

Abstract
The role of integrins in leukocyte apoptosis is unclear, some studies suggest enhancement, others inhibition. We have found that β2-integrin engagement on neutrophils can either inhibit or enhance apoptosis depending on the activation state of the integrin and the presence of proapoptotic stimuli. Both clustering and activation of αMβ2 delays spontaneous, or unstimulated, apoptosis, maintains mitochondrial membrane potential, and prevents cytochrome c release. In contrast, in the presence of proapoptotic stimuli, such as Fas ligation, TNFα, or UV irradiation, ligation of active αMβ2 resulted in enhanced mitochondrial changes and apoptosis. Clustering of inactive integrins did not show this proapoptotic effect and continued to inhibit apoptosis. This discrepancy was attributed to differential signaling in response to integrin clustering versus activation. Clustered, inactive αMβ2 was capable of stimulating the kinases ERK and Akt. Activated αMβ2 stimulated Akt, but not ERK. When proapoptotic stimuli were combined with either αMβ2 clustering or activation, Akt activity was blocked, allowing integrin activation to enhance apoptosis. Clustered, inactive αMβ2 continued to inhibit stimulated apoptosis due to maintained ERK activity. Therefore, β2-integrin engagement can both delay and enhance apoptosis in the same cell, suggesting that integrins can play a dual role in the apoptotic progression of leukocytes.
Keywords: apoptosis, beta2 integrin, mitochondria, cytochrome c, neutrophil
Introduction
Neutrophils have a short lifespan both in vivo and in vitro, undergoing spontaneous apoptosis with up to 80% death by 16–20 h of in vitro culture (Savill et al. 1989). This process is characterized by cell rounding, nuclear condensation, internucleosomal DNA fragmentation, and exposure of phosphatidylserine on the cell surface. Neutrophil apoptosis can be accelerated by various stimuli, such as UV irradiation, Fas, or tumor necrosis factor α (TNFα) receptor ligation (Takeda et al. 1993; Liles et al. 1996; Frasch et al. 1998). The signaling that drives spontaneous and accelerated apoptosis in neutrophils appears to be similar to other cell types, in that it involves effector caspases (Frasch et al. 1998), and may be modulated by the Bcl-2 family members A1, Mcl-1, and Bcl-xL (Chuang et al. 1998; Moulding et al. 1998; Weinmann et al. 1999), although a role for mitochondria has yet to be established. It is likely that in vivo, neutrophil apoptosis is important in the homeostatic control of circulating neutrophil numbers and in the resolution of acute inflammation. In this latter situation, engulfment of apoptotic neutrophils by phagocytes both eliminates potentially damaging proinflammatory substances and stimulates the production of antiinflammatory mediators (Fadok et al. 1998). Therefore, an understanding of factors modulating neutrophil apoptosis is critical to elucidating the regulation of inflammatory processes.
β2-integrins are important molecules for neutrophil function playing roles in cell activation, homing, and phagocytosis. In constitutively adherent cells, integrins play a critical role in supporting growth and survival (reviewed in Giancotti and Ruoslahti 1999). Some evidence exists to support a similar role for integrins in leukocytes. Antibody cross-linking of the αM integrin chain on neutrophils was shown to inhibit spontaneous and Fas-mediated apoptosis (Watson et al. 1997a,Watson et al. 1997b) and activated endothelial cells inhibited neutrophil apoptosis in an ICAM-dependent manner (Ginis and Faller 1997). In addition, engagement of β7 integrins was shown to enhance the survival of eosinophils (Meerschaert et al. 1999). In conflict with this, however, are reports that β2-integrins are involved in enhancing or stimulating apoptosis. The cell death of thioglycollate-elicited peritoneal neutrophils from αM integrin chain deficient mice was delayed in vitro relative to control mice (Coxon et al. 1996) and cross-linking of β2-integrins with antibody, or adhesion to components of the extracellular matrix has been shown to enhance TNFα-induced apoptosis (Walzog et al. 1997; Kettritz et al. 1999). Based on the apparent contradiction evident in these studies, a role could be proposed for β2-integrins either in accelerating or delaying apoptosis. Therefore, the objective of the current study was to elucidate the role of β2-integrins in regulating apoptosis in controlled in vitro conditions with or without integrin activation and/or proapoptotic stimuli, and to determine the signaling pathways involved in these responses.
The β2-integrin family consists of three αβ dimers: αLβ2 (LFA-1), αMβ2 (Mac-1), or αXβ2 (p150,95). β2-integrins are capable of interacting with a diverse array of ligands, but exist predominantly in an inactive, low affinity state (Diamond and Springer 1993). However, when activated, they increase their affinity for multivalent ligands, which leads to stable cell–cell or cell–matrix adhesion. It is evident that leukocyte integrins, like their anchorage-dependent counterparts, are capable of activating complex signaling networks upon engagement. The initial link between integrins and downstream signaling events appears to be provided by several protein tyrosine kinases, including members of the Src, Syk, and focal adhesion kinase families (Lowell and Berton 1999). Recruitment of adapter molecules and kinases leads to the recruitment and activation of phosphatidylinositol-3 kinase (PI-3K; Meng and Lowell 1998) and Ras/Raf/mitogen-activated protein kinase (MAPK) pathways (McGilvray et al. 1998). The adhesion-dependent activation of these pathways is important because they contribute to optimal cell activation, including increased respiratory burst and cytokine expression (Nathan et al. 1989; Walzog et al. 1999).
The integrin-mediated downstream signals responsible for survival in anchorage-dependent cells are similar or identical to those used by growth factor receptors. Two kinase pathways that have received considerable attention are the PI-3K/Akt and extracellular signal-regulated MAPK (ERK) pathways. Both Akt and ERK are serine/threonine kinases that are invoked by numerous growth and survival factors. It may be possible, however, to override these antiapoptotic stimuli by potential proapoptotic insults. PI-3K and Akt activity is dramatically inhibited in fibroblasts exposed to stress stimuli, an effect attributed to the generation of ceramide (Zundel and Giaccia 1998). In addition, insulin-activated PI-3K and ERK are inhibited by oxidative stress (Hansen et al. 1999; Tirosh et al. 1999). This type of switch-off mechanism could affect integrin-mediated survival signals and potentially explain the apparent contradiction in the role of β2-integrins in leukocyte apoptosis, wherein the survival outcome of integrin engagement may depend not only on the degree and type of integrin engagement, but also on the presence or absence of proapoptotic factors.
Our data demonstrate different cellular responses upon clustering of inactive β2-integrins vs. activation of β2-integrins on neutrophils. We show that either αMβ2 clustering or activation is differentially capable of activating Akt and/or MAPK-driven signaling pathways that prevent mitochondrial changes and inhibit spontaneous apoptosis. However, β2-integrin activation, but not clustering, can enhance apoptosis stimulated by Fas ligation, TNFα, or UV irradiation. This combination of stimuli (integrin activation and proapoptotic stimuli) shifts the balance in the cell from an antiapoptotic to a proapoptotic posture by eliminating both Akt activation and mitochondrial protection. In contrast, αMβ2 clustering, in the absence of activation, continues to inhibit apoptosis in the presence of proapoptotic stimuli due to the activation of MAPK-driven signals.
Therefore, we propose a novel model, whereby the state of integrin activation and the environment in which the cell resides results in different survival outcomes. Clustered αMβ2, in the absence of integrin activation, is universally antiapoptotic either during spontaneous or stimulated apoptotic progression. Downstream signals from activated β2-integrins are also potentially antiapoptotic, however, in the presence of death stimuli, these survival signals are abrogated leading to enhanced cell death.
Materials and Methods
Antibodies and other Reagents
The following mAbs were obtained from commercial sources and were confirmed to bind to human PMN by flow cytometry. Anti-αM 2LPM19c, anti-αX KB90, and anti-HLA-ABC W6/32 were obtained from DAKO. Anti-αM Mo1 was obtained from Coulter Immunotech. Anti-αL MEM25, anti-αM VIM12, and anti-β2 CLB-LFA1/1 were obtained from Caltag. Anti-β1 MAR4, anti-β3 VI-PL2, and anti-CD45 HI30 were obtained from BD PharMingen. Unless specified otherwise, mAbs were used at a final concentration of 1 μg/ml. All antibodies were tested as dialyzed preparations to rule out nonspecific effects of commercial solutions. Formyl-methionine-leucine-phenylalanine (fMLP) was obtained from Sigma-Aldrich and used at a final concentration of 10−7 M. TNFα was obtained from R&D and was used at a final concentration of 1,000 U/ml. Anti-Fas IgM clone CH11 was obtained from Upstate and used at a concentration of 0.5 or 1 μg/ml to induce apoptosis. Inhibitors of MEK (PD98059, 2′-amino-3′methoxyflavone and UO126, 1,2-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene) and PI-3K (wortmannin and LY294002, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one) were obtained from Calbiochem.
rhICAM-1–coated Bead Preparation
Sulfate beads (1 μm; Molecular Probes) were washed in PBS and incubated with 10 μg of soluble recombinant human intracellular adhesion molecule 1 (rhICAM-1) in 0.9% saline with 20 mM Tris-HCL, pH 7.4, for 30 min at room temperature. We used rhICAM from two different sources (R&D and Alexa) with identical results. Beads were sonicated for 5 min, and resuspended in HBSS with 10 mg/ml LPS-free human serum albumin (HSA) for 30 min at room temperature. Beads were washed in HBSS and then resuspended in RPMI plus 1% BSA. This was added to PMN to give an approximate final concentration of 1 μg/ml rhICAM-1. Beads coated with HSA alone were used as controls in some experiments without effect.
Neutrophil Isolation
Human neutrophils were purified from whole blood as described previously (Haslett et al. 1985). Neutrophil purity was >95% with typically <2% eosinophils.
Assessment of Apoptosis in Suspension Neutrophils
After isolation neutrophils were suspended at 2 × 106 cells/ml in RPMI supplemented with 1% LPS-free BSA (Sigma-Aldrich, A-1933) and incubated in conical polypropylene tubes (Sarstedt). Neutrophils, with or without β2-integrin reagents, were either allowed to spontaneously apoptose over 10 h or were exposed to anti-Fas or TNFα for 4 h. Experiments carried out with PI-3K and MEK inhibitors used a 30-min preincubation before addition of stimuli. After the requisite incubation 2 × 105 cells were diluted and cytocentrifuged at 600 rpm for 3 min on to glass slides and stained with a modified Wright-Giemsa stain. Cells were counted as apoptotic if their nuclei lost segmentation and appeared round and darkly stained. At least 200 cells were counted per slide in a blinded fashion. The accuracy of morphological assessment was checked periodically by flow cytometric analysis of hypodiploid DNA in propidium iodide stained neutrophils. Apoptotic percentages obtained by these two methods agreed within 5%.
Assessment of Apoptosis in Adherent Neutrophils
5,000,000 cells were suspended in 1 ml Ca2+/Mg2+-free Krebs-Ringer phosphate buffer, pH 7.2, containing 0.2% dextrose (KRPD) plus 0.25% HSA and labeled with a 111-Indium (111In; New England Nuclear) labeling mix consisting of KRPD/HSA and 4 × 10−3 M tropolonate. After a 5 min incubation at room temperature, cells were washed with KRPD/HSA and resuspended at 6 × 104 cells/ml in RPMI plus 1% BSA. This labeling procedure had no discernible effect on apoptotic progression. Labeled neutrophils were added to culture tubes or plated onto 15-mm glass coverslips coated overnight with 2.5 mg/ml fibrinogen (Calbiochem). Triplicate samples for each condition were set up to determine the total amount of radioactivity present. After a 4–6 h incubation, cells were gently fixed with dropwise addition of 0.5% glutaraldehyde. Cells were fixed for 30 min at 37°C or overnight at 4°C. Nonadherent cells were removed, washed, and cytocentrifuged. Cells in suspension were treated identically. Cytospin preparations and coverslips (adherent cells) were stained with 2 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Calbiochem) in 50% methanol/PBS for 20 min. The slides and coverslips were washed gently in PBS and coverslipped or mounted using o-phenylenediamine. Cells were examined for apoptotic morphology by fluorescence microscopy with UV excitation, with 200–210 cells counted per slide. The overall percentage of apoptotic cells was determined by multiplying the number of normal and apoptotic cells by the percentage of nonadherent versus adherent cells measured as radioactivity present in replicate wells. These adjusted numbers were then added to determine an overall percentage of normal and apoptotic cells.
Determination of Integrin Clustering or Activation
For visualization of αM clustering, PMN were incubated with the mAbs 2LPM19 or VIM12 at 4 or 37°C for 45 min as above, centrifuged at 1,000 g at 4°C, and resuspended in isotonic 2% paraformaldehyde/sucrose/PBS. After a 20 min fixation and washing with KRPD/HSA, cells were incubated with 1:200 Cy3-conjugated goat anti–mouse (Jackson ImmunoResearch) for 30 min on ice, washed, resuspended in 5–10 μl, mounted onto glass slides with Gel Mount (Biomeda), and coverslipped. Stained cells were examined with a 63× water lens on a Vanox-T Olympus microscope. For αM activation, stimulated PMN were incubated 1 h on ice with 1.5 μg biotinylated CBRM1/5 (biotinylation was carried out using a commercial kit (Pierce Chemical Co.) or biotinylated anti-CD45 (BD PharMingen). Cells were washed and counterstained with 1:50 phycoerythrin-labeled streptavidin. Cells were analyzed by flow cytometry using FACscalibur (Becton Dickinson). Total β2-integrin levels were measured on the same cells using FITC-labeled anti-CD18 (Caltag).
Fab and F(ab′)2 Antibody Preparation
Digestion of the anti-αM clone 2LPM19c and the anti-HLA-ABC clone W6/32 was accomplished using kits according to manufacturers instructions (Pierce Chemical Co,). Fab fragments were produced at 37°C using an immobilized pepsin slurry. F(ab′)2 fragments were produced at 37°C using immobilized Ficin columns. Optimal digestion times were 12 h for Fab and 15 h for F(ab′)2 fragments. Fab and F(ab′)2 fragments were purified with protein A and fractions collected, assayed for protein content by absorbence at 280 nm, pooled, and dialyzed extensively against PBS over a period of 24–36 h. The integrity of the fragments was assessed by nonreducing SDS-PAGE and silver stain, as well as Western blotting with HRP-labeled anti-mouse antibody. Bands of ∼110 kD and 50-kD bands were seen corresponding to F(ab′)2 and Fab fragments, respectively. Neutrophils were stained with antibody fragments to confirm binding, and in addition, 2LPM19c fragments were tested for their ability to block fMLP-induced adhesion to confirm functionality of these fragments. All 2LPM19c fragments used in this study were able to inhibit adhesion as well as whole antibody.
ERK Immunoprecipitation and Kinase Assay
Lysates from isolated PMN, incubated with the appropriate stimulus at 37°C, were assayed for ERK activity using a enzyme activity reagent kit (Upstate) with slight modification of the provided protocol. After stimulation, 7.5 × 106 cells were centrifuged at 4°C and lysed with ice cold RIPA buffer (supplemented with 15 μg/ml leupeptin and aprotinin, 1 mM PMSF and 0.2 mM sodium orthovanadate). Lysates were centrifuged at 12,000 rpm at 4°C for 10 min. Supernatants were transferred to protein A–Sepharose (Zymed) beads containing 1 μg of rabbit anti-ERK2 antibody (Santa Cruz) and incubated for 2 h at 4°C with rotation. After incubation, beads were washed twice with cold lysis buffer and once with assay buffer (Upstate). Beads were resuspended in 50 μl assay buffer containing myelin basic protein, inhibitor cocktail, 1 μCi γ[32P]ATP (Amersham Pharmacia) and Mg/ATP cocktail. Beads were incubated for 10 min at 30°C with agitation. A portion of the reaction mix was transferred to P81 phosphocellulose paper, washed three times with 0.75% phosphoric acid, and analyzed by liquid scintillation counting.
Akt Immunoprecipitation and Kinase Assay
As with the ERK assays, Akt activity was measured using a enzyme activity kit (Upstate) with some modification of the provided protocol. PMN were lysed as in ERK assays. Supernatants were immunoprecipitated with protein–G Sepharose (Zymed) and 1 μg rabbit anti-Akt antibody (Santa Cruz). A purified rabbit polyclonal antibody to the unrelated kinase Rsk1 (Santa Cruz) was used as a control for specific immunoprecipitation of both Akt and ERK activity. Beads were incubated for 2 h at 4°C with rotation. Beads were washed as above and resuspended in 40 μl assay buffer containing substrate peptide (RPRAATF), protein kinase A inhibitor, 1 μCi γ[32P]ATP (1 μCi; Amersham Pharamcia), and Mg/ATP cocktail. Samples were incubated for 10 min at 30°C with agitation, centrifuged, and supernatants transferred to 20 μl 40% TCA, mixed, and incubated for 5 min at room temperature. A portion of the reaction mix was transferred to P81 phosphocellulose paper, washed as above and analyzed by liquid scintillation counting. Optimal activation times for Akt and ERK were determined for all appropriate stimuli.
Staining of Mitochondria
After stimulation, PMN were incubated with 10 μM JC-1 (Molecular Probes) at 37°C for 10 min, centrifuged, resuspended in cold PBS, and analyzed by flow cytometry with an excitation wavelength of 480 nm and 585/42-nm emission filter. Decreases in mean fluorescence intensity (FL-2) correspond to a loss of fluorescent JC-1 J aggregates in the mitochondria that occurs upon decreased mitochondrial membrane potential (Cossarizza et al. 1993). For fluorescence microscopy, cells were stained as above, washed, resuspended, and gel mounted on to glass slides. MAB1273 (Chemicon) staining was carried out in permeabilized cells (0.1% Tween20 in PBS/BSA) with MAB1273 for 30 min at 37°C, followed by washing (twice with PBS/BSA) and detection with 1:1,000 Cy3-conjugated F(ab′)2 anti-mouse IgG (Jackson ImmunoResearch). Cells were washed, mounted, and visualized as above.
Cytochrome c (CytC) Immunostaining
Human PMN were incubated with stimuli for 1 or 1.5 h and CytC staining was performed as described elsewhere (Kennedy et al. 1999). The specificity of CytC localization was determined using mitotracker red CXC (Mito trk; Molecular Probes) as a costain. After fixation and CytC staining, cells were incubated with 1 μM Mito trk for 10 min, washed (twice) and mounted. Stained cells were examined as above using the FITC (CytC) and Cy3 filters (Mito trk).
Data Analysis
Averages and SD values were calculated from at least three experiments. Statistical analysis was carried out using the JMP statistical program (SAS Institute, Cary, NC). The Tukey-Kramer and Dunnett's parametrical tests were used for single and multiple comparisons, respectively. Parametric tests were determined to be appropriate based on apparent normal distribution and equal variances (O'Brein test) of apoptosis data.
Results
Clustering or Activation of β2-Integrins Can Inhibit Neutrophil Apoptosis
Clustering of integrins in cell membranes contributes to the activation of survival signals (Giancotti and Ruoslahti 1999). Therefore, our initial objective in this study was to determine if αMβ2 clustering on neutrophils by direct engagement could modulate apoptosis. The experiments were performed under defined conditions with cells incubated in suspension, thus minimizing surface interaction. In this circumstance, integrin engagement with antibodies to either the αM or β2 subunits delayed the apoptosis of human neutrophils (Fig. 1 A). Anti-αM antibodies (2LPM19c, VIM12, and Mo1), an anti-β2 antibody, and an anti-αL antibody, all inhibited neutrophil apoptosis by 30–60% at 10 h. In contrast, antibody to αx, a component of the β2-integrin p150,95, was largely ineffective, as were antibodies to β1 and β3 integrin chains. Antibodies to the nonintegrin surface molecules HLA and CD45 were also ineffective. A F(ab′)2 fragment of 2LPM19c (hereafter referred to as 19c) was very effective at inhibiting neutrophil apoptosis indicating lack of FcγR involvement (Fig. 1 B). A monovalent Fab fragment of the same antibody had no effect, demonstrating that the apoptosis inhibition seen with 19c was not due to blocking cell–cell proapoptotic adhesive signals (19c Fab and F(ab′)2 were equally efficient at inhibiting adhesion), but rather a direct result of bivalent antibody engagement and β2-integrin clustering or cross-linking (Fig. 1 B). β2-integrin–mediated survival was not limited to antibody engagement as incubation of neutrophils with rhICAM-1, a natural ligand for β2-integrins, immobilized on 1 μm beads also suppressed apoptosis (Fig. 2). This suppression was due to ICAM-αMβ2 interactions since blocking anti-αM Fab (itself noninhibitory) completely reversed this suppression (Fig. 2). Like antibody-mediated inhibition, ICAM-induced inhibition also required multivalent binding and clustering since soluble rhICAM-1, which exists largely in monomeric form (Reilly et al. 1995), had no effect on apoptosis (data not shown). The inhibitory effect of αMβ2 clustering was still evident late in culture (20 h), suggesting the induction of long-term survival signals in neutrophils (Fig. 1 C).
Figure 1.
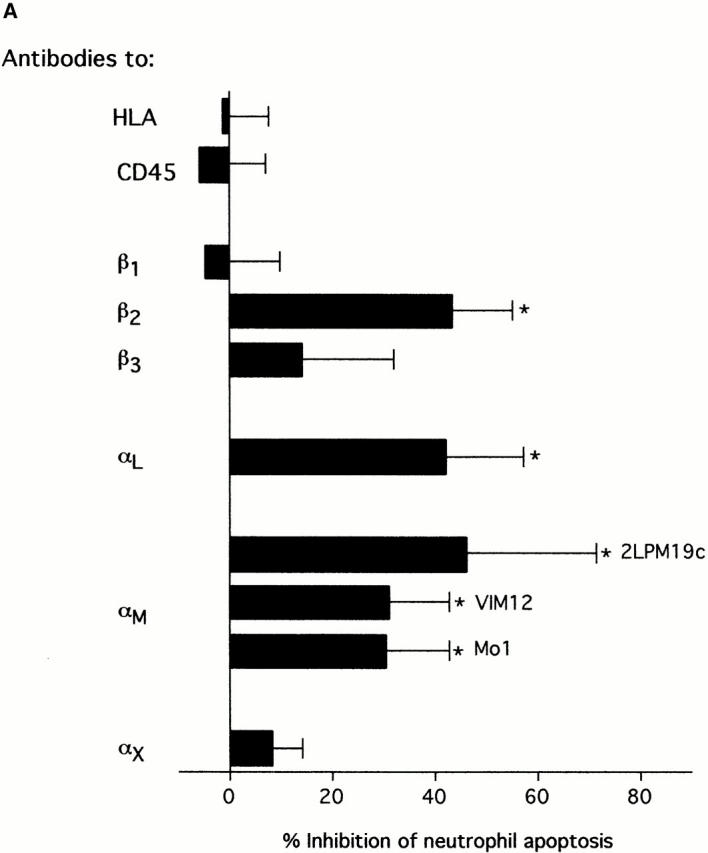
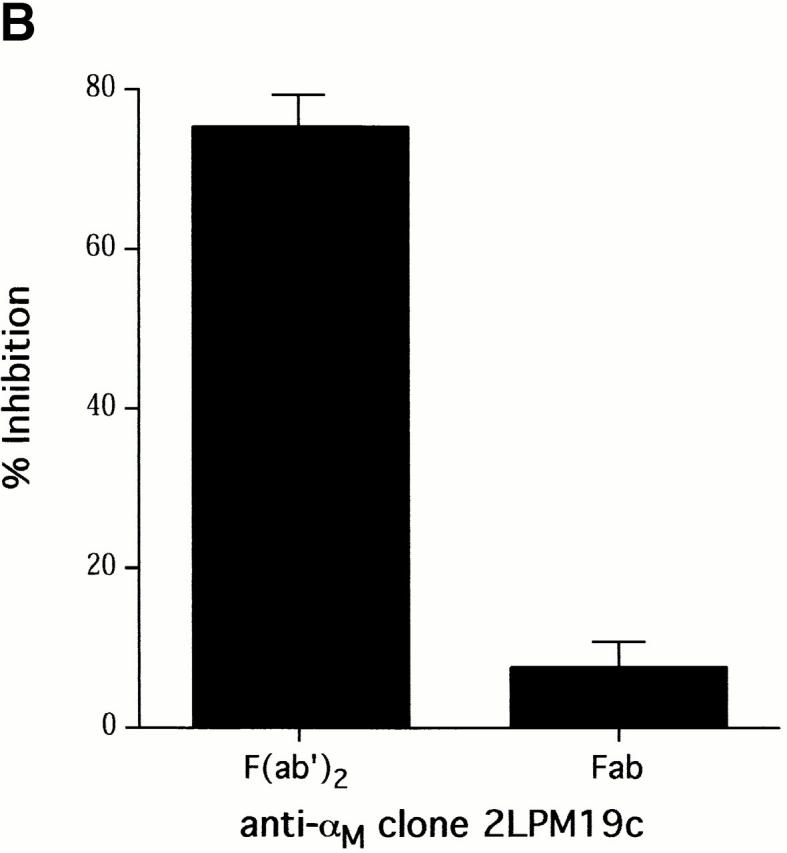

Inhibition of spontaneous neutrophil apoptosis by blocking or activating antibodies to β2-integrins. (A) Inhibition of apoptosis was specific for antibodies to αL, αM and β2-integrin chains. Neutrophils (2 × 106 cells/ml) were incubated at 37°C in polypropylene tubes with 1 μg/ml of antibody and apoptosis assessed by cellular morphology at 10 h. The data represent mean percent inhibition of apoptosis ± SD of four experiments. The average control apoptosis at 10 h was 39 ± 11% and the means of all antibody treatments were compared with this value using the multiple comparison Dunnett's Test (*P < 0.05). (B) F(ab′)2 fragments, but not monovalent Fab, of anti-αM mAb 2LPM19c inhibited apoptosis. Mean percent inhibition ± SD of three experiments. (C) Inhibition time course of blocking anti-αM clone (2LPM19c) or activating anti-αM clone (VIM12). Anti-CD45 (HI30) is shown as a negative control. Neutrophils were treated with 1 μg/ml of antibody and apoptosis determined at 2, 10, and 20 h. Graph is representative of five experiments.
Figure 2.

Inhibition of neutrophil apoptosis after β2-integrin activation or clustering is reversed by anti-αM Fab. β2-integrins were either activated with Mn2+, activating antibody (VIM12), or fMLP or clustered with immobilized rhICAM. In all cases blockade of apoptosis was reversed by a 10-min preincubation with 1 μg 2LPM19c Fab. Preincubation with anti-HLA Fab (clone W6/32) had no effect. Data represents mean percent apoptosis ± SD of three experiments.
Since β2-integrins exist largely in an inactive state and do not exhibit high affinity for ligands unless activated (Diamond and Springer 1993), it was important to distinguish between the effects of αMβ2 clustering and activation on apoptosis. In contrast to 19c, which blocks αMβ2-mediated adhesion by interacting with the major ligand binding domain (I-domain) of αM (Diamond et al. 1993), VIM12 is an outside-in activator of αMβ2-dependent adhesion (Stöckl et al. 1995). Like VIM12, Mn2+ addition can also activate β2-integrins via outside-in mechanisms and stimulate leukocyte adhesion and aggregation (Altieri 1991; Lynman et al. 1998). As demonstrated in Fig. 2, both VIM12 and Mn2+ were effective in suppressing apoptosis from 40–60%. Importantly, VIM12 and Mn2+ inhibition of apoptosis was reversed by blocking anti-αM Fab, but not a control Fab, suggesting dependence on ligand binding by αMβ2 in these suspension cultures.
Before further elucidating the mechanism of apoptosis inhibition via clustered or activated integrins, it was important to confirm αM clustering and/or activation in our culture system. Both 19c, as well as the activating antibody VIM12, were capable of punctate membrane clustering of αMβ2 during 37°C incubation with antibodies, whereas a ring-like, uniform distribution of αMβ2 was seen at 4°C (Fig. 3 A). Incubation of cells at 37°C in the presence of rhICAM beads and Mn2+ also resulted in αMβ2 clustering (data not shown). To determine the activation state of αM, we used the activation epitope antibody CBRM 1/5 kindly supplied by Dr. T. Springer (Harvard Medical School, Boston, MA; Diamond and Springer 1993). As expected, VIM12 incubation resulted in an increase in CBRM 1/5 binding with no increase in a control surface marker (CD45) and only a slight increase in total β2-integrin levels (FITC-labeled anti-CD18; Fig. 3 B, top). Mn2+ exposure also resulted in a consistent increase in CBRM 1/5 binding (data not shown). Unfortunately, the activation state of αMβ2 in the presence of 19c could not be determined due to steric hindrance between this antibody and CBRM1/5, as seen by the complete abrogation of fMLP-stimulated upregulation of CBRM1/5 binding (Fig. 3 D). However, 19c does not appear to activate αMβ2, insofar as it does not stimulate cell adhesion or aggregation (Diamond et al. 1993; data not shown). In addition, cells treated with rhICAM beads did not show increased CBRM1/5 binding or increased total β2-integrin levels (Fig. 3 B, bottom) indicating that rhICAM beads also were not directly activating αMβ2. This apparent lack of activation was not due to a masking of αMβ2 activation, like that seen with 19c, since fMLP-stimulated neutrophils coincubated with rhICAM beads and CBRM1/5 showed unaffected upregulation of CBRM1/5 epitope expression (Fig. 3 C). The lack of β2-integrin activation by rhICAM is further supported by the observation that the binding of rhICAM beads did not increase significantly over time (up to 4 h) suggesting that the binding of rhICAM did not lead to any measurable increase in adhesion (data not shown). Overall, our data support that 19c and rhICAM beads elicit β2-integrin clustering without leading to any substantial activation; whereas VIM12 and Mn2+ are capable of activating β2-integrins leading to increased neutrophil adhesiveness.
Figure 3.



Clustering and activation of αMβ2 integrin on neutrophils. (A) Activating (VIM12) and blocking (2LPM19c) antibodies to αM are capable of integrin clustering at 37°C, but not at 4°C. Fresh neutrophils were incubated with 2 μg/ml of either VIM12 or 2LPM19c at 4 or 37°C for 45 min and stained as in Materials and Methods. Figures are representative of four experiments. Bar, 5 μm. (B) VIM12 treatment, but not rhICAM-1 beads, induced αM-integrin activation as measured by mAb CBRM 1/5 binding. Neutrophils were treated as above with either VIM12 or rhICAM beads (1 μg of total ICAM) and were then stained as in Materials and Methods. To assess total β2-integrin levels, cells were stained with FITC–anti-CD18. Histograms are representative of six experiments. (C and D) Blocking of fMLP-stimulated CBRM1/5 binding by 2LPM19c (D), but not rhICAM-1 beads (C). Neutrophils were treated with 2LPM19c or rhICAM-1 beads as above, followed by a 5 min incubation with fMLP and staining with biotinylated CBRM1/5. Histograms are representative of four experiments.
Differential Activation of PI-3 Kinase and MAPK (ERK) Pathways after αMβ2 Clustering or Activation
Studies in adherent cells have implicated PI-3K and ERK pathways in mediating integrin survival signaling (Giancotti and Ruoslahti 1999). As shown in Fig. 4 A, inhibitors of the ERK activator MEK (PD98059), or PI-3K (wortmannin) were each able to reverse the antiapoptotic effect of αMβ2 clustering with either 19c F(ab′)2 or rhICAM-1 coated beads (Fig. 4 A). This pattern of reversal was also seen with the MEK inhibitor UO126 and the PI-3K inhibitor LY294002 (data not shown) indicating likely specificity for MEK and PI-3K. This nearly complete reversal of integrin clustering-mediated survival suggested major involvement of PI-3K and MEK driven pathways in the inhibition of apoptosis. In contrast, αMβ2 activation-mediated inhibition with either VIM12 or Mn2+ was reversed by wortmannin or LY294002, but was unaffected by the MEK inhibitors (Fig. 4; LY294002 and UO126, data not shown), indicating involvement of PI-3K pathways only. Thus, a clear difference between signals induced by β2-integrin activation and clustering is suggested.
Figure 4.

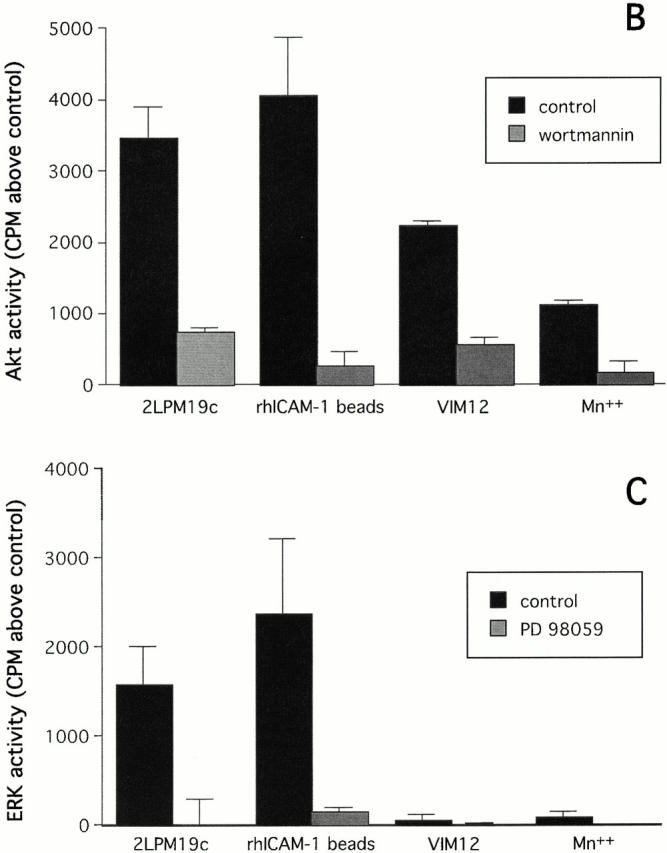
Inhibition of apoptosis by β2-integrin activation involves PI-3K/Akt, whereas inhibition by integrin clustering involves both PI-3K/Akt and MAPK pathways. (A) Neutrophils were preincubated for 10 min with either 30 μM PD98059 or 100 nM wortmannin before the addition of stimuli that activate (MnCl2, VIM12, or fMLP) or cluster (2LPM19c F(ab′)2 or immobilized rhICAM-1) β2-integrins. Concentrations of PD98059 and wortmannin used were chosen based on values reported elsewhere (Alessi et al. 1995; Arcaro and Wynman 1993) and dose response curves for the reversal of apoptosis inhibition. Cells were incubated for 10 h and apoptosis assessed by morphology. Bars represent mean percent inhibition of apoptosis ± SD of three experiments. (B) Activation of Akt by αM integrin clustering (2LPM19c or rhICAM-1 beads) or αM activation (VIM12 or MnCl). Neutrophils were incubated with each stimulus without or with 100 nM wortmannin for 20 min. (C) Activation of ERK by αM integrin clustering (2LPM19c), but not αM activation (VIM12). Cells were treated as in A, except without or with 30 μM PD 98059. Akt and ERK activity was measured as the incorporation of 32P above untreated cells (cpm ± SD; n = 3).
The chemotactic peptide fMLP was included in these experiments because of its known, characterized, ability to activate β2-integrins in neutrophils (Fig. 3 B; Jones et al. 1998). It is clear from Fig. 2 that fMLP, like VIM12 and Mn2+, inhibits apoptosis via αMβ2-dependent mechanisms. However, fMLP activates via signaling from the fMLP receptor to the integrin (inside-out activation). Unlike outside-in activation with VIM12 and Mn2+, suppression by fMLP was reversed by both wortmannin and PD98059 (Fig. 4 A). This is consistent with the known ability of fMLP to activate MEK driven pathways in neutrophils (Worthen et al. 1994). Therefore, the reversal of fMLP-mediated neutrophil survival by blocking MEK/ERK-driven pathways could be due to a blockade in a pathway leading to integrin activation rather than, or in addition to, inhibition of a survival pathway originating from αMβ2.
Due to the possible difference between the survival pathways elicited by the clustering of inactive integrins and integrin activation, we postulated differences in downstream kinase activation. Akt and ERK are serine/threonine kinases that are downstream of PI-3K and MEK1, respectively, and are known to be involved in survival signaling by maintaining an antiapoptotic balance among Bcl-2 family members (Datta et al. 1999; Scheid et al. 1999; Townsend et al. 1999). Clustering of αMβ2 by 19c (intact antibody or F(ab′)2), or rhICAM beads, activated both Akt and ERK as measured by in vitro kinase assays with immunoprecipitated enzymes at 20 min of stimulation (Fig. 4B and Fig. C). As expected, the activation of Akt was inhibited by wortmannin and ERK by PD98059. Fab fragments of 19c were inactive (data not shown), thus correlating with their lack of effect on apoptosis. By contrast, VIM12 and Mn2+ stimulated Akt activity, without activating ERK (Fig. 4B and Fig. C). These observations, combined with the inhibitor experiments (Fig. 4 A), provide good evidence for a difference in the antiapoptotic signaling generated via αMβ2 clustering versus activation.
While both αMβ2 clustering and activation stimulated Akt activity, the kinetics was somewhat different. Clustering-induced Akt activity appeared at 10 min, peaked at 15–20 min, and was undetectable by 30 min. After integrin activation, however, Akt activation continued to increase over the entire time course (30 min).
αMβ2-mediated Protection Against Mitochondrial Damage
One of the hallmarks of apoptotic changes is the loss of mitochondrial membrane potential and release of CytC. Although neutrophils do not contain large numbers of mitochondria (Edwards 1994), it seemed reasonable to hypothesize a similar response in these cells. The cationic, lipophilic dye JC-1 forms fluorescent J aggregates in the mitochondria of healthy, nonapoptotic cells. As mitochondrial membrane potential decreases during apoptosis, the J aggregate formation (fluorescence) also decreases (Cossarizza et al. 1993). Intracellular localization of JC-1 to mitochondria in neutrophils was verified by comparing localization of JC-1 staining with the staining pattern of a mitochondrial marker antibody (MAB1273; Fig. 5 A). (Costaining with JC-1 and MAB1273 could not be performed on the same cell because of the broad emission spectrum of JC-1 and filter limitations.) As expected, JC-1 fluorescence decreased during spontaneous apoptosis (compare control histograms at 0 and 4 h) and this was blocked by both αMβ2 clustering and activation (Fig. 5 B). This block in fluorescence decrease is shown graphically in Fig. 5 C, where the data are shown as percent fluorescence increase over control cells at 4 h. Both 19c and VIM12 yield an ∼50% increase in JC-1 fluorescence over that of control (4 h) cells indicating a maintenance of mitochondrial membrane potential. In addition, we examined the presence of CytC in neutrophils via immunofluorescence to confirm a possible correlation with the loss of mitochondrial potential. As seen in the top of Fig. 5 D, CytC could be detected in fresh neutrophils, and costaining with a mitochondrial dye (MitoTracker Red) revealed a cytoplasmic punctate pattern (localization that agrees well with JC-1- and MAB1273 staining; Fig. 5 A). The amount of CytC detected after 1.5 h in culture was significantly decreased (Fig. 5 D, bottom, Control), indicating a spontaneous release of CytC from the mitochondria. However, integrin clustering or activation with 19c or VIM12 prevented the apparent release of CytC. Combined, these data provide good evidence that mitochondrial protection is a downstream mechanism for integrin-mediated inhibition of spontaneous neutrophil apoptosis.
Figure 5.

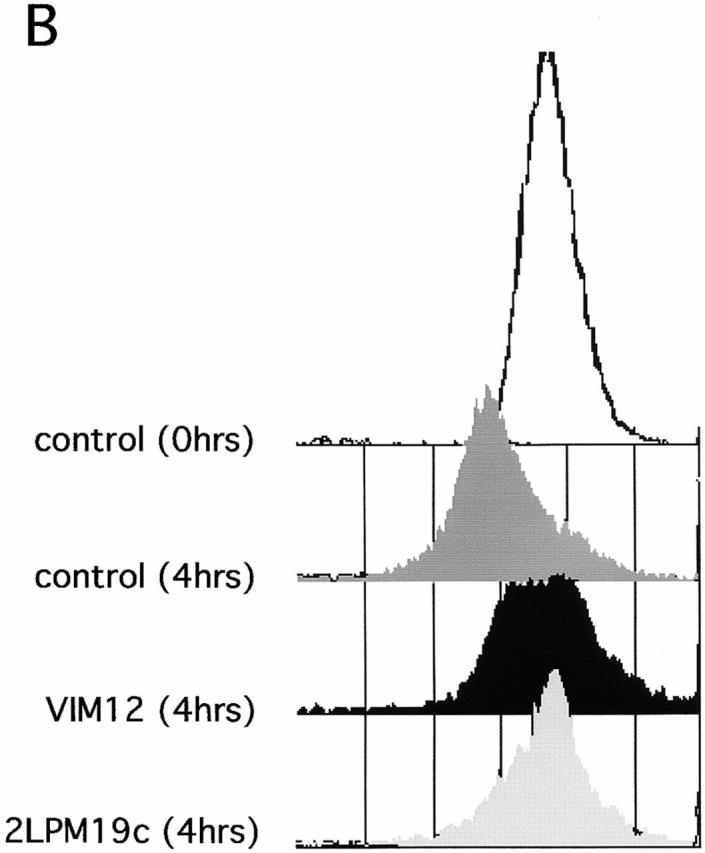


Both β2-integrin activation and clustering maintains mitochondria membrane potential and prevents cytochrome c release. (A) Identification and localization of mitochondria in neutrophils. Fresh cells were stained with either a mitochondria marker MAB1273 (1 μg) or the mitochondria permeable dye JC-1 (10 μM), and analyzed by fluorescent microscopy. Two cells stained with each reagent are shown. Bar, 5 μm. (B and C) αMβ2 integrin-mediated maintenance of mitochondria membrane potential. Fresh neutrophils (0 h), or neutrophils incubated for 4 h without or with VIM12 or 2LPM19c, were stained with 10 μM JC-1 and analyzed by flow cytometry. B shows representative histograms from 3 individual experiments. (C) Bars represent the mean percent increase in JC-1 fluorescence over cells at 4 h ± SD (n = 3). (D) αMβ2 integrin-mediated prevention of CytC release. Neutrophils were incubated for 1.5 h as above, permeabilized, and stained with anti-CytC antibody as in Kennedy et al. 1999. Mitochondria specific staining of anti-CytC was confirmed in fresh neutrophils by co-staining with mitotracker red (Mito trk) with colocalization verified by overlaid green/red fluorescence (CytC/Mito trk). Bar, 5 μm.
The Antiapoptotic Effect of β2-integrin Activation Is Switched to Apoptosis Enhancement and Mitochondrial Damage in the Presence of Death Stimuli
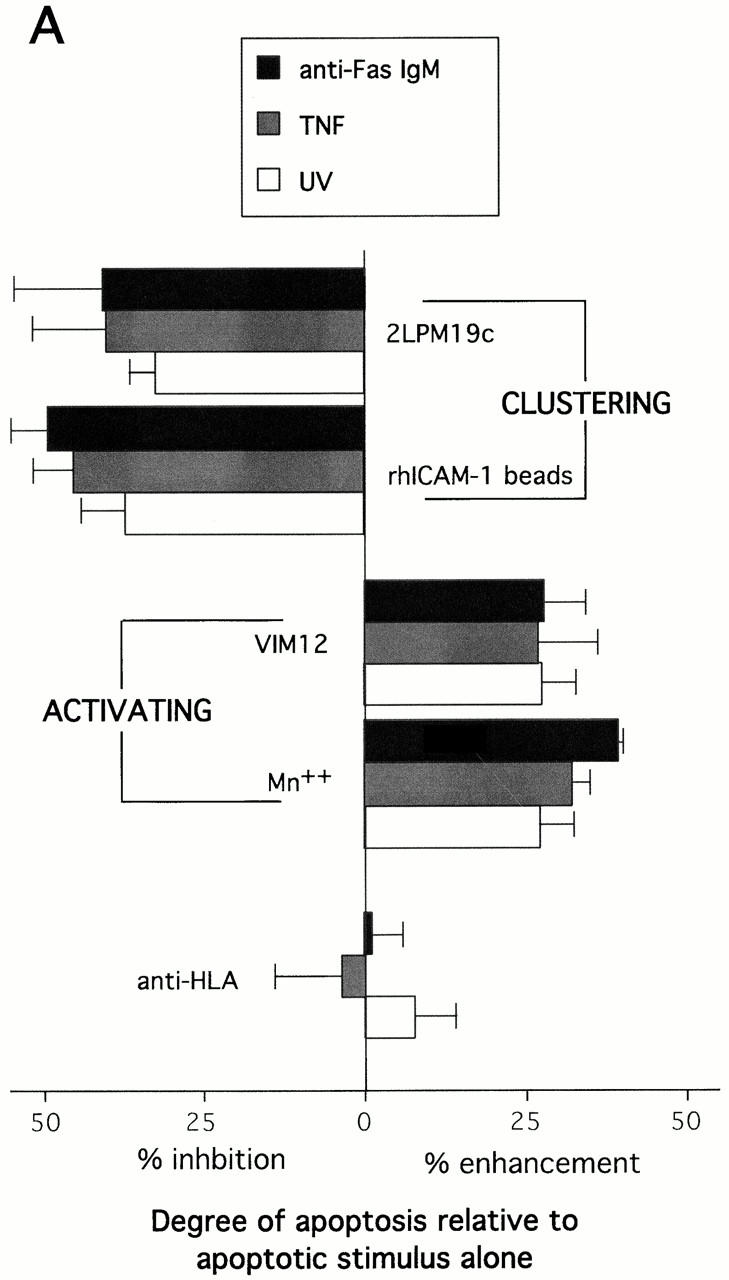
While the experiments described above show that αMβ2 engagement, either by clustering, or by activation/ligand interaction, inhibit neutrophil apoptosis, several reports suggest that β2-integrins may also be involved in enhancing apoptosis (Coxon et al. 1996; Walzog et al. 1997; Kettritz et al. 1999). However, these studies were performed with inflammatory neutrophils (Coxon et al. 1996) or with the addition of TNFα (Walzog et al. 1997; Kettritz et al. 1999). This raises the possibility that αMβ2 engagement could have differential effects on survival, depending on the integrin activation state and the presence or absence of other stimuli. The data shown in Fig. 6 A support this concept. The activating antibody, VIM12, as well as Mn2+ enhanced apoptosis induced by anti-Fas, TNFα, or UV irradiation. By contrast, clustering of αMβ2 with 19c or rhICAM-1 beads before the addition of anti-Fas, TNFα, or UV irradiation continued to suppress apoptosis.
Figure 6.
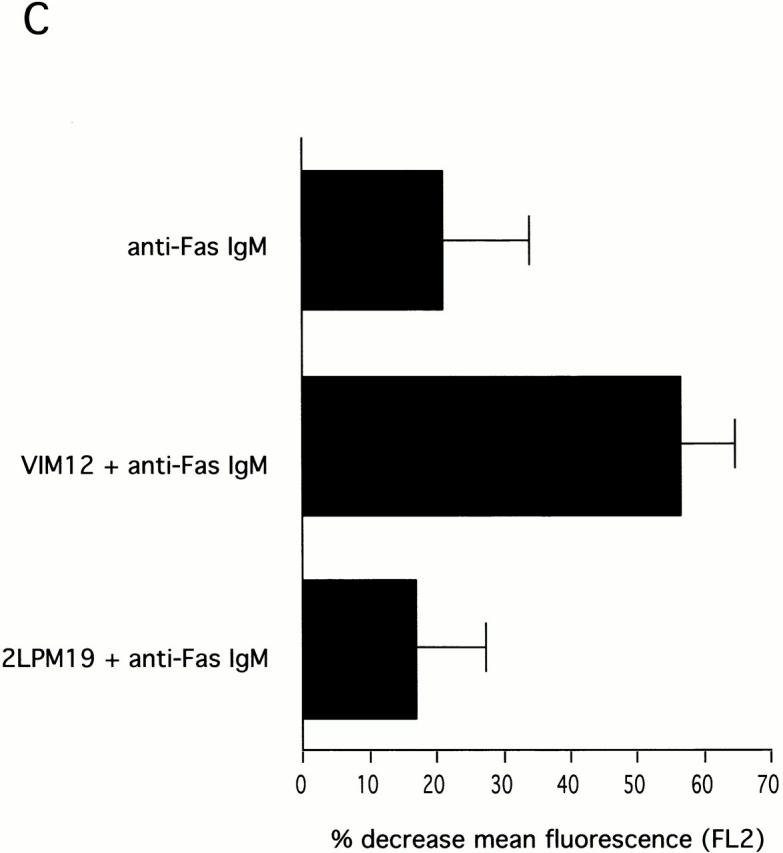


Stimulus-induced apoptosis and mitochondrial changes were enhanced by αMβ2 activation. (A) Activated αMβ2, but not clustered, inactivated αMβ2, enhanced apoptosis induced by TNF α, anti-Fas IgM, or UV irradiation. VIM12 and 2LPM19c were added simultaneously with anti-Fas IgM or TNF α and allowed to incubate for 4 h. UV treated cells were allowed to incubate with antibodies for 1 h before a 5 min UV exposure; cells were then incubated for 2 h. Bars represent the mean percent change in apoptosis compared with untreated cells ± SD (n = 3). Average control apoptosis levels were 46% for anti-Fas, 27% for TNFα, and 49% for UV-treated cells. (B and C) αMβ2 activation in combination with anti-Fas resulted in increased reduction of mitochondria membrane potential. Cells were incubated for 4 h with anti-Fas IgM without or with antibodies. JC-1 staining and analysis was carried out as in Fig. 5. (B) Histograms are representative of three experiments. (C) Bars represent the mean percent decrease in FL2 compared with 4 h cells ± SD (n = 3). (D) αMβ2 activation in combination with anti-Fas resulted in greater CytC release from mitochondria. Cells were treated and stained for CytC as above. The intensity of these images was increased relative to Fig. 5 D to illustrate loss of CytC staining upon incubation with anti-Fas and anti-Fas plus VIM12. Bar, 10 μm.
This observation suggested that we may see an enhancement of induced mitochondrial damage in the presence of integrin activation. Fig. 6 B shows JC-1 fluorescence as measured in neutrophils incubated for 4 h with no stimulus or with anti-Fas in the presence and absence of either αMβ2 clustering (19c) or activation (VIM12). Treatment with anti-Fas resulted in a decrease in JC-1 fluorescence compared with control neutrophils (Fig. 6b and Fig. c). The combination of anti-Fas plus VIM12-induced αMβ2 activation dramatically enhanced this decrease. In contrast, αMβ2 clustering with 19c showed no change in anti-Fas induced mitochondrial changes, even though 19c suppresses anti-Fas–induced apoptosis. TNFα and UV-induced loss of mitochondrial membrane potential was also increased by VIM12 (data not shown), correlating with the enhancement of TNFα and UV-induced apoptosis. In addition, mitochondrial CytC release was also enhanced in the combined presence of anti-Fas and VIM12. Fig. 6 D shows CytC in control (1 h) and anti-Fas treated neutrophils. The CytC immunostaining correlates with JC-1 fluorescence and is decreased in the presence of anti-Fas. An even greater loss of CytC staining is seen with the anti-Fas/VIM12 combination with no change seen with the anti-Fas/19c combination.
The data contained in Fig. 6 is indicative of a major difference between the functional outcome of either β2-integrin clustering or β2-integrin activation and strongly suggest that activated αMβ2 can serve a dual role, preventing mitochondrial damage and cell death in one situation, but aiding proapoptotic stimuli by enhancing mitochondrial damage and cell death in another.
Proapoptotic Stimuli Abrogate αMβ2-induced Akt Activation
The enhancement of stimulus-induced neutrophil apoptosis seen in the presence of αMβ2 activation suggested that the integrin-activated survival signals might be disrupted. To investigate this, β2-integrin–mediated Akt activation, common to clustering and activation, was examined in the absence or presence of anti-Fas IgM (Fig. 7). VIM12 and Mn2+ stimulated Akt activity was entirely inhibited in the presence of Fas ligation, as was 19c and rhICAM bead stimulated Akt activity. Therefore, the switch from integrin-mediated survival and mitochondrial protection to integrin-mediated death enhancement may result from an active downregulation of Akt survival mechanisms.
Figure 7.

Fas ligation plus β2-integrin activation or clustering prevented Akt activation. Cells were preincubated with or without anti-Fas IgM for 5 min before incubation with antibodies, rhICAM-1 beads, or MnCl2 for 20 min. Akt activity was measured as in Fig. 4. Bars represent 32P incorporated above untreated cells (cpm ± SD; n = 3). Similar effects were seen with TNFα (data not shown).
Surprisingly, anti-Fas alone activated Akt to a comparable degree to that of αMβ2 clustering or activation (Fig. 7). It was the combination of these stimuli that lowered Akt activity to control levels. This strongly suggests that signals originating from Fas and αMβ2 can synergize to prevent the activation of Akt.
αMβ2 Clustering Continues to Inhibit Stimulated Apoptosis in the Absence of Akt Activation Due to MAPK Activation
As noted above, both αMβ2 clustering and αMβ2 activation can synergize with Fas to eliminate Akt activation. However, unlike β2-integrin activation, clustering of β2-integrins continued to inhibit stimulated apoptosis in the absence of mitochondrial protection (Fig. 6) or Akt activity (Fig. 7). As indicated in Fig. 4, clustering of αMβ2 via 19c or rhICAM beads activated ERK in addition to Akt. Thus, αMβ2 clustering may still be capable of activating ERK in the presence of Fas ligation. We tested this hypothesis and found that ERK activity was unaffected by coincubation with anti-Fas (Fig. 8 A). In conjunction with these experiments we found that the sustained inhibitory effect of 19c or rhICAM beads on anti-Fas–induced apoptosis could be reversed by the MEK inhibitor PD98059 (Fig. 8 B). This observation suggests that ERK can compensate for the loss of PI-3K–dependent Akt activity in neutrophils with clustered, inactive αMβ2 and inhibit stimulated cell death in a manner that appears to be independent of mitochondrial protection (Fig. 6b, Fig. c, and Fig. d).
Figure 8.
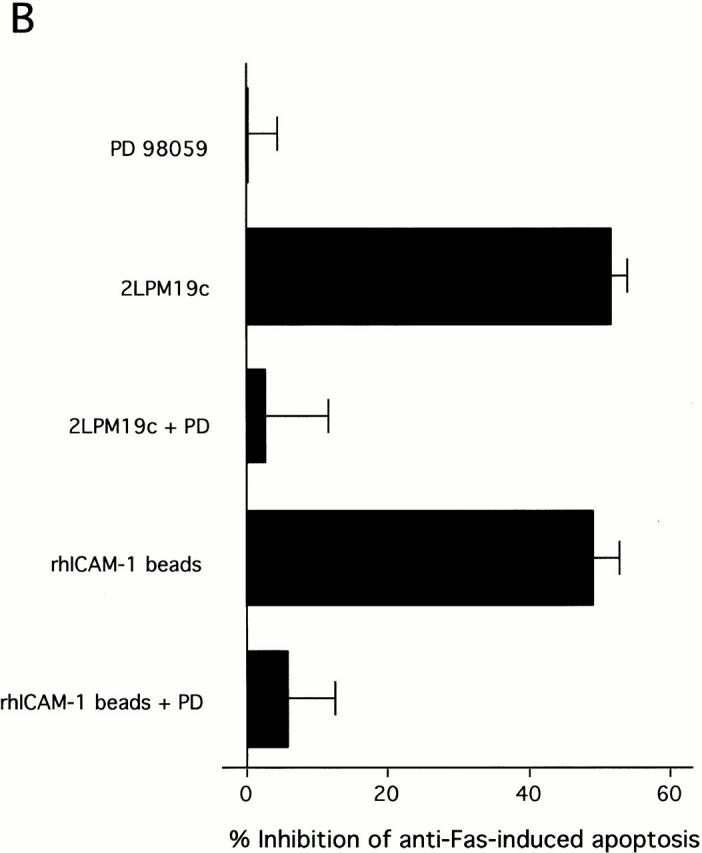

Clustering of β2-integrins inhibits Fas-induced apoptosis (in the absence of Akt) via continued ERK activation. (A) Cells were preincubated without or with anti-Fas IgM for 5 min before incubation with 2LPM19c or rhICAM beads for 20 min. ERK activity was measured as in Fig. 4. (B) Cells were treated with PD98059 as in Fig. 3, followed by the addition of 2LPM19c or rhICAM-1 beads and anti-Fas IgM. Apoptosis was assessed at 4 h. Bars represents mean percent inhibition ± SD (n = 3).
Adherence of Neutrophils to Fibrinogen Inhibits Apoptosis in a Manner Identical to β2-integrin Clustering
All of the experiments to this point were carried out on neutrophils in suspension. The fact that β2-integrins are the primary adhesion molecules on neutrophils and can mediate adhesion to a wide variety of ligands raised the likelihood that β2-integrin mediated surface engagement would also modulate neutrophil apoptosis.
To clearly determine surface effects on apoptosis, a special adhesion system was established. The experiments were performed on fibrinogen-coated glass coverslips using a very low cell number (6 × 104 cells/ml compared with 2 × 106 cells/ml in suspension incubations) to maximize cell-surface interaction and minimize cell–cell interaction. At more normal cell concentrations, and as in most studies of these phenomena (Ginis and Faller 1997; Kettritz et al. 1999), only a portion of the neutrophils could conceivably adhere to the available surface, many would adhere to each other. A further problem in these systems is the likelihood that when the neutrophils become apoptotic, their surface adhesion is lost. To assure assay of the entire neutrophil population, 111In-tagged cells were used and cells that lifted off the surface, as well as those that remained adherent, were included in direct assessment of nuclear condensation by the nuclear dye DAPI.
Using these methods, neutrophils on fibrinogen were shown to exhibit decreased spontaneous apoptosis compared with those (at an equal cell concentration) incubated in suspension (Fig. 9). As expected, the surface mediated-survival was reversed by wortmannin and PD98059 in a fashion identical to the αMβ2 clustering agents 19c and rhICAM-1 beads. Since neutrophils interact with ligands like fibrinogen with high affinity upon activation, it was important to examine this phenomenon in the presence of fMLP. Like unstimulated neutrophils, fMLP-stimulated neutrophils exhibited significant increased survival on fibrinogen, an effect that was greater than unstimulated cells and was significantly reversed by wortmannin and PD98059 (Fig. 9). This agrees with the inhibitory effects of fMLP in suspension and the ability of fMLP to directly activate MAP kinase pathways. The increase in inhibition seen in the presence of fMLP plus fibrinogen was also seen in combination with rhICAM beads (data not shown). Therefore, these data further support a role for β2-integrin engagement in the inhibition of neutrophil apoptosis and suggest that the interaction of both inactive and active β2-integrins with different surface bound natural ligands, in the absence of proapoptotic signals, are capable of significantly inhibiting apoptosis.
Figure 9.

Neutrophil apoptosis is inhibited on fibrinogen in a PI-3K and MAPK dependent manner. 111In-labeled neutrophils (6 × 104 cells/ml) were incubated either in suspension or on fibrinogen-coated glass coverslips. After a 4 h incubation, cells were fixed, nonadherent and adherent cells were separated and stained with DAPI to measure apoptosis. PD 98059 (30 μM) and wortmannin (100 nM) were added after labeling 15 min before plating. Apoptotic percentages were calculated as outlined in the Materials and Methods. Graph represents the percent inhibition of apoptosis on fibrinogen compared with suspension culture. Bars represent the mean ± SD (n = 5). The significance of reversal of inhibition by PD98059 and wortmannin was determined by single mean comparisons to control percent inhibition using the Tukey-Kramer test (*P < 0.05, compared with control inhibition; † P < 0.05, compared with fMLP-stimulated inhibition).
Discussion
We conclude that a significant function for β2-integrins on neutrophils is the modulation of cell survival. As outlined in the introduction, some studies have implicated integrins in either promoting neutrophil survival or enhancing apoptosis, depending on the type of integrin engagement and/or the presence of proinflammatory or proapoptotic stimuli. The current study explains these apparent contradictions by establishing a dual role for β2-integrins in cell survival and provides a potential signaling explanation for this observation. This dual role is a result of a clear difference between the functional outcome and signals induced by β2-integrin clustering versus activation (Fig. 10).
Figure 10.

The role of β2-integrin clustering and activation in neutrophil survival. Signaling model shows pathways activated upon αMβ2 clustering or activation with arrows indicating stimulatory cascades and gray bars indicating potential points of inhibition of apoptotic signaling. β2-integrin structures are modeled after Stewart and Hogg 1996.
The engagement of inactive αMβ2 with 19c, a mAb that interacts at or near the I-domain of αM (Fig. 1), was capable of inhibiting the spontaneous apoptosis of human neutrophils via activation of ERK and Akt, followed by mitochondrial protection. We believe that this requires some degree of αMβ2 clustering since bivalent molecules (whole or F(ab′)2) were required to elicit this effect. In addition, VIM12, an antibody capable of activating αMβ2 (Stöckl et al. 1995), was also capable of inhibiting spontaneous apoptosis, protecting mitochondria and activating Akt, but not ERK. Therefore, while definite differences exist in the signaling response of neutrophils to either β2-integrin clustering or activation (Fig. 4 and Fig. 6), both are capable of inhibiting spontaneous apoptosis (Fig. 2).
Like the mAb 19c, the natural αMβ2 ligands ICAM and fibrinogen, when immobilized, were capable of inhibiting spontaneous neutrophil apoptosis (Fig. 2 and Fig. 9), a phenomenon that we attribute to integrin clustering. We can detect a small, consistent percentage of resting neutrophils bound to ICAM-1 beads and fibrinogen-coated coverslips in the absence of measurable CBRM1/5 binding (data not shown), suggesting that unactivated αMβ2 is capable of binding immobilized ligands to some degree. A low percentage of adhering cells is not surprising given the low affinity of unactivated αMβ2 for ligands like ICAM. However, a recent report indicates that low affinity αLβ2/ICAM interactions on resting T cells are capable of initiating clustering of inactive αLβ2 on the cell surface (Bleijs et al. 2000). A similar type of mechanism could be acting in neutrophils where low affinity interactions with ICAM or fibrinogen could promote the clustering of inactive αMβ2 and subsequent activation of Akt and ERK signaling pathways in the absence of a large degree of ligand binding. In addition, inhibition was observed, and antiapoptotic signaling pathways activated, in neutrophils stimulated with fMLP in the presence of immobilized ICAM or fibrinogen (Fig. 9; ICAM data not shown). This observation is important because it indicates that irregardless of the state of αMβ2 activation, binding of natural ligands by β2-integrins can inhibit spontaneous apoptosis to a high degree.
VIM12, Mn2+, and fMLP-mediated inhibition was blocked by anti-αM Fab (Fig. 2) indicating that ligand engagement by activated αMβ2 was necessary to inhibit apoptosis. Since these experiments were conducted in suspension cultures (minimal surface engagement), it is probable that this inhibition is a result of homotypic neutrophil aggregation (Lynman et al. 1998). VIM12, Mn2+, and fMLP are all capable of stimulating some degree of neutrophil aggregation (Stöckl et al. 1995; Lynman et al. 1998) leading to ligand-mediated αMβ2 cross-linking. Therefore, while we distinguish between activation and clustering as two separate physical states of β2-integrins (Fig. 10), it is likely that activated αMβ2 are present as clusters in the membrane. Indeed, immunofluorescence confirms that VIM12 and Mn2+ clusters αMβ2 (Fig. 3 A; Mn2+ data not shown). Unlike clustering of inactive αMβ2, apoptosis suppression mediated by activated αMβ2 was unaffected by MAPK (ERK) inhibition, but was reversed by PI-3K blockade (Fig. 4 A). The activation of downstream effectors correlated with this, in that activation of Akt, but not ERK, was seen. It is interesting that activated αMβ2 is capable of inhibiting neutrophil apoptosis in the absence of ERK activation. This suggests that the activation of PI-3K pathways alone can, under certain circumstances, compensate for the loss of concomitant MAPK activation. It is possible that augmented signals from activated αMβ2 are capable of sustained activation of PI-3K pathways. As mentioned above, Akt remained active for a longer period upon activation of αMβ2 (still high at 30 min). This may allow for a greater degree of Akt-mediated mechanisms capable of maintaining mitochondrial membrane potential and inhibiting CytC release.
While αMβ2 clustering and αMβ2 activation induces survival signals capable of inhibiting spontaneous apoptosis, differential effects were seen on apoptosis stimulated by anti-Fas, TNFα, or UV exposure. In this scenario, activated αMβ2 enhanced apoptosis, while inactive, but clustered, αMβ2 continued to inhibit apoptosis. A mechanistic basis for this switch from an antiapoptotic tone to a proapoptotic one is provided by the data showing that αMβ2 mediated Akt activation was abrogated in the presence of Fas ligation and TNFα (Fig. 7; TNFα data not shown). The absence of this pathway potentially explains why activated αMβ2, which does not activate ERK, no longer inhibits in the presence of proapoptotic stimuli. In addition, the effect on mitochondrial membrane potential and CytC release further supports the notion that Akt may protect the neutrophil by stabilizing mitochondria; without this mechanism clustered, inactive αMβ2 no longer confers mitochondrial protection and activated αMβ2 enhances mitochondrial damage (Fig. 6). It is important to note that Fas ligation alone activated some Akt (Fig. 7). (The possibility that Fas can activate PI-3K or Akt is not unprecedented; Datta et al. 1999.) This, therefore, supports that synergistic signals are responsible for turning off Akt. It is likely that combined signals from Fas and αMβ2 can limit the activity of PI-3K stimulated pathways either directly, by not allowing activation and recruitment of molecules involved in PI-3K activation; or indirectly, by activating molecules that can inhibit the downstream signaling from PI-3K such as the inositol phosphatases SH2 domain-containing inositol 5-phosphate (SHIP) and PTEN (Stambolic et al. 1998; Liu et al. 1999). SHIP and PTEN are of particular interest since they have been implicated in integrin signaling (Giurato et al. 1997; Tamura et al. 1999).
Clustering of αMβ2 with either 19c or ICAM-1 continued to inhibit stimulated apoptosis in the apparent absence of PI-3K/Akt mediated antiapoptotic signals (Fig. 6 A). We attribute this to αMβ2 clustering-mediated activation of MAPK (MEK/ERK; Fig. 4 and Fig. 8). This suggests that a PI-3K/Akt-independent ERK pathway acts to inhibit anti-Fas–induced neutrophil apoptosis. This observation appears to disagree with a previous study from our laboratory (Frasch et al. 1998); however, the duration of ERK activation seen with αMβ2 clustering is likely to be significantly longer than that seen with other stimuli and could allow for more antiapoptotic pathway activation. Blocking ERK activity with PD98059 did not cause αMβ2 clustering induced signals to become proapoptotic (Fig. 8 B) as was the case with ERK-deficient αMβ2 activation (Fig. 6 A). Activated αMβ2 (but not clustered, inactive αMβ2) leads to the production of oxidants that may directly damage mitochondria and therefore enhance apoptosis in the absence of ERK activity (B. Whitlock, unpublished observation). This is supported by the observation that apoptosis was significantly enhanced by TNFα on fibrinogen (data not shown), a scenario known to induce massive oxidant production (Nathan et al. 1989).
Our data support an important role for Akt and ERK in neutrophils; however, downstream targets in these cells remain to be identified. Akt can modulate apoptosis through several potential targets, including the phosphorylation of proapoptotic family members such as Bad (Datta et al. 1999). This phosphorylation contributes to an antiapoptotic balance among Bcl-2 family members and, thus, prevents the alteration of mitochondrial membrane potential and release of CytC. We have been unable to detect Bad in human neutrophils (S. Gardai, unpublished observation) so the significance of this pathway is unclear. However, a recent report described potential Bad-independent Akt mechanisms that operate to prevent CytC release (Kennedy et al. 1999). Other potential targets for activated Akt are the transcription factors CREB and NF-κB (Datta et al. 1999). Their activation may result in the upregulation of antiapoptotic molecules like A1, an NF-κB inducible gene (Chuang et al. 1998).
Unlike Akt, and with the exception of fMLP stimulation, ERK was only activated by integrin clustering, not activation. The fact that αMβ2 clustering-mediated inhibition was completely reversed either by inhibition of PI-3K or MEK (Fig. 4 A) suggests that both Akt and ERK must be activated and may have common targets, including those listed above. However, because ERK activity is responsible for the continued inhibition of stimulated apoptosis in the apparent absence of PI-3K/Akt activity and mitochondrial protection, it is likely that other MAPK targets exist downstream of Bcl-2-family regulation and CytC release. In support of this, a recent study found that B-Raf overexpression and constitutive ERK activation in fibroblasts prevented apoptosis via a mechanism independent of PI-3K/Akt, downstream of CytC release, and upstream of caspase 3 activation (Erhardt et al. 1999). A similar role for ERK may be present in neutrophils.
Our data raise the intriguing possibility that repetitive engagement of active or inactive β2-integrins on circulating leukocytes as they pass through the microvasculature could contribute to their in vivo lifespan by delaying their intrinsic death program. We have earlier shown the need for cell deformation during microvascular transit (Worthen et al. 1989), a circumstance that could provide an ideal opportunity for transient integrin engagement. The noncirculating pool of relatively mature granulocytes, known to be available for immediate release into the circulation, may also be prevented from undergoing cell death by integrin ligation in the bone marrow (Palframan et al. 1998). This type of apoptotic control could ultimately dictate the homeostatic levels of circulating leukocytes, as well as contribute to maintenance of cell number during a systemic immune response.
On the other hand, integrin-mediated cellular survivals might be counterproductive in the more permanent adhesive environment of an inflammatory reaction, but here, as we show, the presence of TNFα and other potentially proapoptotic stimuli (e.g., Fas ligand) would combine to reverse the protective effects of integrin signaling. Experiments in αM deficient mice support the notion of a likely role for β2-integrins in the apoptotic progression of elicited inflammatory neutrophils (Coxon et al. 1996). In that report, the authors speculate that this is due to the ability of these cells to phagocytose via αMβ2, which could, in turn, stimulate apoptosis. Based on our data, however, we would propose an alternate interpretation, whereby extravasated neutrophils undergo apoptosis more quickly due to engagement of activated αMβ2 and a death signal (e.g., TNFα) at the inflammatory site. This enhanced death could provide, along with apoptotic cell uptake, an effective mechanism to resolve inflammation. Therefore, this dual role of integrins in leukocyte apoptosis suggests that β2-integrins can influence survival throughout the life of the cell and, therefore, need to be considered in any discussion of the role of apoptosis in the control of leukocyte numbers and the resolution of immune responses.
Acknowledgments
We thank P. McDonald, D. Richter, P. Hoffman, and A. de Cathelineau for technical advice, and S.C. Frasch and C.A. Ogden for advice and discussion.
This work was supported by the National Institutes of Health grants HL34303, GM48211, and HL60980. B.B. Whitlock is supported by a National Research Service Award (NRSA) training grant (HL07670-10).
Footnotes
Ben B. Whitlock and Shyra Gardai contributed equally to the completion of this work.
Abbreviations used in this paper: CytC, cytochrome c; ERK, extracellular signal-regulated kinase; fMLP, formyl-methionine-leucine-phenylalanine; MAPK, mitogen-activated protein kinase; PI-3K, phopshatidylinositol-3 kinase; rhICAM-1, recombinant human intracellular adhesion molecule 1; TNFα, tumor necrosis factor α.
References
- Alessi D.R., Cuenda A., Cohen P., Dudley D.T., Saltiel A.R. PD 98059 is a specific inhibitor of the activation of mitogen-activated protein kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Altieri D.C. Occupancy of CD11b/CD18 (Mac-1) divalent ion binding site(s) induces leukocyte adhesion. J. Immunol. 1991;147:1891–1898. [PubMed] [Google Scholar]
- Arcaro A., Wynman M.P. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitorthe role of phosphatidylinositol 3,4,5-trisphophate in neutrophil responses. Biochem. J. 1993;296:297–301. doi: 10.1042/bj2960297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleijs D.A., Binnerts M.E., van Vliet S.J., Figdor C.G., van Kooyk Y. Low-affinity LFA-1/ICAM-3 interactions augment LFA-1/ICAM-1-mediated T cell adhesion and signaling by redistribution of LFA-1. J. Cell Sci. 2000;113:391–400. doi: 10.1242/jcs.113.3.391. [DOI] [PubMed] [Google Scholar]
- Chuang P.I., Yee E., Karsan A., Winn R.K., Harlan J.M. A1 is a constitutive and inducible Bcl-2 homologue in mature human neutrophils. Biochem. Biophys. Res. Comm. 1998;249:361–365. doi: 10.1006/bbrc.1998.9155. [DOI] [PubMed] [Google Scholar]
- Cossarizza A., Baccarani-contri M., Kalashnikova G., Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloride-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1) Biochem. Biophys. Res. Comm. 1993;197:40–45. doi: 10.1006/bbrc.1993.2438. [DOI] [PubMed] [Google Scholar]
- Coxon A., Rieu P., Barkalow F.J., Askari S., Sharpe A.H., von Adrian U.H., Arnaout M.A., Mayadas T.N. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosisa homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Brunet A., Greenberg M.E. Cellular survivala play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Diamond M.S., Springer T.A. A subpopulation of Mac-1 (CD11b/Cd18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J. Cell Biol. 1993;120:545–556. doi: 10.1083/jcb.120.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond M.S., Garcia-Aguilar J., Bickford J.K., Corbi A.L., Springer T.A. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J. Cell Biol. 1993;120:1031–1043. doi: 10.1083/jcb.120.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S.W. Biochemistry and Physiology of the Neutrophil 1994. Cambridge University Press; New York: pp. 299 pp [Google Scholar]
- Erhardt P., Schremser E.J., Cooper G.M. B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol. Cell. Biol. 1999;19:5308–5315. doi: 10.1128/mcb.19.8.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok V.A., Bratton D.L., Konowal A., Freed P.W., Westcott J.Y., Henson P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2 and PAF. J. Clin. Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasch S.C., Nick J.A., Fadok V.A., Bratton D.L., Worthen G.S., Henson P.M. p38 Mitogen-activated protein kinase-dependent and -independent signal transduction pathways leading to apoptosis in human neutrophils. J. Biol. Chem. 1998;273:8389–8397. doi: 10.1074/jbc.273.14.8389. [DOI] [PubMed] [Google Scholar]
- Giancotti F.G., Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Ginis I., Faller D.V. Protection from apoptosis in human neutrophils is determined by the surface of adhesion. Am. J. Physiol. 1997;272:C295–C309. doi: 10.1152/ajpcell.1997.272.1.C295. [DOI] [PubMed] [Google Scholar]
- Giurato S., Payrastre B., Drayer A.L., Plantavid M., Woscholski R., Parker P., Erneux C., Chap H. Tyrosine phosphorylation and relocation of SHIP are integrin-mediated in thrombin-stimulated human blood platelets. J. Biol. Chem. 1997;272:26857–26863. doi: 10.1074/jbc.272.43.26857. [DOI] [PubMed] [Google Scholar]
- Hansen L.L., Ikeda Y., Olsen G.S., Busch A.K., Mosthaf L. Insulin signaling is inhibited by micromolar concentrations of H2O2 . J. Biol. Chem. 1999;274:25078–25084. doi: 10.1074/jbc.274.35.25078. [DOI] [PubMed] [Google Scholar]
- Haslett C., Guthrie L.A., Kopaniak M.M., Johnston R.B., Jr., Henson P.M. Modulation of multiple neutrophil functions by preparative methods of trace concentrations of bacterial lipopolysaccharide. Am. J. Pathol. 1985;119:101–110. [PMC free article] [PubMed] [Google Scholar]
- Jones S.L., Knaus U.G., Bokoch G.M., Brown E.J. Two signaling mechanisms for activation of αMβ2 avidity in polymorphonuclear neutrophils. J. Biol. Chem. 1998;273:10556–10566. doi: 10.1074/jbc.273.17.10556. [DOI] [PubMed] [Google Scholar]
- Kennedy S.G., Kandel E.S., Cross T.K., Hay N. Akt/protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol. Cell. Biol. 1999;19:5800–5810. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettritz R., Xu Y.-X., Kerren T., Quass P., Klein J.B., Luft F.C., Haller H. Extracellular matrix regulates apoptosis in human neutrophils. Kidney Int. 1999;55:562–571. doi: 10.1046/j.1523-1755.1999.00280.x. [DOI] [PubMed] [Google Scholar]
- Liles W.C., Kiener P.A., Ledbetter J.A., Aruffoe A., Klebanoff S.J. Differential expression of Fas (CD95) and Fas ligand on normal human phagocytesimplication for the regulation of apoptosis in neutrophils. J. Exp. Med. 1996;184:429–440. doi: 10.1084/jem.184.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Sasaki T., Kozieradzki I., Wakeham A., Itie A., Dumont D.J., Penninger J.M. SHIP is a negative regulator of growth factor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999;13:786–791. doi: 10.1101/gad.13.7.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell C.A., Berton G. Integrin signal transduction in myeloid leukocytes. J. Leuk. Biol. 1999;65:313–320. doi: 10.1002/jlb.65.3.313. [DOI] [PubMed] [Google Scholar]
- Lynman E., Sklar L.A., Taylor A.D., Neelamegham S., Edwards B.S., Smith C.W., Simon S.I. β2-Integrins mediate stable adhesion in collisional interactions between neutrophils and ICAM-1-expressing cells. J. Leuk. Biol. 1998;64:622–630. doi: 10.1002/jlb.64.5.622. [DOI] [PubMed] [Google Scholar]
- McGilvray I.D., Lu Z., Wei A.C., Rotstein O.D. MAP-kinase dependent induction of monocytic procoagulant activity by beta2-intregrins. J. Surg. Res. 1998;80:272–279. doi: 10.1006/jsre.1998.5319. [DOI] [PubMed] [Google Scholar]
- Meerschaert J., Vrtis R.F., Shikama Y., Sedgwick J.B., Busse W.W., Mosher D.F. Engagement of α4β7 integrins by monoclonal antibodies or ligands enhances survival of human eosinophils in vitro. J. Immunol. 1999;163:6217–6227. [PubMed] [Google Scholar]
- Meng F., Lowell C.A. A beta1 integrin signaling pathway involving Src-family kinases, Cbl and PI-3 kinase is required for macrophage spreading and migration. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:4391–4403. doi: 10.1093/emboj/17.15.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulding D.A., Quayle J.A., Hart C.A., Edwards S.W. Mcl-1 expression in human neutrophilsregulation by cytokines and correlation with cell survival. Blood. 1998;92:2495–2502. [PubMed] [Google Scholar]
- Nathan C., Srimal S., Farber C., Sanchez E., Kabbash L., Asch A., Gailit J., Wright S.D. Cytokine-induced respiratory burst of human neutrophilsdependence on extracellular matrix proteins and CD11/CD18 integrins. J. Cell. Biol. 1989;109:1341–1349. doi: 10.1083/jcb.109.3.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palframan R.T., Collins P.D., Severs N.J., Rothery S., Williams T.J., Rankin S.M. Mechanisms of acute eosinophilia mobilization from the bone marrow stimulated by interleukin 5the role of specific adhesion moleules and phosphatidylinositol 3-kinase. J. Exp. Med. 1998;188:1621–1632. doi: 10.1084/jem.188.9.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly P.L., Woska J.R., Jr., Jeanfavre D.D., McNally E., Rothlein R., Bormann B.-J. The native structure of inercellular adhesion molecule-1 (ICAM-1) is a dimercorrelation with binding to LFA-1. J. Immunol. 1995;155:529–532. [PubMed] [Google Scholar]
- Savill J.S., Wylie A.H., Henson J.E., Walport M.J., Henson P.M., Haslett C. Macrophage phagocytosis of aging neutrophils in inflammationprogrammed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid M.P., Schubert K.M., Duronio V. Regulation of Bad phosphorylation and association with Bcl-xL by the MAPK/Erk kinase. J. Biol. Chem. 1999;274:31108–31113. doi: 10.1074/jbc.274.43.31108. [DOI] [PubMed] [Google Scholar]
- Stambolic V., Suzuki A., de la Pompa J.L., Brothers G.M., Mirtsos C., Sasaki T., Ruland J., Penninger J.M., Siderovski D.P., Mak T. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Stewart M., Hogg N. Regulation of leukocyte integrin functionaffinity vs. avidity. J. Cell. Biochem. 1996;61:554–561. doi: 10.1002/(sici)1097-4644(19960616)61:4<554::aid-jcb8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Stöckl J., Majdic O., Pickl W.F., Rosenkranz A., Prager E., Gschwantler E., Knapp W. Granulocyte activation via a binding site near the C-terminal region of complement receptor type 3 α-chain (CD11b) potentially involved in intramembrane complex formation with glycosylphosphatidylinositol-anchored FcγRIIIB (CD16) molecules. J. Immunol. 1995;154:5452–5463. [PubMed] [Google Scholar]
- Takeda Y., Watanabe H., Yonehara S., Yamashita T., Saito S., Sendo F. Rapid acceleration of neutrophil apoptosis by tumor necrosis factor-α. Intl. Immunol. 1993;5:691–694. doi: 10.1093/intimm/5.6.691. [DOI] [PubMed] [Google Scholar]
- Tamura M., Gu J., Tran H., Yamada K.M. PTEN gene and integrin signaling in cancer. J. Natl. Cancer Inst. 1999;91:1820–1828. doi: 10.1093/jnci/91.21.1820. [DOI] [PubMed] [Google Scholar]
- Tirosh A., Potashnik R., Bashan N., Rudich A. Oxidative stress disrupts insulin-induced cellular redistribution on insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. A putative cellular mechanism for impaired protein kinase B activaion and GLUT4 translocation. J. Biol. Chem. 1999;274:10595–10602. doi: 10.1074/jbc.274.15.10595. [DOI] [PubMed] [Google Scholar]
- Townsend K.J., Zhou P., Qian L., Bieszczad C.K., Lowrey C.H., Yen A., Craig R.W. Regulation of MCL1 through a serum response factor/ELK-1-mediated mechanism links expression of a viability-promoting member of the BCL2 family to the induction of hematopoietic cell differentiation. J. Biol. Chem. 1999;274:1801–1813. doi: 10.1074/jbc.274.3.1801. [DOI] [PubMed] [Google Scholar]
- Walzog B., Jeblonski F., Zakrzewicz A., Gaehtgens P. β2 integrins (CD11/CD18) promote apoptosis of human neutrophils. FASEB J. 1997;11:1177–1186. doi: 10.1096/fasebj.11.13.9367353. [DOI] [PubMed] [Google Scholar]
- Walzog B., Weinmann P., Jeblonski F., Scharffetter-Kochanek K., Bommert K., Gaehtgens P. A role for beta(2) integrins (CD11/CD18) in the regulation of cytokine gene expression of polymorphonuclear neutrophils during the inflammatory response. FASEB J. 1999;13:1855–1865. doi: 10.1096/fasebj.13.13.1855. [DOI] [PubMed] [Google Scholar]
- Watson R.W.G., Rotstein O.D., Jimenez M., Parodo J., Marshall J.C. Augmented intracellular glutathione inhibits Fas-triggered apoptosis of activated human neutrophils Blood 89 1997. 4175 4181a [PubMed] [Google Scholar]
- Watson R.W.G., Rotstein O.D., Nathens A.B., Parodo J., Marshall J.C. Neutrophil apoptosis is modulated by endothelial transmigration and adhesion molecule engagement J. Immunol. 158 1997. 945 953b [PubMed] [Google Scholar]
- Weinmann P., Gaehtgens P., Walzog B. Bcl-Xl- and Bax-α-mediated regulation of apoptosis of human neutrophils via caspase-3. Blood. 1999;93:3108–3115. [PubMed] [Google Scholar]
- Worthen G.S., Avdi N., Buhl A.M., Suzuki N., Johnson G.L. FMLP activates ras and raf in human neutrophilspotential role in activation of MAP kinases. J. Clin. Invest. 1994;94:815–823. doi: 10.1172/JCI117401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthen G.S., Schwab B., Elson E.L., Downey G.P. Mechanics of stimulated neutrophilscell stiffening induces retention in capillaries. Science. 1989;245:183–186. doi: 10.1126/science.2749255. [DOI] [PubMed] [Google Scholar]
- Zundel W., Giaccia A. Inhibition of the antiapoptotic PI(3)K/Akt/Bad pathway by stress. Genes Dev. 1998;12:1941–1946. doi: 10.1101/gad.12.13.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]