

Note: Dr. Steinman received the prestigious 2007 Albert Lasker Award for Basic Medical Research for the discovery of dendritic cells. The award committee called him “a scientific sleuth, who without cloak and dagger, uncovered the intelligence-gathering component of our immune system—a monumental achievement in the 200-year history of immunology.” Dr. Steinman came to Baylor on September 20, 2007, as part of the celebration of the tenth anniversary of the Baylor Institute for Immunology Research (Figure 1). We were honored to welcome him and appreciate his willingness to publish his well-received presentation in Proceedings.
Figure 1.

Ralph M. Steinman, MD, with Jacques Banchereau, PhD, director of the Baylor Institute for Immunology Research.
—Michael a. e. Ramsay, MD, associate editor and president of Baylor Research Institute
Immunology plays an important role in many clinical conditions. In some instances—such as infectious diseases and cancer—the goal is to improve the immune response to the pathogen or challenge; in other instances—such as transplantation, autoimmunity, and allergy—the goal is to reduce the specific immune response. Already, immunology is providing valuable therapies, mainly in the form of monoclonal antibodies. Anti–tumor necrosis factor (TNF) is being used to treat arthritis and other inflammatory diseases, and anti-CD20 is being used to treat B cell lymphoma. However, the T cell limb of the immune response also needs to be harnessed in a disease- or antigen-specific manner in medicine.
This opportunity in immunology and medicine is significant because of the way T cells are formed. The immune system is blessed with a superb repertoire of clones (Figure 2). Each clone has a specific receptor for antigen, and within that repertoire are clones specific for microbes, tumors, self, the environment, and so forth—many of the relevant antigens to disease. Thus, the repertoire provides the potential for antigen-specific therapies and vaccines, which will mount a broad-based but specific response to the relevant pathogen, in contrast to cyclosporine, for example, which is a general immunosuppressant.
Figure 2.
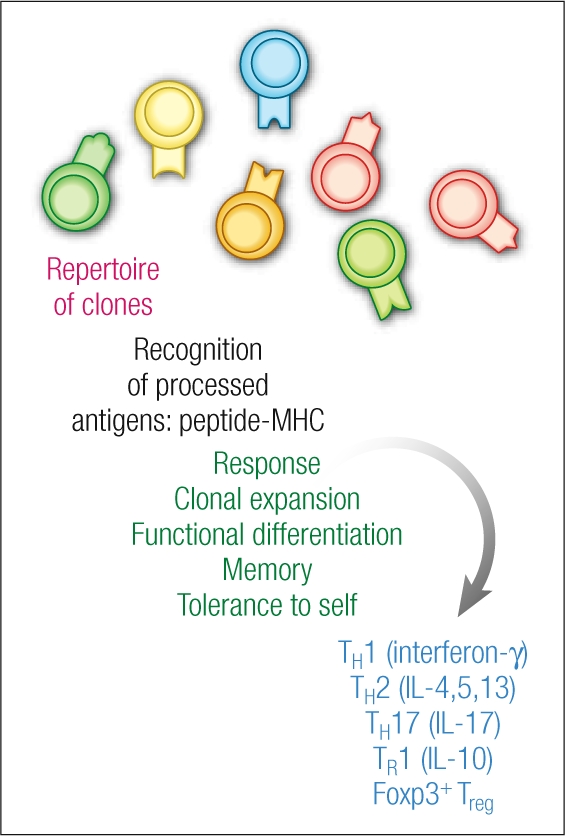
Three elemental R's of T cell biology: repertoire, recognition, response. Reprinted with permission from Nature Medicine (1).
One of the main goals in human immunology research is to establish the principles to manipulate these clones in a patient, so that they will expand and carry out the function that is appropriate to a successful therapy. At the Rockefeller University and the Baylor Institute for Immunology Research, one of the main approaches is to try to harness the biology of dendritic cells, which use their tree-like processes or dendrites to constantly probe their environment (Figure 3).
Figure 3.
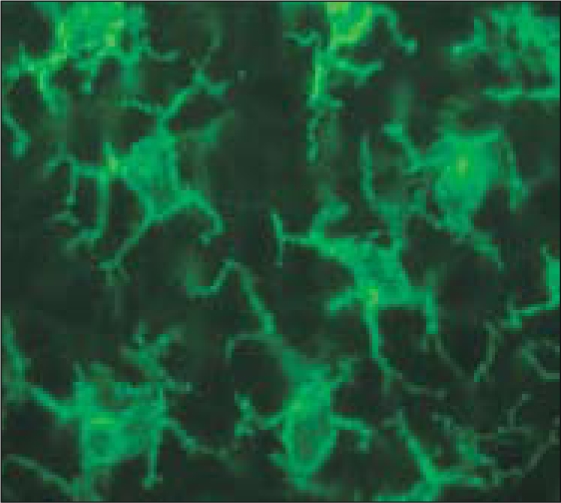
Dendritic cells probing the environment in mouse lymphoid tissue. Photomicrograph courtesy of Dr. Juliana Idoyaga; reprinted with permission from Nature Medicine (1).
FEATURES OF DENDRITIC CELLS
Dendritic cells have several innate features that make them logical targets in dissecting disease and developing new therapies. While dendritic cells interact with all classes of lymphocytes, the information on their interaction with T cells is more developed.
Dendritic cells are found on body surfaces, such as the airway and the gut. They can capture antigens that enter the body, and then they move to the T cell areas of lymphoid organs to find the right clones and start the immune response. Dendritic cells have a battery of uptake receptors to capture antigens in a specific and efficient way, by absorptive endocytosis. After the capture, they process and present these antigens primarily as peptide–major histocompatibility complex (MHC) complexes. These two features of localization and presentation allow dendritic cells to harness the repertoire of T cell clones in a specific way.
In addition, dendritic cells are very sensitive to environmental stimuli—not only microbial stimuli but also a number of endogenous stimuli, called alarmins. In response to these stimuli, they undergo differentiation, which is called maturation because it is a terminal event in dendritic cell development. Finally, dendritic cells have many forms, called subsets, each with distinct receptors for antigen uptake and response to various stimuli. The features of maturation and subsets allow dendritic cells to dictate the type of response that the repertoire will carry out.
APPROACHES FOR THE HARNESSING OF DENDRITIC CELLS
In an effort to employ dendritic cells to control the immune system, two approaches have been used, especially in the context of cancer treatment: 1) generating dendritic cells ex vivo, loading them with antigens, and then reinfusing them back into the patient; and 2) delivering antigens to dendritic cells directly in vivo.
The ex vivo approach
Niki Romani and Garold Schuler began working on the ex vivo dendritic cell preparation pathway in the laboratory more than 10 years ago, and this method was also developed by Jacques Banchereau and Karolina Palucka and is used at the Baylor Institute for Immunology Research.
The monocyte-derived dendritic cells that are most frequently studied are highly enriched. Researchers are loading the cells with multiple tumor antigens—dying tumor cells, RNA from tumor cells, collections of defined antigens—with the goal of instigating a very broad immune response to cancer, making it difficult for the tumor to escape by further mutation. The ex vivo method also allows control of the maturation process and the type of subset to be given to the patient.
The antigen-loaded dendritic cell preparations have been shown to be safe in a number of different institutions, and patients have responded with an immune response. While some progress has been made and the method has great potential, it also faces some obstacles. In the experiments that have been done, relatively few cancer antigens have been presented by the dendritic cells. Another problem is that the injected dendritic cells have migrated poorly to the T cell areas to initiate selection from the repertoire. Only a percentage of the dendritic cells that are injected make it to the lymphoid organ, which is a severe limitation. For the most part, this therapy has been used in the setting of advanced metastatic disease. No studies to date have proven that dendritic cell therapy can improve survival, and regressions of metastatic lesions are seen infrequently. Finally, there has been very little coordination and relatively little support for this research at this stage of its development. Well-designed studies are greatly needed.
In vivo approach
Another approach being used in the cancer setting is to try to coax dendritic cells in the patient to capture and present the patient's own tumor cells. Groups around the world are refining this effort so that the dendritic cells are more likely to induce an appropriate immune response.
At Rockefeller University, we have found that the intravenous route is much more efficient than the subcutaneous route in the injection of irradiated tumor cells, at least in mice. In addition, the issue of maturation must be addressed. The dendritic cell in vivo is specialized to bring about tolerance; therefore, one has to provide it with additional information to mature and start an immune response. We have been using an innate cell, called a natural killer T (NKT) cell, to mature the dendritic cell. I believe the preclinical evidence is sufficient in the mouse system to apply this method for clinical hematologic malignancies.
Recent work by Laurence Zitvogel's group in France has shown that some chemotherapies seem to make tumor cells more immunogenic following cell death and also favor uptake of the killed tumor cells by dendritic cells (2). My feeling is that this strategy should be used after a patient has been immunized, i.e., after the immune T cells are in place and can be boosted to kill the tumor cells and test what develops in terms of improved resistance to cancer.
A third effort, now being tested in a phase III study, is GVAX. Glenn Dranoff pioneered this method, which involves transducing a tumor cell with granulocyte-macrophage colony-stimulating factor (GM-CSF) through retroviruses or adenoviruses. When these tumor cells are then irradiated and injected into the skin, they recruit dendritic cells, which pick up the dying GM-CSF–secreting tumor cells and elicit a strong and reliable immune response. Biopsied metastatic lesions have shown an influx of T cells into the tumors following immunizations.
Finally, antitumor antibodies are a popular form of immunotherapy. In vitro, these antibodies have been shown to increase the uptake of tumors into dendritic cells and drive their maturation through the Fc receptor on the dendritic cells. Patients responding to this therapy appear to have an immune response to the antitumor antibody–coded dendritic cell, which is mediating an immune response.
VACCINES: MICROBIOLOGICAL AND IMMUNOLOGICAL APPROACHES
At Rockefeller, our laboratory is focused on vaccines against AIDS and malaria, using the potent antigens present in microbes to develop a strategy to enhance dendritic cell function in patients. Vaccines have been a success story in medicine. We still need vaccines, however, for prevalent infectious diseases, as well as the immune-related conditions mentioned earlier, such as cancer, allergy, and autoimmune diseases. The goal with vaccines is to elicit a long-lived immune response that either enhances immunity or turns it off.
Until recently, the approach to vaccine development has been microbiological. The microbiologists cleverly identified the agent that caused the infectious disease and learned how to attenuate that organism so that when it was administered as a vaccine, a naive individual would gain protective immunity. But what is interesting about this approach, which has been used for over 100 years, is that we still haven't worked out the principles whereby these attenuated microbes elicit this valuable protective immunity. That scenario has set the stage for immunologic approaches to vaccinations, in which the focus is on the identification of the protective antigens, the adjuvants needed to elicit protective immunity, and the types of B cells, T cells, and other immune responses needed to provide protection. In this way, vaccine biology is moving from a more or less empirical approach to a highly immunologic one, in which we try to delineate a number of fascinating pathways.
Louis Pasteur started vaccine biology without knowing anything about immunology; antibodies were discovered 30 years after he did his first vaccine work. Yet, Pasteur left quite a legacy (Table 1). The person who started our laboratory at Rockefeller, yet another Frenchman, René Dubos, wrote one of the best biographies of Pasteur. In it, he quoted a speech Pasteur gave upon being elected to the French Academy of Letters. Pasteur said that the scientific method is a “marvelous experimental method [that] … eliminates certain facts, brings forth others, interrogates nature, compels it to reply, and stops only when the mind is fully satisfied” (3).
Table 1.
The microbial vaccine legacy of Louis Pasteur
| Vaccine | Year developed |
| Live attenuated | |
| Rabies | 1880 |
| Tuberculosis | 1927 |
| Yellow fever | 1938 |
| Measles | 1963 |
| Polio | 1963 |
| Mumps | 1967 |
| Rubella | 1969 |
| Typhoid fever | 1985 |
| Cholera | 1985 |
| Varicella | 1996 |
| Rotavirus | 1998 |
| Inactivated | |
| Plague | 1897 |
| Rabies | 1980 |
| Pertussis | 1914 |
| Influenza | 1936 |
| Hepatitis A | 1971 |
| Polio | 1956 |
| Tickborne and Japanese encephalitis | 1990 |
| Typhoid fever | 1896 |
| Cholera | 1896 |
Most science is terrific at dissecting a particular problem, such as the immune response, but in vaccine biology, you not only have to dissect the pathway, but you have to prove it by compelling the patient to reply to the knowledge that you've generated. This is the goal of our latest research.
HIGHLIGHTS OF VACCINE STUDIES
We are developing vaccines composed of microbial proteins within monoclonal antibodies that target uptake receptors on dendritic cells or their subsets, and these receptors are expressed on the dendritic cells in the lymphoid organs. We also deliver a stimulus to the dendritic cell so as to elicit maturation and an immune response. Within this overall strategy, we still have much to learn about each of these variables: the receptors, the subsets, and the maturation stimulus. The goal is to improve vaccine efficacy by getting the vaccine protein to the dendritic cell, an approach that had not been used before in vaccine design. At the same time, because we are selectively targeting antigen to the dendritic cell in situ, we are starting to learn what the dendritic cell is doing in the intact lymphoid organ, whereas previously we would isolate dendritic cells, manipulate them in culture, and put them back in. Targeting antigens to dendritic cells is a much more direct way to look at their function in vivo.
The first dendritic cell target we have studied is DEC-205 (originally called NLDC-145), an endocytic receptor expressed at high levels on dendritic cells. We take the heavy-and light-chain cDNA from an anti–DEC-205 monoclonal antibody and engineer the heavy chain at the carboxyl terminus to encode a microbial protein of interest to us. We are examining four proteins because of their different immunologic challenges (Table 2). For the maturation stimulus, we initially used a microbial mimic, polyinosinic:polycytidylic acid (poly I:C), and an agonistic anti-CD40 antibody. Because of its potential risks in a naive population, anti-CD40 antibody was subsequently dropped, and now we use poly I:C alone. This work is a collaboration with many people, but primarily Michel Nussenzweig, who heads a molecular immunology laboratory at Rockefeller and was the first to start making these engineered antibodies.
Table 2.
Microbial proteins studied with vaccines
| Protein | Disease | Comments |
| HIV-1 gag p24/p41 and nef | HIV/AIDS | Need for strong Th1-type immunity predicted to have a protective value; first one needs to identify principles to elicit this kind of immunity to test the hypothesis |
| Plasmodia circumsporozoite | Malaria | Need for a very strong antibody response as well as a T cell response to provide protection; the circumsporozoite protein used covers the early-life-cycle parasite that enters the liver |
| Yersinia pestis LcrV | Pneumonia | Study of protection at a mucosal surface |
| Leishmania major LACK | Leishmaniasis | A classical model, in which the immune system, by producing interferon-gamma, activates macrophages to kill a parasite |
Results showed that the antibodies improved antigen presentation, that is, the targeting had an effect in the animal. We showed the effect in experiments with several different proteins as antigens.
Clonal expansion of T cells
First, we examined expansion of T cells, here T cells specific for the Leishmania LACK (Leishmania activated C kinase) antigen, 3 days after injection. We marked the T cells with thy-1.1, so that injected cells could be distinguished from recipient cells, and used a fluorescent dye, carboxyfluorescein succinimidyl ester (CSFE), to allow measurement of T cell proliferation. In the experiment shown, we used 0.3 and 0.03 μg of anti-DEC-LACK; 0.3 and 0.03 μg of 33D1-LACK (a monoclonal antibody targeting a different subset of dendritic cell); 3 and 0.3 μg of LACK without a dendritic cell target; and a control. While LACK by itself stimulated T cells at the high dose of 3 μg, it showed little activity at the 0.3 μg level. In contrast, the two monoclonal antibodies showed extensive cell division at even the 0.03 μg level (Figure 4). T cells can undergo nine or ten divisions in 3 days, a very rapid rate of clonal expansion. Thus, by incorporating the LACK protein into the dendritic cell monoclonal antibodies, we increased antigen presentation by at least 100-fold (4). The proliferation of these T cells marks successful antigen presentation in vivo.
Figure 4.

Hybrid anti–dendritic cell antibodies (αDEC-LACK and 33D1-LACK) efficiently trigger clonal expansion of transgenic T cells in vivo compared with the protein alone (LACK) or the control group (PBS, phosphate-buffered saline). 3 × 106 CFSE-labeled TCR transgenic cells were administered intravenously to thy-1.2+ mice 1 day before immunization intraperitoneally with graded doses of αDEC-LACK, 33D1-LACK, Ig-LACK, or LACK protein. Spleens were harvested at 3 days to enumerate thy-1.1+ T cells that had diluted CFSE. The expansion of transgenic T cells is displayed as representative CFSE dilution plots, gated on thy-1.1+ LACK transgenic CD4+ T cells in response to the indicated doses of fusion monoclonal antibody or LACK protein. Reprinted with permission from Journal of Experimental Medicine (4).
Protective immunity
Another step was to see whether the T cell response led to protective immunity. In this experiment, led by Christine Trumpfheller, we tested a single dose of an anti–dendritic cell antibody vaccine carrying HIV gag protein (5).
To monitor the immune response, we have been using overlapping peptide mixtures as the source of antigen—a method that has been a boon to immunology. The problem in measuring the immune response is that proteins like LACK or HIV gag have to be processed successfully during the immune assay, regardless of the MHC molecules that have to present the processed peptides to the immune T cells. Researchers in Germany found that short peptides, usually 15-mers, could be staggered every 4 amino acids along the length of the protein, and these pools of peptides, rather than the whole protein, could be used as the source of antigen. With this method, the MHC molecules, regardless of the donor, will capture peptides that are appropriate for their polymorphisms and present them to the immune T cells and elicit an immune response.
Once again, delivering the HIV gag p24 through DEC205 enhanced the immune efficacy of the protein vaccine, as measured by the percentage of gag-specific CD4+, interferon-gamma (IFN-γ) T cells (Figure 5). To test for protective immunity, we challenged the treated mice with a recombinant vaccinia virus. We wanted to achieve protection by targeting an HIV protein with poly I:C as a clinical adjuvant, rather than using anti-CD40, since poly I:C was more clinically feasible. After priming the animal with anti-DEC and poly I:C, we waited 6 weeks, since that's been reported to be the desirable interval to acquire T cell memory. Then we boosted with a second dose, waited another 6 to 18 weeks, and challenged the mice with vaccinia-gag. We examined the animals' capacity to deal with that challenge over 6 days. Results showed that the animals who had received the prime-and-boost vaccine treatment maintained their weight and showed about two logs of protection against growth of the virus in the lung.
Figure 5.

DEC-205 enhances the immune efficacy of a protein vaccine. BALB/c mice were immunized subcutaneously with graded doses of αDEC-p24 or control Ig-p24 monoclonal antibodies and maturation stimulus. Nineteen days later, we assessed the percentage of IFN-γ+ CD4+ cells in gated CD3+ splenic T cells using gag p24 peptide pools. One of three similar experiments is shown. Reprinted with permission from Journal of Experimental Medicine (5).
Thus, this experiment showed that low doses of vaccine protein are sufficient to immunize the animal. The vaccine produces high gag-specific T cell frequencies, as well as high amounts of cytokines per T cell relative to other forms of vaccination. The vaccine also showed breadth, priming to many peptides in several MHCs—in contrast with previous vaccines, in which usually only one MHC could be successfully immunized. In terms of quality, the majority of the T cells produce multiple cytokines (including interleukin[IL]-2, IFN-γ, and TNF-α), which is currently thought to be a useful correlate of protection to HIV. Finally, there is protection that is dependent on these immune T cells and IFN-γ. We have made similar observations on the immune response to Plasmodium CSP and Yersinia pestis LcrV proteins, although in these other models we are still working on the protection data.
Cross-presentation of antigen to CD8+ T lymphocytes
An important issue is whether the vaccine strategy would mediate presentation to CD8+ T cells. Humans are very polymorphic for MHC class I. Much of the research on cross-presentation to CD8+ T cells has been done with single peptides and single MHC haplotypes. So, for example, a lot of the clinical studies have been done only with HLA 2.1 and certain peptides that are presented by it, while the vast majority of experiments in mice have been done with a single peptide from ovalbumin in one single MHC. We wanted to see if we could get presentation to CD8+ T cells in many MHC types.
Christine Trumpfheller, Angela Granelli-Piperno, and Leonia Bozzacco prepared fusion antibodies to human DEC-205, MMR, and DC-SIGN and engineered them to deliver HIV gag p24. We showed that these fusion antibodies bound well to dendritic cells. We then prepared either bulk mononuclear cells or T cells that were cocultured with dendritic cells from HIV-infected individuals who were in a good state of immune activity (CD4+ cells >600/μL). After CFSE labeling, we stimulated the T cells for 7 days with the fusion antibody vaccine α-DEC-p24 and then restimulated them for 6 hours with pools of p24 peptides to see if they would make IFN-γ when they were rechallenged with different peptides from the gag protein. We then could identify the peptides that had been extracted and processed from this vaccine and presented on different HLA molecules.
Among the 11 patients we studied, we identified eight different gag peptides that were processed and then recognized via DEC-205. After typing for HLA class I alleles, we identified known peptide sequences from the Los Alamos database that are presented on a corresponding HLA product (6). Based on these results, it appears that dendritic cells are efficient at cross-presenting internalized nonreplicating proteins on to MHC I and that we may be able to deliver at least some peptides to many different human MHC molecules for purposes of vaccination.
T cell differentiation during dendritic cell vaccine design
Differentiation is a particularly demanding issue in vaccine design, because a naive T cell clone can develop among numerous pathways, with very different consequences functionally (see Figure 2). Th1 cells are classically protective in many instances, particularly in activating macrophages. Th2 cells can bring about allergies. Other pathways of differentiation cause T cells to silence the immune response. For HIV vaccines, the goal is to elicit very strong Th1 immunity in addition to killer T cells.
In terms of pathways, IL-12 can influence precursors to become Th1 cells, while IL-4 can influence precursors to become Th2 cells. Th1 cells express T-bet and secrete IFN-γ; Th2 cells express GATA3 and secrete IL-4 (7).
We studied the function of dendritic cell subsets in vivo, knowing that the subset of dendritic cells that is marked by CD8+ and expression of the DEC receptor is known to make IL-12. I'm not fond of the CD8 marker because it does not apply to any non-human species in terms of dendritic cells. Instead, I prefer to use receptor molecules that have a known function: CD205+ for the CD8+ subset and 33D1+ or DCIR2+ for the CD8− subset. Both subsets can process an antigen to make MHC class II peptide complexes, but the CD205+ subset makes a high level of IL-12.
We examined the consequences of dendritic cell subset function in vivo using a protozoan parasite system, Leishmania major. Researchers had shown that BALB/c mice were highly susceptible to this parasite. They were also prone to making the Th2 type of T cell to L. major. B6 mice, in contrast, were genetically resistant to the parasite and were prone to making the IFN-γ–producing cell. However, if a transgene for IL-4 was inserted into the B6 mouse, the mouse became susceptible to L. major, even though it also made IFN-γ.
Our results assessed immune responses through the percentage of CD4+ T cells that were antigen specific and secreted IFN-γ or made IL-4 (Figure 6). Infection with L. major resulted in an IL-4 response in the BALB/c mouse, the species susceptible to the parasite. Immunization with DEC-targeted LACK, however, resulted entirely in the induction of IFN-γ–producing cells, not IL-4–producing cells. This change in the usual response of BALB/c mice did not require IL-12, the classical instructing cytokine. In contrast, with 33D1 targeted, there were some IFN-γ–producing cells, though less than with DEC, and an IL-4–producing response was clearly made. If we knocked out the IL-12 in this group, the IFN-γ–producing cells were lost, but the IL-4 producers were intact. Thus, with one subset of cells, the response was IL-12-dependent; with the other subset, the response was qualitatively different and surprising: a Th1 response that was IL-12–independent.
Figure 6.

DEC+ (33D1−) dendritic cells polarize T cells (even in BALB/c mice) to produce IFN-γ, but IL-12 p40 is not required. IL-12 p40−/− mice and control littermates were immunized intraperitoneally as indicated on the x-axis. Three weeks later, (a) the frequency of IFN-γ+ CD4+ CD3+ splenic T cells was determined by intracellular staining or (b) CD4+ splenocytes were restimulated in vitro with CD11c+ cells, medium, or LACK peptide for 36 to 48 hours to measure IL-4–secreting cells by ELISPOT. Reprinted with permission from Journal of Experimental Medicine (4).
With free, nontargeted antigen, the result was mixed: partial reduction of the IFN-γ response and upregulation of IL-4 upon ablation of IL-12. The phosphate-buffered saline (PBS) column was the control group, with no immune response. This is the thesis work of Helena Soares, done in collaboration with Nick Glaichenhaus in France (4).
We then examined the mechanism employed by DEC-205+ to induce CD4+T cells to produce IFN-γ in the absence of IL-12 p40 and in an otherwise Th2-prone BALB/c mouse. We found that the CD70 molecule seems to be critical for this immune response. We injected anti-CD70 or control monoclonal antibody into naive mice. A day later we immunized the mice with different forms of LACK, i.e., targeting to the DEC subset, the 33D1 subset, or no target, together with a maturation stimulus (poly I:C and anti-CD40). Three weeks later, we measured the immunity.
The control group had a strong immune response, with numerous CD4+ T cells producing IFN-γ. With anti-CD70, however, the immune response to anti-DEC-LACK was totally blocked. Targeting the antigen to the other subset caused reduction of the number of IFN-γ–producing cells, but this subset was totally resistant to anti-CD70. Therefore, the subset targeted determines the quality of the response as well as the mechanism used to induce that response. Blocking anti-CD70 following administration of a nontargeted antigen had only modest inhibitory effects, since the LACK went to both subsets (Figure 7) (4).
Figure 7.

αCD70 treatment (FR70, H.Yagita) in vivo blocks immunity to LACK in naive mice, after targeting of antigen to DEC+ but not DEC− dendritic cells. BALB/c mice were injected intraperitoneally with 50 μg αCD70 monoclonal antibody 1 day before intraperitoneal immunization with 10 μg αDEC-LACK, 10 μg 33D1LACK, 50 μg LACK protein, and 10 μg Ig-LACK in the presence of αCD40 and poly I:C, or PBS. Three weeks later, the frequency of IFN-γ+ CD4+ CD3+ splenic T cells was determined by intracellular staining. One of two similar experiments. Reprinted with permission from Journal of Experimental Medicine (4).
Seeing the critical role of CD70 during an in vivo helper T cell response leads to another question: whether TNF superfamily members are the decision makers used by dendritic cells to control the quality of T cell immunity. Yong-Jun Liu in Houston has shown that OX40L, another TNF family member, can pivotally control a Th2 response. Other studies have shown that GiTR-L, yet another TNF family member, might be a way for the dendritic cell to avoid a suppression type of response. These TNF superfamily members may be key correlates of immunogenicity.
Use of the targeting concept for DNA vaccines
The targeting concept may be able to be extended to existing vaccines in research that has just been submitted for publication. Some HIV vaccines are in extensive phase IIb studies, and there are concerns that they lack sufficient immunizing power. A postdoctoral fellow, Godwin Nchinda, has designed DNA vaccines that target dendritic cells. He has engineered the DNA vaccines that are currently under testing in the field so that they have a targeting single-chain antibody to the DEC-205 receptor. When we make these constructs, we express them in tissue culture and show that the single-chain vaccine fusion protein is secreted and binds to a cell line that expresses the DEC-205 receptor but not to a control cell line.
To test the targeted DNA vaccine in mice, we examined the protection after airway challenge with a recombinant vaccinia-gag virus. At all test doses, 0.3, 3, and 30 μg of DNA vaccine, mice were able to retain their weight after receiving the vaccine and being challenged. In contrast, unvaccinated mice lost body weight, and mice receiving the untargeted vaccine likewise lost 20% to 25% of their weight. Virus titers were also lower in the mice who received the DEC-205–targeted DNA vaccine.
CONCLUSION
Vaccines composed of an anti–dendritic cell hybrid monoclonal antibody and a maturation stimulus can improve the quantity, breadth, and quality of the T cell response. Targeting of the different types of dendritic cells also allows us to identify mechanisms of dendritic cell function in vivo. With the prevalent interfaces of immunology and medicine, dendritic cells are a desirable target in the development of treatments and vaccines for a variety of conditions.
References
- 1.Steinman RM. Dendritic cells: versatile controllers of the immune system. Nat Med. 2007;13(10):vii–xi. doi: 10.1038/nm1643. [DOI] [PubMed] [Google Scholar]
- 2.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 3.Dubos R. Pasteur and Modern Science. Washington, DC: American Society for Microbiology; 1995. [Google Scholar]
- 4.Soares H, Waechter H, Glaichenhaus N, Mougneau E, Yagita H, Mizenina O, Dudziak D, Nussenzweig MC, Steinman RM. A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J Exp Med. 2007;204(5):1095–1106. doi: 10.1084/jem.20070176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trumpfheller C, Finke JS, Lopez CB, Moran TM, Moltedo B, Soares H, Huang Y, Schlesinger SJ, Park CG, Nussenzweig MC, Granelli-Piperno A, Steinman RM. Intensified and protective CD4+ T cell immunity in mice with anti-dendritic cell HIV gag fusion antibody vaccine. J Exp Med. 2006;203(3):607–617. doi: 10.1084/jem.20052005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bozzacco L, Trumpfheller C, Siegal FP, Mehandru S, Markowitz M, Carrington M, Nussenzweig MC, Piperno AG, Steinman RM. DEC-205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+T cells in a spectrum of human MHC I haplotypes. Proc Natl Acad Sci U S A. 2007;104(4):1289–1294. doi: 10.1073/pnas.0610383104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2(12):933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]