

Abstract
The properties of keratin intermediate filaments (IFs) have been studied after transfection with green fluorescent protein (GFP)-tagged K18 and/or K8 (type I/II IF proteins). GFP-K8 and -K18 become incorporated into tonofibrils, which are comprised of bundles of keratin IFs. These tonofibrils exhibit a remarkably wide range of motile and dynamic activities. Fluorescence recovery after photobleaching (FRAP) analyses show that they recover their fluorescence slowly with a recovery t 1/2 of ∼100 min. The movements of bleach zones during recovery show that closely spaced tonofibrils (<1 μm apart) often move at different rates and in different directions. Individual tonofibrils frequently change their shapes, and in some cases these changes appear as propagated waveforms along their long axes. In addition, short fibrils, termed keratin squiggles, are seen at the cell periphery where they move mainly towards the cell center. The motile properties of keratin IFs are also compared with those of type III IFs (vimentin) in PtK2 cells. Intriguingly, the dynamic properties of keratin tonofibrils and squiggles are dramatically different from those of vimentin fibrils and squiggles within the same cytoplasmic regions. This suggests that there are different factors regulating the dynamic properties of different types of IFs within the same cytoplasmic regions.
Keywords: keratin, tonofibrils, squiggles, dynamics, motility
Introduction
Intermediate filaments (IFs) belong to a superfamily of ∼50 proteins that are expressed in cell type–specific fashions. Based upon their primary sequences, these proteins have been subdivided into six types (Coulombe et al. 2000; Herrmann and Aebi 2000). Type I, acidic keratins, and type II, neutral-basic keratins, comprise the largest and most complex group of IF proteins. Although IFs can be assembled as heteropolymers from any combinations of type I and II keratins in vitro, specific pairs of keratin are expressed in epithelial cells in vivo (Fuchs and Weber 1994). For example, simple epithelia express K8/K18 with variable levels of K19 and K20, whereas stratified epithelia express K5/K14 basally and K1/K10 suprabasally (Fuchs and Weber 1994). Keratin IFs are usually organized into bundles known as tonofibrils, which form cage-like structures around the nucleus and extend from the perinuclear region to the cell periphery (Franke et al. 1978a,Franke et al. 1978b; Goldman et al. 1985, Goldman et al. 1986).
It has been demonstrated that IFs play crucial roles in maintaining the structural integrity and mechanical properties of cells. For example, skin blistering is induced by dominant-negative mutations of K5/K14 and K1/K10 in patients with epidermolysis bullosa simplex and epidermolytic hyperkeratosis (Fuchs and Cleveland 1998). These blisters appear to be related to the structural alteration of networks of tonofibrils, which in turn causes cytolysis upon mechanical stress. Furthermore, transgenic mice expressing a dominant-negative mutation of K18 display hepatocyte fragility and necrosis, accompanied by the disruption of tonofibril networks (Ku et al. 1995).
Based upon their proposed mechanical roles, IFs have been considered to be relatively stable cytoskeletal structures. However, there is increasing evidence that IFs have dynamic properties in vivo. For example, transfected or microinjected keratins are incorporated into endogenous tonofibrils in epithelial cells (Albers and Fuchs 1987; Miller et al. 1991). Furthermore, tonofibrils disassemble into nonfilamentous aggregates during prometaphase and reassemble in daughter cells after cytokinesis in numerous types of epithelial cells (Franke et al. 1982; Lane et al. 1982).
Recent studies using green fluorescent protein (GFP)-tagged IF proteins have provided new insights into their motility and assembly properties in living cells (Ho et al. 1998; Prahlad et al. 1998; Yoon et al. 1998; Windoffer and Leube 1999; Wang et al. 2000). Time-lapse observations of GFP-vimentin show that vimentin IF networks constantly change their configurations in fibroblasts (Ho et al. 1998; Yoon et al. 1998). Detailed analyses of individual vimentin fibrils demonstrate that they change their shapes; they extend and shorten over relatively short time intervals and translocate bidirectionally (Yoon et al. 1998). Furthermore, rapid movements of IF precursor complexes, termed particles and squiggles, are observed in spreading and well-spread fibroblasts (Prahlad et al. 1998; Yoon et al. 1998). In the case of keratin IFs, time-lapse observations of epithelial cells expressing GFP-K13 show that centrally located tonofibrils exhibit slow oscillations or undulations, observed by conventional epifluorescence optics (Windoffer and Leube 1999). In addition, there are unidirectional fluctuations of diffuse and particulate keratin from the cell surface towards the nucleus. These observations are very different from the bidirectional movements of individual vimentin fibrils, which were analyzed by the movements of photobleach zones made perpendicular to their long axes using high resolution confocal microscopy (Yoon et al. 1998). Similar detailed analyses of the movements of individual tonofibrils have not been carried out.
In this study, we describe for the first time quantitative analyses of the motile properties of individual tonofibrils in live epithelial cells. We have also been able to compare directly the motility and the rates of subunit exchange of individual keratin tonofibrils and vimentin fibrils in PtK2 epithelial cells. Fluorescence recovery after photobleaching (FRAP) analyses of PtK2 cells expressing GFP-K18 (type I keratin), GFP-K8 (type II keratin), or both show that there is a steady-state equilibrium between subunits and polymerized tonofibrils. During recovery, individual tonofibrils spaced as closely as 0.3–1 μm can translocate in opposite directions, that is, towards or away from the cell periphery. These movements are inhibited by nocodazole but not by cytochalasin B, indicating a dependence upon microtubules (MTs). In addition, individual tonofibrils can change their shapes, frequently appearing to propagate waveforms along their long axes. The propagation of these waveforms continues in both nocodazole and cytochalasin B, suggesting that these movements represent inherent motile properties of tonofibrils. Preliminary experiments suggest that these properties are related to the state of phosphorylation of keratin. Furthermore, time-lapse observations demonstrate that short filamentous structures, termed keratin squiggles, can translocate mainly from the cell periphery towards the cell center. These movements are inhibited to a great extent by nocodazole but not by cytochalasin B. Interestingly keratin and vimentin IFs possess strikingly different properties within the same cytoplasmic regions. For example, movements of individual tonofibrils and keratin squiggles are significantly slower than those of their vimentin counterparts, and FRAP analyses demonstrate that keratin tonofibrils recover their fluorescence ∼20 times slower than vimentin fibrils.
Materials and Methods
GFP-Keratin cDNA Constructs
All restriction enzymes and DNA modifying enzymes were purchased from GIBCO BRL. Other reagents were from Sigma-Aldrich, unless otherwise noted.
GFP- and yellow fluorescent protein (YFP)-tagged keratin cDNA constructs were prepared by fusing GFP and YFP to the NH2 termini of keratin cDNAs. Human K8–pUC18 and human K18–pBluescript SK+ were provided by Dr. Bishr Omary (Stanford University, Palo Alto, CA). The complete human keratin 8 cDNA sequence (Hofmann and Franke 1997) was amplified by PCR using primers that create the restriction enzyme sites for BamHI at the 5′ and 3′ ends. The resulting fragment was subcloned into the BamHI site of pEGFP-C1 and pEYFP-C1 (CLONTECH Laboratories, Inc.) to generate GFP-K8 and YFP-K8, respectively. GFP-K8–FLAG was created by inserting the epitope sequence of FLAG (also known as octapeptide; GACTACAAAGACGATGACGACAAG) to the COOH terminus of GFP-K8. The complete human K18 cDNA sequence (Oshima et al. 1986) was also amplified by PCR using primers that create the restriction enzyme sites for EcoRI at the 5′ end and XbaI at the 3′ end. The resulting fragment was subcloned into EcoRI/XbaI sites of pEGFP-C1 to generate GFP-K18. K18-myc was made by PCR to generate the restriction enzyme sites for HindIII at the 5′ end and for XhoI at the 3′ end of K18 and to subclone into HindIII/XhoI sites of pcDNA3 containing the myc epitope sequence (CAAGAGCAAGAAGAGGACTTGAATGTC) at the COOH terminus of the XhoI site (Invitrogene). GFP-K18–myc was prepared by subcloning the HindIII-XbaI fragment containing K18-myc into HindIII/XbaI sites of pEGFP-C3. GFP-vimentin cDNA construct was described elsewhere (Yoon et al. 1998). Cyan fluorescent protein (CFP)-vimentin was generated by subcloning the BamHI-BamHI fragment containing the complete human vimentin cDNA (Chou et al. 1996) into the BamHI site of pECFP-C1. All cDNA constructs were purified by CsCl2 density–gradient centrifugation and verified by DNA sequencing (Sambrook et al. 1989).
Cell Cultures and Transient Transfection
SW13 vim− cells (a gift from Dr. Robert Evans, University of Colorado, Denver, CO) were grown in DME with 10% FCS and antibiotics (100 U/ml penicillin and streptomycin). HeLa, MCF-7, and PtK2 epithelial cells were grown in MEM with 10% FCS, 5 mM sodium pyruvate, 0.1 mM nonessential amino acid solution (GIBCO BRL), and antibiotics.
GFP-, YFP-, and CFP-tagged keratin and vimentin cDNA constructs were transiently transfected according to Huang et al. 1998. Subconfluent cells grown in 100-mm dishes were trypsinized and suspended in 250 μl of culture medium containing 10 mM Hepes, pH 7.1. The suspension was mixed with 7 μg of target DNA and 13 μg of sheared salmon sperm DNA (Amresco Inc.). In cotransfection experiments, 5 μg of each target DNA and 10 μg of sheared salmon sperm DNA were used. Subsequently, the suspension was electroporated at 0.25 V/960 μFD (Gene Pulser II; Bio-Rad Laboratories) and plated onto 35-mm dishes. It should be noted that the results obtained in cells transfected with GFP-keratin cDNA constructs with or without the myc or FLAG tag were identical (data not shown).
Microinjection
A monoclonal antibody against dynein intermediate chain (clone 70.1; Sigma-Aldrich) was dialyzed against PBS and concentrated to a final concentration of 1 mg/ml (Burkhardt et al. 1997). PtK2 cells grown onto locator coverslips (Bellco Glass, Inc.) were microinjected using an automated microinjection system (Eppendorf Scientific, Inc.) and then returned to a CO2 incubator at 37°C, as described previously (Goldman et al. 1996). At 3–4 h after microinjection, cells were fixed for immunofluorescence or maintained live for time-lapse observations.
Immunofluorescence
Indirect immunofluorescence was carried out as described by Miller et al. 1993. The primary antibodies used were rabbit polyclonal anti–bovine tongue keratin (Jones et al. 1988), polyclonal antioctapeptide (anti-FLAG; Zymed Laboratories), mouse monoclonal anti–vimentin (V9; Sigma-Aldrich), monoclonal anti–β-tubulin (Amersham Pharmacia Biotech), monoclonal antitransferrin receptor (Zymed Laboratories), or monoclonal anti-myc (9E10; Evan et al. 1985). The secondary antibodies used were Alexa 488– or Alexa 633–conjugated goat anti–mouse IgG, Alexa 633–conjugated goat anti–mouse IgM, or Alexa 568–conjugated goat anti–rabbit IgG (Molecular Probes Inc.). Alexa 568–conjugated phalloidin (Molecular Probes Inc.) was also used to label microfilaments (MFs).
Live Cell Studies
Transfected cells were trypsinized at 48 h after transfection and replated onto coverslips to achieve 70% confluence in culture medium containing 10 mM Hepes, pH 7.1. For time-lapse observations and FRAP analyses, coverslips were placed on slides with glass feet, sealed with a mixture of vaseline, beeswax, and lanolin (1:1:1), and maintained at 37°C with an air-stream stage incubator (ASI 400; Nevtek) (Yoon et al. 1998).
In some cases, transfected cells were treated with nocodazole and/or cytochalasin B at final concentrations of 10 and 2 μM, respectively. In other cases, calyculin A (Calbiochem) was used at a final concentration of 0.5 nM. For energy depletion studies, 50 mM 2-deoxy-glucose and 0.05% sodium azide were added to glucose-deficient culture medium (Yoon et al. 1998).
Time-Lapse Observations
Time-lapse observations were made with a ZEISS LSM 510 confocal microscope equipped with a 100×, 1.4 NA oil immersion objective. GFP images were acquired by excitation at 488 nm and emission at 515–545 nm. YFP and CFP images were simultaneously collected by excitation at 458 and 514 nm and emission at 475–525 and 530 nm, respectively. Images of GFP-keratin were collected every 2–15 min for periods up to 3 h, and images of GFP-vimentin were collected every 30 s for up to 40 min in the same focal plane (see Yoon et al. 1998). Phase–contrast images of cells were taken before, during, and immediately after time-lapse observations to ensure that there were no significant changes in cell shape and position during periods of observation.
Fluorescence Recovery after Photobleaching Analyses
FRAP analyses were carried out with the confocal microscope as described by Yoon et al. 1998. For PtK2 cells expressing both YFP-K8 and CFP-vimentin, bar-shaped regions were bleached at 458 and 514 nm for 3 s, and recovery was monitored at 2-min intervals. For PtK2 cells expressing either GFP-K8/K18 or GFP-vimentin, bar-shaped regions were bleached at 488 nm for 3 s, and recovery was monitored at 15-min intervals for GFP-K8/K18 tonofibrils and at 2-min intervals for GFP-vimentin fibrils. Phase–contrast images of cells were taken before and immediately after FRAP analyses to ensure that there were no significant changes in cell shape and position.
Shear Stress Experiments
For shear stress studies, PtK2 cells grown on Bioptics coverslips were subjected to uniform laminar (two-dimensional) flow in a temperature-controlled parallel plate flow chamber (FCS2; Bioptics, Inc.) (Helmke et al. 2000) mounted on the stage of the confocal microscope.
Shear stress, τ (dyn/cm2) was calculated from the following relation: τ = 3Qv/2a 2ω, where Q = volumetric flow rate (ml/s), v = culture medium viscosity (Poise), a = channel half height (cm), and ω = channel width (cm). A peristaltic pump was calibrated and used to generate known rates of flow. The viscosity of the culture medium measured at 37°C was 7.339 × 10−3 Poise using an Ostwald viscometer. The channel width is assumed to be the same as the dimension of the T-bar in the chamber (1.3 cm). The channel half height is governed by the thickness of the gasket (0.2 mm).
Image Analysis
As indicated above, analyses of the motile properties of GFP-tagged IFs were restricted to cells that exhibited no obvious shape changes as determined by phase–contrast microscopy. If the average displacement of a cell's margins was >0.08 μm in 40 min (0.02 μm/min), it was excluded from the analyses. This ensured that any recorded movements of IFs were due to their intrinsic properties and not passive reflections of significant changes in cell shape (see Yoon et al. 1998).
Position, length, and grayscale pixel values were measured on digitized confocal images using the Metamorph image analysis program (Universal Imaging Corp.). Pixel values were converted to distance using the confocal scale bars. The average rate of movement was determined by calculating distance versus time. To adjust for sample fading during FRAP analyses, the average grayscale pixel value of the prebleach image was measured, and this value was used to normalize the intensity level of subsequent images (Yoon et al. 1998). Fluorescence recovery t 1/2 were estimated by measuring the average grayscale pixel values across bleached fibrils at each time point using the line-scan function. All data were reported as mean ± SDs.
Preparation of IF-enriched Cytoskeletons and Immunoblotting Analyses
IF-enriched cytoskeletal preparations (Zackroff and Goldman 1979) were made from PtK2 cells after treatment with 0.5 nM calyculin A for 30 min. These preparations were separated by SDS-PAGE (Laemmli 1970). Immunoblotting was carried out according to Towbin et al. 1979. The primary antibodies used were mouse monoclonal K8-pSer431 (5B3; Ku and Omary 1997), monoclonal K18-pSer33 (IB4; Ku and Omary 1998), and rabbit polyclonal anti-K8/K18 (R275). The secondary antibodies used were peroxidase-conjugated goat anti–mouse IgG or peroxidase-conjugated goat anti–rabbit IgG (Jackson ImmunoResearch Laboratories).
Online Supplemental Material
The QuickTime videos from which images of Fig. 3 (FRAP analyses), Fig. 5 (shape change and wave propagation), and Fig. 6 (translocation of squiggles) were taken can be found at http://www.jcb.org/cgi/content/full/153/3/503/DC1.
Figure 3.

FRAP analyses of tonofibrils containing GFP-K8 (A–D) and GFP-K18 (E–H) in live PtK2 cells. Bar-shaped regions are photobleached, and fluorescence recovery is monitored at 15-min intervals. Bleached tonofibrils recover their fluorescence within 2–3 h. During recovery, some bleach zones (A–D) are relatively stationary and others (E–H) move. Frequently, bleach zones made across individual tonofibrils move at different speeds and in different directions during recovery (arrows). Elapsed time (h:min:s) is indicated at the lower left of each confocal image. Neither the fluorescence recovery rate nor the rate of bleach zone movement appear to be related to tonofibril thickness. Video available at http://www.jcb.org/cgi/content/full/153/3/503/DC1. Bars, 5 μm.
Figure 5.

Time-lapse observations of tonofibrils in live PtK2 cells transfected with GFP-K18. These fibrils exhibit extensive bending or wave-like movements. In many instances, these waveforms appear to be propagated along individual tonofibrils (arrows 2 and 3). In other cases, these waveforms disappear (arrows 1 and 4). Elapsed time (min:s) is indicated at the lower left of each confocal image. Video available at http://www.jcb.org/cgi/content/full/153/3/503/DC1. Bar, 5 μm.
Figure 6.

(A–C) The overall distributions of keratin and vimentin squiggles are compared in an untransfected PtK2 cell after fixation and processing for double immunofluorescence. Keratin squiggles (A) and vimentin squiggles (B) are frequently visible in the same peripheral regions. However, the overlay (C) indicates that keratin and vimentin squiggles are distributed differently. (D–G) Time-lapse observations of GFP-K18 squiggles indicate that the majority move towards the cell center (see arrows). Elapsed time (min:s) is indicated at the lower left of each confocal image. Video available at http://www.jcb.org/cgi/content/full/153/3/503/DC1. Bars, 5 μm.
Results
GFP-Keratin Is a Faithful Reporter of Endogenous Tonofibrils In Vivo
To characterize the properties of GFP-keratins in live cells, we first determined whether GFP-tagged keratin IFs could assemble in cells devoid of cytoskeletal IF proteins. To this end, SW13 vim− cells were transfected with GFP-K18 (type I keratin) and GFP-K8 (type II keratin) cDNA constructs singly or in combination. When these cells were transfected with either GFP-K8 or GFP-K18 alone, ∼6% (118/1846) displayed only diffuse and/or punctate fluorescence (Fig. 1 A). When GFP-K8 and GFP-K18 were cotransfected, ∼27% (284/1047) were fluorescent, displaying extensive networks of tonofibrils (Fig. 1 B) or short filamentous structures termed squiggles (Fig. 1 C). Similar results were obtained when GFP-K8 and K18-myc were cotransfected into SW13 vim− cells (Fig. 1 C). These observations demonstrate that GFP-tagged type I/II keratins assemble as heteropolymers into filamentous structures in a null background.
Figure 1.
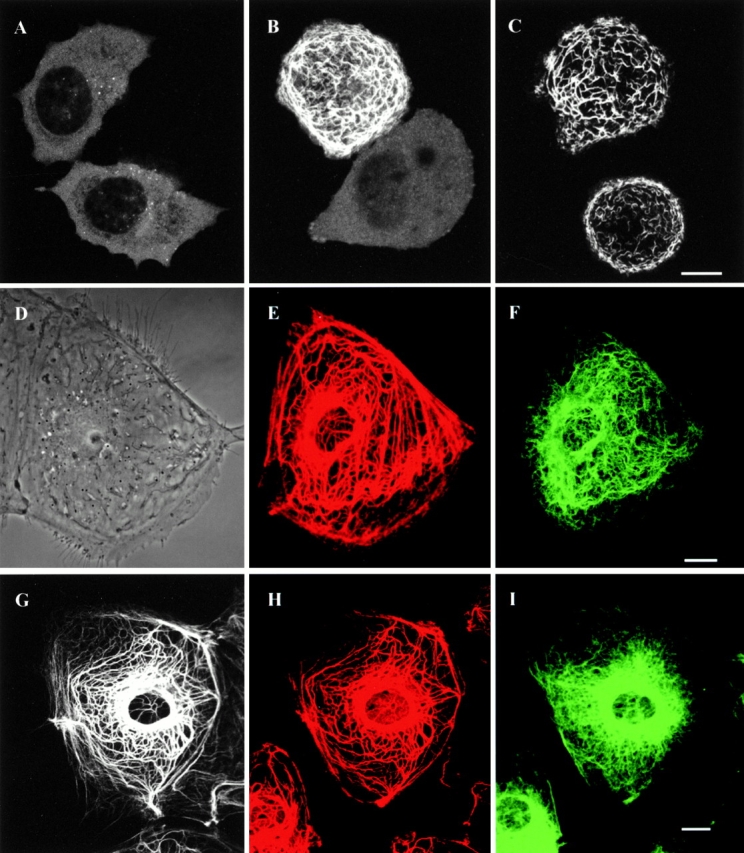
(A) No filamentous networks are seen in live SW13 vim− cells expressing GFP-K8 alone. Transfected cells display mainly diffuse and punctate fluorescence patterns. (B) Cotransfection with GFP-K8 and GFP-K18 frequently produces filamentous networks in live SW13 vim− cells. (C) When live SW13 vim− cells are observed after cotransfection with GFP-K8 and K18-myc, GFP-K8 coassembles with K18-myc to form squiggles and filamentous networks. (D–F) Phase–contrast image (D) of a live PtK2 cell expressing YFP-K8 (E) and CFP-vimentin (F). YFP-K8 displays a typical network of tonofibrils, which is distinct from the CFP-vimentin IF network. (G–H) GFP-K18 (G) also shows a typical tonofibril network in a live PtK2 cell. The same cell was processed for fixation and double immunofluorescence. Keratin is visualized with rabbit polyclonal anti–bovine tongue keratin and Alexa 568-labeled anti–rabbit IgG; and vimentin is visualized with mouse monoclonal anti–vimentin and Alexa 633-labeled anti–mouse IgG. GFP-K18 (G) is only incorporated into the endogenous network of tonofibrils (H) and not into the endogenous vimentin IF network (I). Bars, 10 μm.
We have also studied whether GFP- or YFP-tagged keratins incorporate into endogenous tonofibrils in PtK2 epithelial cells that express both endogenous keratin and vimentin IF networks (Osborn et al. 1980). After cotransfection with YFP-K8 and CFP-vimentin, YFP-K8 displayed typical networks of tonofibrils in live PtK2 cells (Fig. 1 E). These networks were distinct from CFP-vimentin IF networks, which were also found in the same cell (Fig. 1 F). These results show that YFP-keratin and CFP-vimentin become incorporated only into their respective endogenous IF networks. Furthermore, GFP-K18 colocalized exclusively with the endogenous tonofibrils and not with the endogenous vimentin fibrils as demonstrated by the direct observation of a live PtK2 cell (Fig. 1 G), followed by fixing and processing the same cell for double immunofluorescence (Fig. 1H and Fig. I). These observations demonstrate that GFP- and YFP-tagged type I/II keratins are specifically targeted to preexisting tonofibrils in epithelial cells that also express type III vimentin IF networks.
FRAP Analyses of Keratin Tonofibrils in Live Epithelial Cells
We used FRAP analyses of GFP- and YFP-tagged keratin tonofibrils in order to determine whether there is an exchange between subunits and polymerized keratin IFs in live epithelial cells. To ensure that photobleaching did not damage tonofibrils, PtK2 cells expressing GFP-K18–myc were fixed 1 min after photobleaching and processed for immunofluorescence with a rabbit polyclonal antibody directed against bovine tongue keratin and a mouse monoclonal anti-myc. The continuous antikeratin and anti-myc staining patterns across bleach zones indicated that photobleaching did not disrupt the structure of tonofibrils containing GFP-keratin (data not shown; see Yoon et al. 1998).
We were also able to take advantage of the fact that PtK2 cells express both keratin and vimentin. This provided us with a convenient internal standard for monitoring the dynamic properties of keratin IFs, since these properties have been determined in detail for vimentin IFs in fibroblasts (Yoon et al. 1998). To this end, we carried out FRAP analyses of PtK2 cells expressing both YFP-K8 and CFP-vimentin. Analyses were restricted to cells that showed no obvious changes in cell shape and position (described in Materials and Methods). Bleached CFP-vimentin fibrils recovered their fluorescence within 30 min, as described previously (Fig. 2, A–D; Yoon et al. 1998). However, no recovery was observed for bleached YFP-K8 tonofibrils within the same time period (Fig. 2, E–H). To more precisely determine the fluorescence recovery rates of keratin tonofibrils and vimentin fibrils in PtK2 cells, it was necessary to carry out FRAP analyses of cells expressing GFP-K8, GFP-K18, or GFP-vimentin rather than coexpressing YFP-K8 and CFP-vimentin. This was due to the finding that both YFP and CFP fusion proteins bleached faster than GFP fusion proteins, making it impossible to capture a sufficient number of images to calculate the average recovery t 1/2 for both types of IFs within the same cells.
Figure 2.
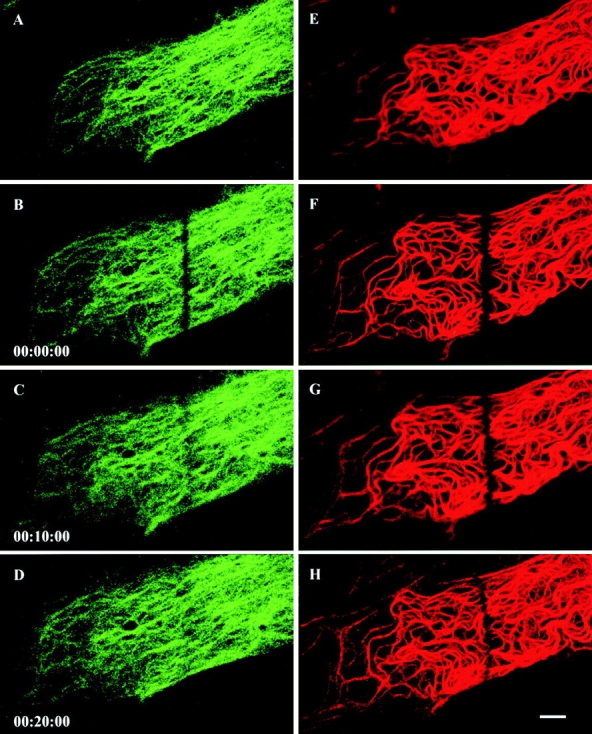
FRAP analyses of CFP-vimentin fibrils and YFP-K8 tonofibrils in the same live PtK2 epithelial cell. Bar-shaped regions are photobleached, and recovery of fluorescence is monitored at 2-min intervals for each fluorophore. Bleached CFP-vimentin fibrils (A–D) completely recover their fluorescence within 20–30 min. However, no recovery is observed for bleached YFP-K8 tonofibrils (E–H) during this same time period. Elapsed time (h:min:s) is indicated at the lower left of each confocal image. Bar, 5 μm.
During fluorescence recovery, some bleached tonofibrils remained relatively stationary (Fig. 3, A–D) and others moved (Fig. 3, E–H). Only stationary tonofibrils were used to estimate t 1/2. The fluorescence intensity measurements showed that recovery rates were the same across entire bleach zones (Fig. 4), indicating that there is no preferential recovery from either end of these zones. FRAP analyses of GFP-K18 (with or without the myc tag) showed that bleached tonofibrils recovered their fluorescence with an average t 1/2 of 109 ± 36 min (n = 24). FRAP analyses of GFP-K8 (with or without the FLAG tag) showed an average t 1/2 of 106 ± 36 min (n = 13). Fluorescence recovery in PtK2 cells coexpressing GFP-K18–myc and GFP-K8–FLAG exhibited a t 1/2 of 103 ± 29 min (n = 20). The coexpression of GFP-K18–myc and GFP-K8–FLAG within the same tonofibrils was confirmed by fixation and processing the same cells for double immunofluorescence using mouse monoclonal anti-myc and rabbit polyclonal anti-FLAG after FRAP analyses (data not shown). These observations demonstrate that the average t 1/2 values are virtually identical for type I and II keratins expressed either individually or together in live PtK2 cells. Furthermore, it appears that the fluorescence recovery rates are not related to the thickness of tonofibrils (data not shown; Fig. 3). We also determined that the average t 1/2 for GFP-vimentin fibrils was 6 ± 2 min (n = 26). This indicates that the turnover rate of keratin tonofibrils is ∼18-fold slower than vimentin fibrils in live PtK2 cells.
Figure 4.

Fluorescence intensity measurements along photobleached tonofibrils in live PtK2 cells. Grayscale pixel values are measured along the bleach zone of a tonofibril containing GFP-K18 at each time point using the line-scan function of the Metamorph image analysis program. In this case, the complete recovery takes place in 150 min with a recovery t1/2 of ∼105 min.
We have also determined whether the recovery rates for keratin IFs are cell type specific. To this end, we compared the t 1/2 of GFP-K8 tonofibrils in PtK2 (kidney epithelial cells) with those in MCF-7 (mammary epithelial cells) and HeLa (cervical epithelial cells). FRAP analyses showed an average t 1/2 of 92 ± 14 min (n = 14) in HeLa cells and a t 1/2 of 85 ± 13 min (n = 10) in MCF-7 cells. These results demonstrate that the turnover rates obtained for keratin IFs are similar in kidney, mammary, and cervical epithelial cells.
Motile Properties of Tonofibrils and Keratin Squiggles in Live Epithelial Cells
As described above, numerous bleach zones moved during fluorescence recovery. Frequently, tonofibrils moved at different rates and in opposite directions (Fig. 3, E–H). Parallel tonofibrils spaced only 0.3–1 μm apart were seen to move either towards or away from the cell periphery. The average rate of translocation of bleach zones was 0.06 ± 0.02 μm/min (n = 14), regardless of the thickness of tonofibrils and the direction of movements (data not shown; Fig. 3, E–H). Bleach zones made on vimentin fibrils also moved in PtK2 cells, averaging 0.15 ± 0.11 μm/min (n = 29). These results demonstrate that neighboring tonofibrils move independently at rates that are approximately threefold slower than vimentin fibrils.
Time-lapse observations also revealed that tonofibrils frequently exhibited bending or wave-like movements (Fig. 5). In some cases, these waveforms appeared to be propagated along the long axes of tonofibrils. In other cases, they disappeared, resulting in the formation of straight fibrils. Shape changes and waveforms differed dramatically on closely spaced tonofibrils, indicating that their movements were independent of each other. Although shape changes were also seen in vimentin fibrils in PtK2 cells, propagated wave-like movements were not observed (data not shown; see Yoon et al. 1998).
In addition to long tonofibrils, short filamentous structures, termed keratin squiggles, were frequently observed in the peripheral regions of untransfected (Fig. 6 A) and transfected (Fig. 6 D) PtK2 cells. Time-lapse observations of GFP-K18 squiggles showed that ∼18% (57/310) moved a distance of ≥0.5 μm in 10 min. The average rate of these movements was 0.24 ± 0.16 μm/min, ranging from 0.11 to 0.62 μm/min (n = 37). Interestingly, the majority (48/57 = 84%) of motile keratin squiggles translocated towards the cell center (Fig. 6, D–G). Vimentin squiggles were also seen in the peripheral regions of untransfected (Fig. 6 B) and transfected PtK2 cells (data not shown), but these showed no obvious overlap with keratin squiggles (Fig. 6 C). Approximately 43% (205/475) of vimentin squiggles were motile and the majority (143/205 = 70%) moved towards the cell periphery at higher speeds (3.5 ± 2.3 μm/min, with a range of 0.34–8.73 μm/min, n = 32) than keratin squiggles. These data show that there is an ∼15-fold difference in the rates of keratin and vimentin squiggle motility and that they tend to move in opposite directions. It is also of interest to note that keratin and vimentin squiggles frequently change their shapes, even when they are not translocating (data not shown).
The Motile Properties of Tonofibrils and Keratin Squiggles Require Metabolic Energy
To determine whether the various types of movements exhibited by tonofibrils and keratin squiggles require metabolic energy, time-lapse observations were made in PtK2 cells after treatment with 50 mM 2-deoxy-glucose and 0.05% sodium azide for 30 min (Yoon et al. 1998). None of the movements described above were detected in the presence of these inhibitors (data not shown), demonstrating that energy is required for these movements.
Effects of Cytochalasin B on the Motility of Tonofibrils and Squiggles
To determine whether the motile properties of tonofibrils and keratin squiggles are dependent on MFs, time-lapse observations and FRAP analyses were carried out in live PtK2 cells after treatment with cytochalasin B. When PtK2 cells were treated with 20 μM cytochalasin B for 30 min, the overall cell morphology became arborized, and the majority of MF networks were disrupted as determined by staining with Alexa 568-phalloidin (data not shown; see Osborn et al. 1977). Under these conditions, numerous tonofibrils and keratin squiggles remained in all regions of the cytoplasm including the arborized processes (Fig. 7A and Fig. B). FRAP analyses of tonofibrils yielded a t 1/2 of 105 ± 32 min (n = 15), and bleach zones frequently moved at the average rate of 0.05 ± 0.01 μm/min (n = 22). These rates were indistinguishable from controls. Tonofibrils continued to bend and frequently appeared to propagate waveforms (not shown). It was also found that ∼15% (36/237) of keratin squiggles continued to move at the average rate of 0.25 ± 0.18 μm/min with a range of 0.08–0.74 μm/min (n = 36; Fig. 7C and Fig. D). These observations demonstrate that the motile properties of tonofibrils and keratin squiggles in cytochalasin B–treated cells are similar to those observed in untreated cells. Similar analyses of GFP-vimentin fibrils and squiggles were also carried out in cytochalasin B–treated PtK2 cells. Bleached fibrils exhibited slower recovery with a t 1/2 of 9 ± 5 min (n = 28), an increase of ∼33% compared with controls. Bleach zones often moved at the average rate of 0.09 ± 0.07 μm/min (n = 17), representing ∼42% decrease compared with controls. However, ∼43% (135/314) of vimentin squiggles continued to move at the average rate of 3.3 ± 1.6 μm/min (ranging from 0.68 to 6.06 μm/min, n = 25), which was similar to controls.
Figure 7.

(A–D) Time-lapse observations of GFP-K18 are made in a live PtK2 cell after treatment with 20 μM cytochalasin B for 30 min. Cell margins are retracted, and the overall morphology is arborized as seen by phase–contrast (A). Keratin squiggles (B) are present in the arborized regions of the same cell (arrow). (C and D) Two motile keratin squiggles are indicated by arrows. (E–H) PtK2 cells were treated with 6 μM nocodazole for 5 h to disassemble MTs. GFP-K18 tonofibrils show extensive wave-like movements. Elapsed time (min:s) is indicated at the lower left of each confocal image. Bars, 5 μm.
Effects of Nocodazole and Antidynein on the Motility of Tonofibrils and Squiggles
When PtK2 cells were treated with 6 μM nocodazole for 5 h, MT networks were depolymerized as determined by indirect immunofluorescence (data not shown). Under these conditions, networks of tonofibrils appeared relatively normal, whereas the majority but not all of the vimentin fibrils were reorganized into perinuclear aggregates (data not shown; see Osborn et al. 1977, Osborn et al. 1980). Under these conditions, tonofibrils retained their ability to bend and engage in wave-like movements (Fig. 7, E–H). FRAP analyses of tonofibrils yielded a t 1/2 of 113 ± 33 min (n = 13), which was similar to controls. However, the translocation of bleach zones was too slow to measure (using the parameters described in Materials and Methods). There was also a threefold reduction in the number of motile keratin squiggles (31/570 = 5%). The average rate for these squiggles was 0.11 ± 0.05 μm/min with a range of 0.05–0.24 μm/min (n = 31), an ∼54% decrease relative to controls. In the presence of nocodazole, bleached vimentin fibrils recovered their fluorescence more slowly with a t 1/2 of 15 ± 4 min (n = 12), representing an increased time of ∼60%. In addition, bleach zones along vimentin fibrils often moved with an average rate of 0.08 ± 0.06 μm/min (n = 15), which was ∼47% slower relative to controls. The majority (625/630 = 99%) of vimentin squiggles did not move in the presence of nocodazole, indicating a greater dependence on MTs relative to keratin squiggles.
Since there was a significant reduction in the motility of keratin squiggles in the presence of nocodazole, we examined whether keratin squiggles were associated with MTs in the peripheral regions of PtK2 cells. The majority of keratin squiggles did not coalign with MTs, although some appeared to cross over or under MTs, as determined by immunofluorescence (Fig. 8, A–C). Since the majority of keratin squiggles moved towards the cell center, we also attempted to determine whether dynein, an MT minus end–directed motor, was involved. For this purpose, we used an antibody (clone 70.1) directed against the 74-kD intermediate chain of cytoplasmic dynein, which has been shown to block dynein-driven organelle movements in HeLa cells (Burkhardt et al. 1997). After microinjection of this antibody, early endosomes (labeled with antitransferrin receptor) accumulated at the cell periphery in PtK2 cells (Fig. 8 D, arrows), which was similar to the results reported by Burkhardt et al. 1997. Under these conditions, no obvious accumulation of keratin squiggles could be detected at the cell periphery (Fig. 8E and Fig. F, arrows). In PtK2 cells transfected with GFP-K18, time-lapse observations made at 3–4 h after microinjection showed that squiggles continued to move at the average rate of 0.24 ± 0.12 μm/min with a range of 0.1–0.61 μm/min (n = 15; Fig. 8, I–K). These results were similar to those obtained for uninjected cells, suggesting that dynein is not involved in the retrograde movement of keratin squiggles.
Figure 8.
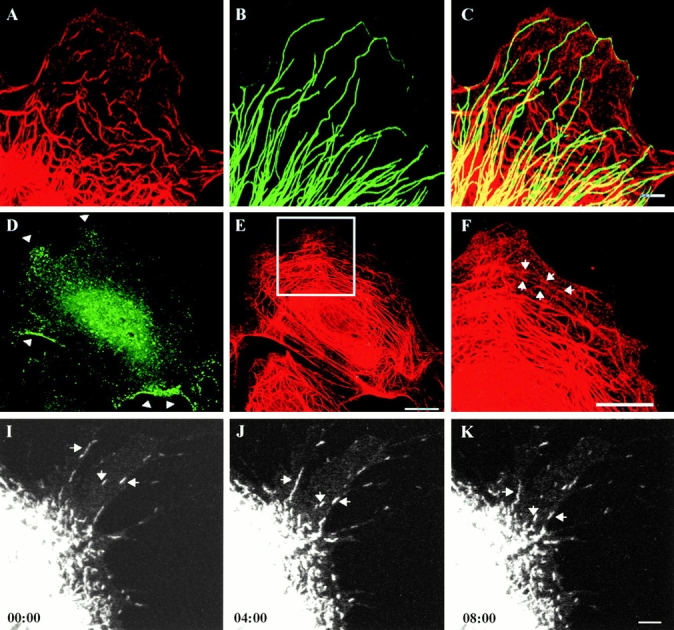
(A–C) The relationship between keratin squiggles and MTs in an untransfected PtK2 cell. Keratin squiggles (A) are visualized with rabbit anti–bovine tongue keratin and MTs (B) with a monoclonal anti–β-tubulin in the peripheral cytoplasm. Overlay (C) shows that keratin squiggles do not coalign with MTs, although a significant number cross over or under the MTs. (D–F) A nontransfected PtK2 cell was processed for double immunofluorescence 3 h after microinjection of antidynein intermediate chain (clone 70.1). The expected accumulation of early endosomes (labeled with antitransferrin receptor) is seen at the cell periphery (D, arrows). Under these conditions, keratin squiggles are visible at the cell periphery in the same region of the same cell (E and F, arrows). Keratin squiggles are stained with anti–bovine tongue keratin. (I–K) GFP-K18 squiggles continue to move in a live PtK2 cell after the microinjection of antidynein intermediate chain. Elapsed time (min:s) is indicated at the lower left of the confocal images. Bars: (A–C and I–K) 5 μm; (D–F) 10 μm.
Further Insights into the Motile Properties of Tonofibrils
Since the bending and wave-like movements of tonofibrils continued in the presence of either cytochalasin B or nocodazole, we also determined whether these movements continued in the presence of both of these inhibitors (described in Materials and Methods). Under these conditions, tonofibrils retained their capacity to bend, straighten, form wave-like configurations, and propagate waveforms (data not shown). These observations suggest that the bending and wave-like movements are independent of both MT and MF systems.
Although the mechanisms underlying these bending and wave-like movements are unknown, it is possible that posttranslational modifications are involved. Since it is well established that IFs are major phosphoproteins of cells (Inagaki et al. 1996), we have initiated studies to determine whether phosphorylation and dephosphorylation might be involved in regulating these types of movements. To this end, PtK2 cells were treated with 0.5 nM calyculin A, a phosphatase inhibitor. Under these conditions, the majority of cells retained normal networks of tonofibrils. However, the phosphorylation levels of both K8 and K18 increased, as determined by phospho-epitope–specific antibodies directed against K8-pSer431 (5B3; Fig. 9, lanes 5 and 6) and K18-pSer33 (IB4; Fig. 9, lanes 7 and 8). Time-lapse observations of GFP-K18 transfected cells after 30 min of calyculin treatment showed that straight tonofibrils can bend and disappear (Fig. 9, B–E, arrows). However, we could not detect the propagation of waveforms along individual tonofibrils (n = 15 cells), indicating that phosphorylation/dephosphorylation plays a role in these movements.
Figure 9.

(A) PtK2 cells were cultured in the absence or presence of calyculin A (0.5 nM for 30 min). Subsequently, IF-enriched cytoskeletal preparations were made, and equal amounts of protein were loaded per lane for SDS-PAGE followed by either staining with Coomassie blue (lanes 1 and 2) or immunoblotting (lanes 3–8). Lanes 1, 3, 5, and 7 are controls, and lanes 2, 4, 6, and 8 are from cells treated with calyculin A. Immunoblotting analyses of the cytoskeletal preparations with the polyclonal antibody R275 (which recognize K8/K18) are shown in lanes 3 and 4. The results of immunoblotting with the monoclonal antibody 5B3 (which recognizes K8-pSer431) are shown in lanes 5 and 6 and with IB4 (which recognizes K18-pSer33) in lanes 7 and 8. Significant increases in K8 and K18 phosphorylation are seen after calyculin A treatment. (B–D) Time-lapse observations of tonofibrils containing GFP-K18 were made after treatment with 0.5 nM calyculin A for 30 min. New bends are formed in straight tonofibrils (arrows). However, propagation of these waveforms could not be detected along the long axes of tonofibrils for periods up to 2 h. Bar, 10 μm.
The bending and wave-like movements of tonofibrils may also be related to their roles in regulating the mechanical properties of epithelial cells. To this end, we investigated the effects of shear stress on tonofibrils in live cells. To investigate whether these movements are influenced by mechanical stress, PtK2 cells were subjected to changes in shear forces, while being observed by confocal microscopy. Time-lapse observations of GFP-K18 showed the deformation of tonofibrils within the first 2–4 min of the onset of shear forces of 15 dyn/cm2 (Fig. 10B, Fig. C, Fig. F, and Fig. G). Without flow, the majority of tonofibrils did not change their configuration over a 2-min interval (Fig. 10A and Fig. E). This was determined by superimposing two color (red and green)-coded images taken 2 min apart (see Helmke et al. 2000). Within 2–4 min after the initiation of flow, the majority of tonofibrils were displaced in the direction of flow (Fig. 10B, Fig. C, Fig. F, and Fig. G). 6 min after flow was induced, no further displacement could be detected (Fig. 10D and Fig. H). In addition to these directional displacements, individual tonofibrils showed bending and waveforms for periods up to 30 min after flow was started (Fig. 10, I–L). These observations imply that networks of tonofibrils might be involved in the initial response to applied mechanical stress but that their shape changes did not appear to be altered under these conditions.
Figure 10.
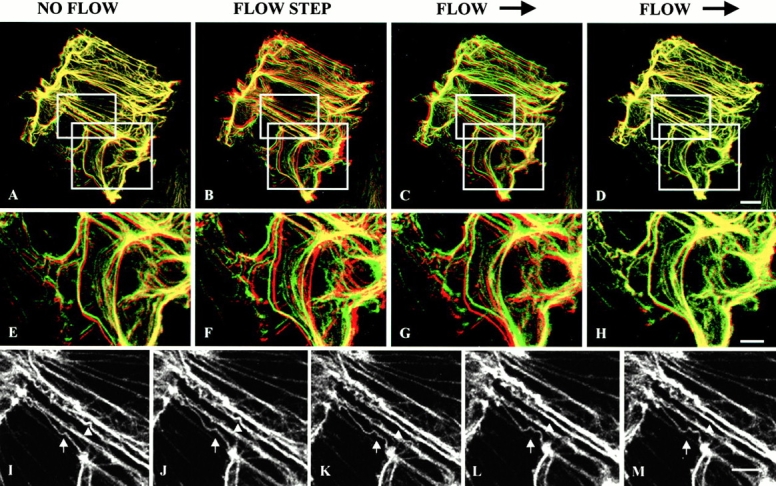
GFP-K18 images are compared before and after a step increase in flow applied to PtK2 cells. Confocal images at 2-min intervals are pseudocolored red and green. Flow direction is left to right. In merged color images, yellow represents zero displacements. (A–D) When live PtK2 cells are subjected to 15 dyn/cm2 shear stress, rapid deformation of networks of tonofibrils are observed during the flow step (B and C) compared with no flow (A) and continued flow (D). (E–H) Higher magnification views show significant directional displacement within 2–4 min of onset of flow. (I–M) Higher magnification views of the same cell also clearly demonstrate that new waveforms can form and existing waveforms can disappear in the presence of flow. Bars: (A–D) 10 μm; (E–M) 5 μm.
Discussion
Previous studies revealed that keratin IF networks are constantly moving in live cells, but the detailed properties of the individual tonofibrils comprising these networks and ultimately responsible for this movement were not analyzed (Windoffer and Leube 1999). In this study, we describe for the first time the properties of individual tonofibrils tagged with GFP-keratin. This has revealed a diverse array of dynamic and motile activities not described previously for the keratin family of proteins.
FRAP analyses of interphase PtK2 cells expressing GFP-keratin demonstrate that there is an equilibrium between exchangeable subunits and the polymerized IFs comprising tonofibrils. Fluorescence intensity measurements during recovery along bleached tonofibrils demonstrate that these are not polarized, since the rate of recovery appears to be the same along entire bleach zones. This can be explained at the molecular level by the antiparallel arrangement of type I/II keratin heterodimers comprising the basic building blocks of keratin IFs (Steinert et al. 1993a,Steinert et al. 1993b, Steinert et al. 1994). Little information exists regarding the possible nature of the exchangeable keratin subunits. However, one candidate may be the small soluble pool of assembly-competent K8/K18 tetramers that have been identified in simple epithelial cells (Chou et al. 1993).
The t 1/2 obtained from FRAP studies of tonofibrils in cells transfected with either GFP-K18 (type I keratin) or GFP-K8 (type II keratin), or both were virtually identical (∼100 min). Therefore, the subunit exchange rates for both types of keratin are the same, which is expected due to the requirement for heterodimers in keratin IF assembly (Fuchs and Weber 1994). In contrast, the t 1/2 for GFP-vimentin is ∼18-fold faster than that of tonofibrils in PtK2 cells (Fig. 2). It is of interest to speculate on the possible mechanisms underlying these dramatically different exchange rates. Previous studies have shown that microinjected type I keratin associates with endogenous type II keratin in seconds (Miller et al. 1993), suggesting that the formation of heterodimers is not a significant rate-limiting step in fluorescence recovery. Thus, recovery depends mainly on the ability of unbleached exchangeable keratin subunits to incorporate into the bleach zones across tonofibrils. This may depend on the number of available assembly sites in tonofibrils. In this regard, the slow fluorescence recovery rates could be related to a limited number of the most readily exchangeable subunits within tonofibrils, and this may be regulated by factors responsible for the packaging of keratin IFs into tonofibrils. For example, filaggrin, an IF-associated protein (IFAP), has been shown to cross-bridge keratin IFs into bundles in terminally differentiated keratinocytes (Dale et al. 1978; Manabe et al. 1991). Although there are no known cross-bridging proteins responsible for the bundling of keratin IFs in simple epithelial cells, such factors are likely to be present, and these could impose significant limitations on subunit exchange rates. On the other hand, vimentin IFs are arranged in loose parallel or criss-crossing arrays, rather than the tightly packed bundles typical for keratin IFs (Franke et al. 1978a,Franke et al. 1978b; Goldman et al. 1986), which may help to explain their faster fluorescence recovery. Another interesting difference between the exchange rates for keratin and vimentin IFs lies in their sensitivity to MT and MF inhibitors. The t 1/2 is significantly increased (∼33–60%; see Yoon et al. 1998) for vimentin fibrils in the presence of nocodazole or cytochalasin B, but this is not the case for keratin tonofibrils. Therefore, it appears that the maintenance of the steady-state exchange between subunits and polymers is somewhat dependent upon MTs and MFs in the case of type III vimentin IFs, but this is not the case for type I/II keratin IFs. However, it is unclear at the present time why these two IF systems are regulated in such different ways.
Analyses of bleach zone translocation demonstrate that neighboring tonofibrils as close as 0.3–1 μm can move slowly in opposite directions, whereas others remain stationary. This emphasizes the independent nature of the movements of individual tonofibrils. Furthermore, these movements are inhibited by nocodazole but not by cytochalasin B, suggesting that tonofibril motility is somewhat dependent on MTs. In support of this, there is ultrastructural evidence suggesting that tonofibrils are associated with MTs (Green and Goldman 1983; Celis et al. 1984), and this association involves IFAPs such as plectin and BPAG1n3 (Svitkina et al. 1996; Fuchs and Yang 1999; Yang et al. 1999). In contrast, movements of vimentin fibrils in PtK2 cells are faster and are inhibited by nocodazole and cytochalasin B, indicating a reliance on both MTs and MFs. Therefore, it is obvious that there are different factors regulating the interactions of these two major types of IF networks with other cytoskeletal systems within the same cell. The specific factors remain largely unknown and therefore represent important targets for future studies.
Tonofibrils frequently exhibit bending movements that often appear as propagated waveforms. In other cases, waveforms disappear, resulting in the straightening of tonofibrils. These movements require energy and continue in the presence of nocodazole and cytochalasin B. Thus, they appear to reflect intrinsic properties of tonofibrils. The mechanisms underlying these bending and wave-like movements are unknown. However, it has been postulated that some of the dynamic properties of IFs are related to the differential removal and/or addition of a subpopulation of readily exchangeable subunits (Stewart 1993; Yoon et al. 1998). In this model, exchangeable and nonexchangeable subunits are distributed at regular intervals along IFs. This could cause local weakenings in the walls of polymerized IFs, which might result in shape changes or waveforms, due to alterations in the interactions between the exchangeable regions and the more stable nonexchangeable regions. Although there is little known about the biochemical basis of IF subunit exchange, there is some evidence suggesting that this process may involve phosphorylation and dephosphorylation (Inagaki et al. 1996). In support of this possibility, the propagation of waveforms along tonofibrils was inhibited in the presence of calyculin A, which increased the phosphorylation state of keratin (Fig. 9). The functional significance of bending or waveform movements is unknown at the present time. However, it is intriguing to speculate that tonofibrils may be capable of propagating waveforms for long distance between the cell and nuclear surfaces. In support of this, ultrastructural studies have demonstrated that tonofibrils form a complex interconnecting network closely associated with both the outer surface of the nuclear envelope and the cell surface, where they are frequently anchored to desmosomes and hemidesmosomes (Goldman et al. 1985, Goldman et al. 1986; Green and Jones 1996). We also show in this study that networks of tonofibrils are rapidly displaced during shear stress, suggesting that they are also involved in the transmission of mechanical forces throughout the cytoplasm (Fig. 10; see Helmke et al. 2000). Therefore, it is conceivable that tonofibrils provide an infrastructure involved in the propagation and transmission of both mechanical and biochemical stimuli throughout the cell.
We also studied the properties of short keratin fibrils or squiggles found primarily near the cell periphery. These squiggles most likely represent early assembly states of tonofibrils (Yoon et al. 1998; Windoffer and Leube 1999). They moved primarily towards the cell center. Remarkably, within these same regions, vimentin squiggles were also seen moving primarily towards the cell membrane. Not only were the majority of the movements for these two types of squiggles opposite to each other, but the keratin squiggles were translocated ∼15 times slower than vimentin squiggles. We have attempted to gain insights into the mechanisms underlying squiggle motility by determining whether these movements are dependent on either MTs or MFs. Keratin squiggle motility was unaltered by cytochalasin B, but in nocodazole there was a significant decrease in their number and rate of movement. This suggests that the translocation of some but not all of the keratin squiggles depend on MTs. On the other hand, the translocation of vimentin squiggles is completely inhibited in the absence of MTs. This is most likely due to a dependence on kinesin, an MT plus end–directed motor (Gyoeva and Gelfand 1991; Prahlad et al. 1998).
There is little information available on the molecular motors responsible for the movements of keratin tonofibrils and squiggles, although it has been shown that the assembly of keratin IFs during Xenopus development is dependent on MTs, possibly through an MT-associated motor (Gard et al. 1997; Gard and Klymkowsky 1998). It is intriguing to speculate that the net movements of keratin squiggles towards the cell center might be related to an MT minus end–directed motor such as dynein (Vallee and Gee 1998). However, in this study keratin squiggles continued to move after microinjection of antidynein intermediate chain, suggesting that dynein is not responsible for the movements of keratin squiggles. There is also the possibility that MT minus end–directed kinesin motors might be involved in the movements of keratin squiggles towards the cell center. These unconventional kinesins have been implicated in moving vesicles towards centrosomes (cell centers), chromosomes to spindle poles, and retrograde axonal transport (Goldstein and Philp 1999).
Supplemental Material
Acknowledgments
We wish to thank Dr. Bishr Omary for providing the K8 and K18 cDNA clones and phospho-epitope–specific keratin antibodies. We also thank Dr. Richard Vallee (University of Massachusetts Medical School, Worcester, MA) for helpful advice for the experiments involving antidynein. We also thank Ms. Laura Davis for assistance in typing this manuscript.
This work has been supported by National Institute of Dental Research (NIDR1 PO1 DE12328).
Footnotes
The online version of this article contains supplemental material.
Drs. K.H. Yoon and M. Yoon contributed equally to this work and should be considered co-first authors.
Abbreviations used in this paper: CFP, cyan fluorescent protein; FRAP, fluorescence recovery after photobleaching; GFP, green fluorescent protein; IF, intermediate filament; IFAP, IF-associated protein; MF, microfilament; MT, microtubule; YFP, yellow fluorescent protein.
References
- Albers K., Fuchs E. The expression of mutant epidermal keratin cDNAs transfected in simple epithelial and squamous cell carcinoma lines. J. Cell Biol. 1987;105:791–806. doi: 10.1083/jcb.105.2.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt J.K., Echeverri C.J., Nilsson T., Vallee R.B. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J. Cell Biol. 1997;139:469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celis J.E., Small J.V., Larsen P.M., Fey S.J., De Mey J., Celis A. Intermediate filaments in monkey kidney TC7 cellsfocal centers and interrelationship with other cytoskeletal systems. Proc. Natl. Acad. Sci. USA. 1984;81:1117–1121. doi: 10.1073/pnas.81.4.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.F., Riopel C.L., Rott L.S., Omary M.B. A significant soluble keratin fraction in ‘simple’ epithelial cells. Lack of an apparent phosphorylation and glycosylation role in keratin solubility. J. Cell Sci. 1993;105:433–444. doi: 10.1242/jcs.105.2.433. [DOI] [PubMed] [Google Scholar]
- Chou Y.H., Opal P., Quinlan R.A., Goldman R.D. The relative roles of specific N- and C-terminal phosphorylation sites in the disassembly of intermediate filament in mitotic BHK-21 cells. J. Cell Sci. 1996;109:817–826. doi: 10.1242/jcs.109.4.817. [DOI] [PubMed] [Google Scholar]
- Coulombe P.A., Bousquet O., Ma L., Yamada S., Wirtz D. The ‘ins’ and ‘outs’ of intermediate filament organization. Trends. Cell Biol. 2000;10:420–428. doi: 10.1016/s0962-8924(00)01828-6. [DOI] [PubMed] [Google Scholar]
- Dale B.A., Holbrook K.A., Steinert P.M. Assembly of stratum corneum basic protein and keratin filaments in macrofibrils. Nature. 1978;276:729–731. doi: 10.1038/276729a0. [DOI] [PubMed] [Google Scholar]
- Evan G.I., Lewis G.K., Ramsay G., Bishop J.M. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke W.W., Grund C., Osborn M., Weber K. The intermediate-sized filaments in rat kangaroo PtK2 cells. I. Morphology in situ Cytobiologie. 17 1978. 365 391a [PubMed] [Google Scholar]
- Franke W.W., Schmid E., Osborn M., Weber K. The intermediate-sized filaments in rat kangaroo PtK2 cells. II. Structure and composition of isolated filaments Cytobiologie. 17 1978. 392 411b [PubMed] [Google Scholar]
- Franke W.W., Schmid E., Grund C., Geiger B. Intermediate filament proteins in non-filamentous structurestransient disintegration and inclusion of subunit proteins in granular aggregates. Cell. 1982;30:103–113. doi: 10.1016/0092-8674(82)90016-2. [DOI] [PubMed] [Google Scholar]
- Fuchs E., Weber K. Intermediate filamentsstructure, dynamics, function and disease. Ann. Rev. Biochem. 1994;63:345–382. doi: 10.1146/annurev.bi.63.070194.002021. [DOI] [PubMed] [Google Scholar]
- Fuchs E., Cleveland D.W. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279:514–519. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- Fuchs E., Yang Y. Crossroads on cytoskeletal highways. Cell. 1999;98:547–550. doi: 10.1016/s0092-8674(00)80041-0. [DOI] [PubMed] [Google Scholar]
- Gard D.L., Klymkowsky M.W. Intermediate filament organization during oogenesis and early development in the clawed frog, Xenopus laevis . Subcell. Biochem. 1998;31:35–70. [PubMed] [Google Scholar]
- Gard D.L., Cha B.J., King E. The organization and animal-vegetal asymmetry of cytokeratin filaments in stage VI Xenopus oocytes is dependent upon F-actin and microtubules. Dev. Biol. 1997;184:95–114. doi: 10.1006/dbio.1997.8508. [DOI] [PubMed] [Google Scholar]
- Goldman R., Goldman A., Green K., Jones J., Lieska N., Yang H.Y. Intermediate filamentspossible functions as cytoskeletal connecting links between the nucleus and the cell surface. Ann. NY Acad. Sci. 1985;455:1–17. doi: 10.1111/j.1749-6632.1985.tb50400.x. [DOI] [PubMed] [Google Scholar]
- Goldman R.D., Goldman A.E., Green K.J., Jones J.C.R., Jones S.M., Yang H.-Y. Intermediate filament networksorganization and possible functions of a diverse group of cytoskeletal elements. J. Cell Sci. Suppl. 1986;5:69–97. doi: 10.1242/jcs.1986.supplement_5.5. [DOI] [PubMed] [Google Scholar]
- Goldman R.D., Khuon S., Chou Y.H., Opal P., Steinert P.M. The function of intermediate filaments in cell shape and cytoskeletal integrity. J. Cell Biol. 1996;134:971–983. doi: 10.1083/jcb.134.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein L.S., Philp A.V. The road less traveledemerging principles of kinesin motor utilization. Ann. Rev. Cell Dev. Biol. 1999;15:141–183. doi: 10.1146/annurev.cellbio.15.1.141. [DOI] [PubMed] [Google Scholar]
- Green K.J., Goldman R.D. The effects of taxol on cytoskeletal components in cultured fibroblasts and epithelial cells. Cell Motil. 1983;3:283–305. doi: 10.1002/cm.970030402. [DOI] [PubMed] [Google Scholar]
- Green K.J., Jones J.C.R. Desmosomes and hemidesmosomesstructure and function of molecular components. FASEB J. 1996;10:871–881. doi: 10.1096/fasebj.10.8.8666164. [DOI] [PubMed] [Google Scholar]
- Gyoeva F.K., Gelfand V.I. Coalignment of vimentin intermediate filaments with microtubules depends on kinesin. Nature. 1991;353:445–448. doi: 10.1038/353445a0. [DOI] [PubMed] [Google Scholar]
- Helmke B.P., Goldman R.D., Davis P.F. Rapid displacement of vimentin intermediate filaments in living endothelial cells exposed to flow. Circ. Res. 2000;86:745–752. doi: 10.1161/01.res.86.7.745. [DOI] [PubMed] [Google Scholar]
- Herrmann H., Aebi U. Intermediate filaments and their associatesmulti-talented structural elements specifying cytoarchitecture and cytodynamics. Curr. Opin. Cell Biol. 2000;12:79–90. doi: 10.1016/s0955-0674(99)00060-5. [DOI] [PubMed] [Google Scholar]
- Ho C.L., Martys J.L., Mikhailov A., Gundersen G.G., Liem R.K. Novel features of intermediate filament dynamics revealed by green fluorescent protein chimeras. J. Cell Sci. 1998;111:1767–1778. doi: 10.1242/jcs.111.13.1767. [DOI] [PubMed] [Google Scholar]
- Hofmann I., Franke W.W. Heterotypic interactions and filament assembly of type I and type II cytokeratins in vitroviscometry and determinations of relative affinities. Eur. J. Cell Biol. 1997;72:122–132. [PubMed] [Google Scholar]
- Huang S., Deerinck T.J., Ellisman M.H., Spector D.L. The perinucleolar compartment and transcription. J. Cell Biol. 1998;143:35–47. doi: 10.1083/jcb.143.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki M., Matsuoka Y., Tsujimura K., Ando S., Tokui T., Takahashi T., Inagaki N. Dynamic property of intermediate filamentsregulation by phosphorylation. Bioessay. 1996;18:481–487. [Google Scholar]
- Jones S.M., Jones J.C.R., Goldman R.D. Fractionation of desmosomes and comparison of the polypeptide composition of desmosomes prepared from two bovine epithelial tissues. J. Cell Biochem. 1988;36:223–236. doi: 10.1002/jcb.240360304. [DOI] [PubMed] [Google Scholar]
- Ku N.O., Omary M.B. Phosphorylation of human keratin 8 in vivo at conserved head domain serine 23 and at epidermal growth factor-stimulated tail domain serine 431. J. Biol Chem. 1997;272:7556–7564. doi: 10.1074/jbc.272.11.7556. [DOI] [PubMed] [Google Scholar]
- Ku N.O., Omary M.B. Phosphorylation of human keratin 18 serine 33 regulates binding to 14-3-3 proteins. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1892–1906. doi: 10.1093/emboj/17.7.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N.O., Michie S., Oshima R.G., Omary M.B. Chronic hepatitis, hepatocyte fragility, and increased soluble phosphoglycokeratins in transgenic mice expressing a keratin 18 conserved arginine mutant. J. Cell Biol. 1995;131:1303–1314. doi: 10.1083/jcb.131.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lane E.B., Goodman S.L., Trejdosiewicz L.K. Disruption of the keratin filament network during epithelial cell division. EMBO (Eur. Mol. Biol. Organ.) J. 1982;1:1365–1372. doi: 10.1002/j.1460-2075.1982.tb01324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe M., Sanchez M., Sun T.T., Dale B.A. Interaction of filaggrin with keratin filaments during advanced stages of normal human epidermal differentiation and in ichthyosis vulgaris. Differentiation. 1991;48:43–50. doi: 10.1111/j.1432-0436.1991.tb00241.x. [DOI] [PubMed] [Google Scholar]
- Miller R.K., Vikstrom K., Goldman R.D. Keratin incorporation into intermediate filament networks is a rapid process. J. Cell Biol. 1991;113:843–855. doi: 10.1083/jcb.113.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R.K., Khuon S., Goldman R.D. Dynamics of keratin assemblyexogenous type I keratin rapidly associates with type II keratin in vivo. J. Cell Biol. 1993;122:123–135. doi: 10.1083/jcb.122.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn M., Franke W.W., Weber K. Visualization of a system of filaments 7-10-nm thick in cultured cells of an epithelioid line (Pt K2) by immunofluorescence microscopy. Proc. Natl. Acad. Sci. USA. 1977;74:2490–2494. doi: 10.1073/pnas.74.6.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn M., Franke W., Weber K. Direct demonstration of the presence of two immunologically distinct intermediate-sized filament systems in the same cell by double immunofluorescence microscopy. Vimentin and cytokeratin fibers in cultured epithelial cells. Exp. Cell Res. 1980;125:37–46. doi: 10.1016/0014-4827(80)90186-x. [DOI] [PubMed] [Google Scholar]
- Oshima R.G., Millan J.L., Cecena G. Comparison of mouse and human keratin 18a component of intermediate filaments expressed prior to implantation. Differentiation. 1986;33:61–68. doi: 10.1111/j.1432-0436.1986.tb00411.x. [DOI] [PubMed] [Google Scholar]
- Prahlad V., Yoon M., Moir R.D., Vale R.D., Goldman R.D. Rapid movements of vimentin on microtubule trackskinesin-dependent assembly of intermediate filament networks. J. Cell Biol. 1998;143:159–170. doi: 10.1083/jcb.143.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Molecular CloningA Laboratory Manual. Cold Spring Harbor Laboratory Press 1989. Cold Spring Harbor, ; New York: pp. 1659 pp [Google Scholar]
- Steinert P.M., Marekov L.N., Fraser R.D., Parry D.A.D. Keratin intermediate filament structure. Crosslinking studies yield quantitative information on molecular dimensions and mechanism of assembly J. Mol. Biol. 230 1993. 436 452a [DOI] [PubMed] [Google Scholar]
- Steinert P.M., Marekov L.N., Parry D.A.D. Diversity of intermediate filament structure. Evidence that the alignment of coiled-coil molecules in vimentin is different from that in keratin intermediate filaments J. Biol. Chem. 268 1993. 24916 24925b [PubMed] [Google Scholar]
- Steinert P.M., North A.C., Parry D.A.D. Structural features of keratin intermediate filaments. J. Invest. Dermatol. 1994;103:19S–24S. doi: 10.1111/1523-1747.ep12398900. [DOI] [PubMed] [Google Scholar]
- Stewart M. Intermediate filament structure and assembly. Curr. Opin. Cell Biol. 1993;5:3–11. doi: 10.1016/s0955-0674(05)80002-x. [DOI] [PubMed] [Google Scholar]
- Svitkina T.M., Verkhovsky A.B., Borisy G.G. Plectin sidearms mediate interactions of intermediate filaments with microtubules and other components of the cytoskeleton. J. Cell Biol. 1996;135:991–1007. doi: 10.1083/jcb.135.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H., Staehelin T., Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheetsprocedure and some applications. Proc. Natl. Acad. Sci. USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee R.B., Gee M.A. Make room for dynein. Trends Cell Biol. 1998;8:490–494. doi: 10.1016/s0962-8924(98)01379-8. [DOI] [PubMed] [Google Scholar]
- Wang L., Ho C.-L., Sun D., Liem R.K.H., Brown A. Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat. Cell Biol. 2000;2:137–141. doi: 10.1038/35004008. [DOI] [PubMed] [Google Scholar]
- Windoffer R., Leube R.E. Detection of cytokeratin dynamics by time-lapse fluorescence microscopy in living cells. J. Cell Sci. 1999;112:4521–4534. doi: 10.1242/jcs.112.24.4521. [DOI] [PubMed] [Google Scholar]
- Yang Y., Bauer C., Strasser G., Wollman R., Julien J.P., Fuchs E. Integrators of the cytoskeleton that stabilize microtubules. Cell. 1999;98:229–238. doi: 10.1016/s0092-8674(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Yoon M., Moir R.D., Prahlad V., Goldman R.D. Motile properties of vimentin intermediate filament networks in living cells. J. Cell Biol. 1998;143:147–157. doi: 10.1083/jcb.143.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zackroff R.V., Goldman R.D. In vitro assembly of intermediate filaments from baby hamster kidney (BHK-21) cells. Proc. Natl. Acad. Sci. USA. 1979;76:6226–6230. doi: 10.1073/pnas.76.12.6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.