002/humu.20310. [DOI] [PubMed] [Google Scholar]
- 38.Yuan ZR, Kohsaka T, Ikegaya T, Suzuki T, Okano S, Abe J, Kobayashi N, Yamada M. Mutational analysis of the Jagged 1 gene in Alagille syndrome families. Hum Mol Genet. 1998;7:1363–9. doi: 10.1093/hmg/7.9.1363. [DOI] [PubMed] [Google Scholar]
- 39.Eldadah ZA, Hamosh A, Biery NJ, Montgomery RA, Duke M, Elkins R, Dietz HC. Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Hum Mol Genet. 2001;10:163–9. doi: 10.1093/hmg/10.2.163. [DOI] [PubMed] [Google Scholar]
- 40.Lafuente MJ, Casterad X, Laso N, Mas S, Panades R, Calleja A, Hernandez S, Turuguet D, Ballesta A, Ascaso C, Lafuente A. Pi*S and Pi*Z alpha 1 antitrypsin polymorphism and the risk for asbestosis in occupational exposure to asbestos. Toxicol Lett. 2002;136:9–17. doi: 10.1016/s0378-4274(02)00283-7. [DOI] [PubMed] [Google Scholar]
- 41.Henry MT, Cave S, Rendall J, O’Connor CM, Morgan K, FitzGerald MX, Kalsheker N. An alpha1-antitrypsin enhancer polymorphism is a genetic modifier of pulmonary outcome in cystic fibrosis. Eur J Hum Genet. 2001;9:273–8. doi: 10.1038/sj.ejhg.5200623. [DOI] [PubMed] [Google Scholar]
- 42.Salvatore F, Scudiero O, Castaldo G. Genotype-phenotype correlation in cystic fibrosis: the role of modifier genes. Am J Med Genet. 2002;111:88–95. doi: 10.1002/ajmg.10461. [DOI] [PubMed] [Google Scholar]
- 43.Rosmorduc O, Hermelin B, Poupon R. MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology. 2001;120:1459–67. doi: 10.1053/gast.2001.23947. [DOI] [PubMed] [Google Scholar]
- 44.Rosmorduc O, Hermelin B, Boelle PY, Parc R, Taboury J, Poupon R. ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology. 2003;125:452–9. doi: 10.1016/s0016-5085(03)00898-9. [DOI] [PubMed] [Google Scholar]
- 45.Nukiwa T, Satoh K, Brantly ML, Ogushi F, Fells GA, Courtney M, Crystal RG. Identification of a second mutation in the protein-coding sequence of the Z type alpha 1-antitrypsin gene. J Biol Chem. 1986;261:15989–94. [PubMed] [Google Scholar]
- 46.Kidd VJ, Wallace RB, Itakura K, Woo SL. Alpha 1-antitrypsin deficiency detection by direct analysis of the mutation in the gene. Nature. 1983;304:230–4. doi: 10.1038/304230a0. [DOI] [PubMed] [Google Scholar]
- 47.Krantz ID, Colliton RP, Genin A, Rand EB, Li L, Piccoli DA, Spinner NB. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet. 1998;62:1361–9. doi: 10.1086/301875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ropke A, Kujat A, Graber M, Giannakudis J, Hansmann I. Identification of 36 novel Jagged1 (JAG1) mutations in patients with Alagille syndrome. Hum Mutat. 2003;21:100. doi: 10.1002/humu.9102. [DOI] [PubMed] [Google Scholar]
- 49.Kano M, Shoda J, Sumazaki R, Oda K, Nimura Y, Tanaka N. Mutations identified in the human multidrug resistance P-glycoprotein 3 (ABCB4) gene in patients with primary hepatolithiasis. Hepatol Res. 2004;29:160–166. doi: 10.1016/j.hepres.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 50.Kohsaka T, Yuan ZR, Guo SX, Tagawa M, Nakamura A, Nakano M, Kawasasaki H, Inomata Y, Tanaka K, Miyauchi J. The significance of human jagged 1 mutations detected in severe cases of extrahepatic biliary atresia. Hepatology. 2002;36:904–12. doi: 10.1053/jhep.2002.35820. [DOI] [PubMed] [Google Scholar]
- 51.Faber JP, Weidinger S, Olek K. Sequence data of the rare deficient alpha 1-antitrypsin variant PI Zaugsburg. Am J Hum Genet. 1990;46:1158–62. [PMC free article] [PubMed] [Google Scholar]
- 52.Balistreri WF, Grand R, Hoofnagle JH, Suchy FJ, Ryckman FC, Perlmutter DH, Sokol RJ. Biliary atresia: current concepts and research directions. Summary of a symposium Hepatology. 1996;23:1682–92. doi: 10.1002/hep.510230652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Panels A–E shows the relationship between amplicon lengths and regions of the target gene (table on the left side) and the appearance of individual amplicons as resolved in 1% agarose gel (picture on the right; MW = molecular weight marker).
Supplementary Figure 2 Relationship between exons/introns, target gene sequences and amplicons in the Jaundice Chip using SERPINA1 as a prototype. The resequencing technology reads out the nucleotide sequence for the SERPINA1 gene (red vertical bars representing exons; red interrupted line representing introns). Using a reference nucleotide sequence, a database is produced containing 300 nucleotides upstream from exon 1, all exons, and 22 nucleotides forming intron/exon boundaries. Collectively, they form non-contiguous “target sequences” (shown in green). Based on the target sequences, 25-mer probes are produced, tiled onto a solid support and inserted into a hybridization cartridge (bottom of the panel and depicted in Figure 1). To produce DNA templates from a patient to hybridize with the chip, short/long range PCR primers, buffer, and thermocycling protocols are designed to amplify a fragment of DNA containing the target sequences (“Amplicons,” shown in blue). Then, amplicons undergo labeling and fragmentation, followed by hybridization with the chip. Scanning of the chip allows for the identification of individual nucleotides based on the high hybridization signal produced by 25-mer probes that fully match complementary sequences.
Supplementary Figure 3 Distribution of consistent “no call” regions along the target sequences read by the Jaundice Chip in the SERPINA1 gene. The “no calls” are depicted by black vertical bars. Target sequences are defined as bases upstream from the first exon, intronic sequence bordering exons, and exons.