

Abstract
Microtubule-associated proteins such as MAP1B have long been suspected to play an important role in neuronal differentiation, but proof has been lacking. Previous MAP1B gene targeting studies yielded contradictory and inconclusive results and did not reveal MAP1B function. In contrast to two earlier efforts, we now describe generation of a complete MAP1B null allele. Mice heterozygous for this MAP1B deletion were not affected. Homozygous mutants were viable but displayed a striking developmental defect in the brain, the selective absence of the corpus callosum, and the concomitant formation of myelinated fiber bundles consisting of misguided cortical axons. In addition, peripheral nerves of MAP1B-deficient mice had a reduced number of large myelinated axons. The myelin sheaths of the remaining axons were of reduced thickness, resulting in a decrease of nerve conduction velocity in the adult sciatic nerve. On the other hand, the anticipated involvement of MAP1B in retinal development and γ-aminobutyric acid C receptor clustering was not substantiated. Our results demonstrate an essential role of MAP1B in development and function of the nervous system and resolve a previous controversy over its importance.
Keywords: microtubule-associated protein 1B, brain development, gene targeting, corpus callosum, myelin
Introduction
Microtubule-associated protein (MAP) 1B is one of the first MAPs to be expressed during embryonic development of the nervous system and the protein has long been suspected to be instrumental in neuronal morphogenesis. Expression can be detected in axons and growth cones as early as day 14 of gestation (Fawcett et al. 1994). In addition, the protein can be phosphorylated by at least two kinases implicated in neuronal development, cdk5 and GSK-3β (García-Pérez et al. 1998; Lucas et al. 1998) and is differentially phosphorylated in distal axons and growth cones (Black et al. 1994; Bush et al. 1996a). Finally, biochemical and cell biological analyses have established that MAP1B can bind not only to microtubules but also to actin filaments (Pedrotti and Islam 1996; Tögel et al. 1998), establishing MAP1B as a potential link between these two components of the growth cone cytoskeleton.
MAP1B purified from mammalian brain constitutes a multimeric protein complex that consists of one 300-kD heavy chain and at least one 32-kD light chain, termed LC1 (Hammarback et al. 1991). Both heavy chain and LC1 are proteolytic cleavage products of a common MAP1B polyprotein precursor that contains LC1 as a 250 amino acid–long carboxy-terminal segment (Hammarback et al. 1991; Tögel et al. 1998, Tögel et al. 1999). This MAP1B precursor protein is encoded by a single gene consisting of seven exons (Lien et al. 1994; Kutschera et al. 1998). However, there exist multiple ways of transcription of this gene to give rise to two types of transcripts (Lien et al. 1994; Kutschera et al. 1998). Type 1, the regular transcripts, represent the majority (≥90%) of MAP1B mRNA and contain exons 1-7. Type 2 transcripts (≤10%) contain noncoding exons upstream of exon 3 and potentially encode an amino-terminally truncated MAP1B precursor lacking the first 95 amino acids of the heavy chain.
In the past few years, several attempts have been made to determine the role of MAP1B in brain development through attenuation of MAP1B expression. However, the results were contradictory. Early studies were carried out in vitro and employed antisense oligonucleotide technology. It was shown that MAP1B is necessary for the growth of axons of PC12 cells and primary neurons in culture (Brugg et al. 1993; DiTella et al. 1996). More recently, two gene-targeting studies were published that yielded contradictory results. In the first, the MAP1B coding region was interrupted at codon 571 by insertion of a stop codon (Edelmann et al. 1996). Mice homozygous for this mutation died before day 10 of gestation. However, even mice heterozygous for this mutation displayed severe neurological phenotypes, raising the possibility that this dominant phenotype was caused or enhanced by expression of the 571 amino acid amino-terminal MAP1B fragment with potential dominant-negative activity (Tögel et al. 1998) encoded by the targeted allele. In contrast, in the second MAP1B knockout study, mice homozygous for the mutation were viable and showed only a slight retardation in postnatal development (Takei et al. 1997). However, the targeting strategy employed did not exclude the synthesis of amino-terminally truncated MAP1B from alternative transcripts (Kutschera et al. 1998). Indeed, a protein reactive with three anti–MAP1B antibodies and slightly smaller than authentic MAP1B was still detected in brain extracts of homozygous mutants (Takei et al. 1997). Thus, the lack of severe phenotypes in these mice might be due to residual MAP1B expression that might be sufficient for proper albeit retarded development.
Here we generated a true null allele of MAP1B by introducing a large deletion in the gene that led to complete ablation of MAP1B expression and prevented synthesis of amino-terminal MAP1B fragments. This allowed us for the first time to assess the effect of MAP1B deficiency in vivo. In contrast to the previous inconclusive and contradictory results, we found that MAP1B is essential for the development and function of the nervous system and is involved in axon guidance of a subset of neurons. We also ruled out an expected essential function of MAP1B in retina development.
Materials and Methods
Gene Targeting
All experiments involving animals were performed in accordance with Austrian Federal Government laws and regulations. The targeting vector was constructed by joining a 4-kb BamHI/KpnI fragment located just upstream of exon 3 and a 2.3-kb SacII/XhoI fragment spanning intron 5 of the mouse MAP1B genomic locus (Kutschera et al. 1998) as the 5′ and 3′ homologous arms, respectively, to a thymidine kinase promoter driven neo cassette (provided by M. Kraus, Institute for Genetics, University of Cologne, Cologne, Germany) in reverse transcriptional orientation (see Fig. 1 A). The linearized targeting vector was electroporated into embryonic stem cells of lines R1 and E14.1 derived from different substrains of mouse strain 129 (Simpson et al. 1997; Threadgill et al. 1997), and G418-resistant colonies of cells heterozygous for the correctly targeted allele were isolated (Hogan et al. 1994) and identified by Southern blot analysis (Sambrook et al. 1989). Germ line chimeras were obtained as described (Hogan et al. 1994) and bred to C57BL/6 females to obtain F1 mice heterozygous for the MAP1B deletion. All results shown here were obtained with F2 and F3 offspring of F1 heterozygotes derived from targeted R1 stem cells. Genotyping of offspring was performed by Southern blot analysis of tail DNA by digestion with EcoRV and hybridization to an exon 7 probe (see Fig. 1) labeled using the AlkPhos Direct™ labeling kit (Amersham Pharmacia Biotech) according to the manufacturer's recommendations.
Figure 1.

Deletion of 93% of the coding region of the mouse chromosomal MAP1B locus leads to MAP1B heavy- and light-chain deficiency. (A) Schematic of the targeting strategy. MAP1B coding exons present in regular transcripts are indicated by filled boxes, noncoding exons (IIIA and IIIU) present in alternative transcripts by open boxes. Exons are numbered (I–VII). Relevant restriction sites, ATG start codons for regular transcripts (exon I), and alternative transcripts (exons IV and V), the heavy chain–light chain cleavage site of the polyprotein precursor, the TGA stop codon, as well as the position of the exon VII probe are indicated. The EcoRV restriction sites of the wild-type and targeted alleles that were used for diagnostic digests are marked (*). The horizontal arrow above the neo cassette indicates the direction of its transcription. For better visibility, the small MAP1B exons and the neo cassette are drawn disproportionately long. (B) Southern blot analysis of tail DNA of wild-type (+/+), heterozygous (+/−), and homozygous (−/−) MAP1B mutant mice using EcoRV and the exon VII probe. The diagnostic EcoRV fragments of the wild-type and targeted allele are 11.5 and 9 kb, respectively. Immunoblot analysis of MAP1B expression. 80 μg of total protein extracted from brains of 1-d-old wild-type (+/+), heterozygous (+/−) or homozygous (−/−) MAP1B mutant mice were fractionated on 7.5% (C) or 12% (D) gels. Blots were incubated with monoclonal antibody MAP5 (C) or polyclonal anti–light-chain antibody (D) to detect heavy chain (HC) and light chain (LC1), respectively. Tubulin (Tb) detected on the same blots with antitubulin antibodies served as internal control. The LC1 doublet detected in wild-type and heterozygous mice was seen consistently and might be due to post-translational modification of the light chain. (E) Immunoblot analysis of expression of an amino-terminal MAP1B fragment potentially encoded by the targeted allele. 80 μg of total protein extracted from brains of 1-d-old wild-type (+/+) or homozygous (−/−) MAP1B mutant mice were fractionated on a 6% gel. Expression of the heavy chain (HC) was analyzed using the monoclonal antibody MAP5 and an affinity purified polyclonal rabbit antipeptide antibody (E2) directed against mouse MAP1B amino acids 45–58 encoded by exon 2 as indicated. Both antibodies detected a protein band of the same size in wild-type but not mutant mice, demonstrating that the antipeptide antibody E2 recognized and was specific for the MAP1B heavy chain. The same samples were fractionated on a 17.5% gel. The resulting blot was cut into three horizontal segments. The segment containing proteins above 46 kD in size was incubated with antitubulin antibodies for the detection of tubulin (Tb; internal control). The segment containing proteins from 20–46 kD was incubated with the polyclonal anti–MAP1B light chain antibody (LC1). The segment containing proteins smaller than 20 kD was incubated with antibody E2 as indicated for the detection of the 95 amino acid–long amino-terminal MAP1B fragment potentially encoded by exons 1 and 2 of the targeted allele with an expected size of 10 kD. No such amino-terminal fragment was detected.
Protein Analysis
For protein expression analysis, freshly collected or frozen tissues were homogenized at 100 mg/ml in a buffer containing 8 M urea, 4% SDS, 150 mM Tris-HCl, pH 6.8, 12 mM EDTA, 0.3% DTT, 10 μM benzamidine, 1 mM PMSF, 2 μM pepstatin, 2 μM aprotinin, 2 μM leupeptin, 20% (vol/vol) glycerol, and 0.002% bromphenol blue with a PT 3000 Kinematica homogenizer. Samples were centrifuged at 10,000 g for 5 min at 4°C. The supernatants were sonicated, centrifuged again, and incubated at 65°C for 10 min. Aliquots were analyzed by SDS-PAGE and immunoblotting was performed as described (Sambrook et al. 1989).
A rabbit polyclonal anti–heavy chain antiserum termed E2 was raised against the synthetic peptide CETVTEEHLRRAIGN (mouse MAP1B exon 2 encoded amino acids 45–58 linked to an amino-terminal cysteine; Gramsch Laboratories), affinity purified as described (Tögel et al. 1998), and used at a dilution of 1:1,000. Other antibodies: anti–MAP1B heavy chain mAb MAP5 clone AA-6 (1:1000; Boehringer), affinity purified polyclonal rabbit anti–MAP1B light chain antibody (1 μg/ml; Tögel et al. 1998), anti–MAP1A heavy chain mAb HM-1 (1:500; Biogenesis), polyclonal rabbit anti–MAP1A light chain serum raised against the synthetic peptide CKGPVDRTSRTVPRPR (rat MAP1A amino acids 2605–2619 linked to an amino-terminal cysteine; 1:1,000), anti–MAP2 mAb HM-2 (1:800; Sigma-Aldrich), anti–tau mAb TAU1 (1:1,000; Boehringer), anti–light chain 3 polyclonal rabbit antibody (1:2,500; provided by J. Hammarback, Wake Forest University, School of Medicine, Department of Neurobiology and Anatomy, Winston Salem, NC), and anti–α-tubulin mAb B-5-1-2 (1:800; Sigma-Aldrich). Alkaline phosphatase-conjugated goat anti–rabbit and anti–mouse antibodies (1:7,500; Promega) were used as secondary antibodies.
Histopathology, Immunocytochemistry, Electron Microscopy, and Histomorphometry
1-d-old mice were anesthetized with ether, the bodywall was opened, and the animals were fixed overnight in 4% paraformaldehyde in PBS, pH 7.4. Older animals were perfused intracardially with 4% paraformaldehyde in PBS. Brains and spinal cords were dissected out, fixed overnight in 4% paraformaldehyde in PBS, and routinely embedded in paraffin. 5-μm-thick paraffin sections were stained with hematoxylin/eosin, Luxol fast blue myelin stain, and Bielschowski Silver impregnation for axons. Immunocytochemistry was performed on paraffin sections with antibodies against glial fibrillary acidic protein (Boehringer) and the macrophage antibody Mac-3 (PharMingen). Binding of primary antibodies was visualized with a biotin-avidine-peroxidase technique. For electron microscopy, animals were perfused with 3% glutaraldehyde in 0.1 M phosphate buffer. Small tissue blocks of brain (corpus callosum, optic nerve, and cerebellum) or sciatic nerve were impregnated in 1% osmic acid and routinely embedded in epoxy resin. 0.5-μm-thick plastic sections were cut on a Reichert Ultracut and stained with toluidine blue. Ultrathin sections were viewed in a Jeol 1010 electron microscope.
For histomorphometric analysis of Toluidin blue–stained semithin sections of sciatic nerves of MAP1B deficient (n = 5, aged 8–11 mo) and wild-type mice (n = 3, aged 8–11 mo) the DIGI-2000 software (version 3.0) was used. The amount of endoneural connective tissue was assessed by subtraction of the sum total of neuronal structures from the entirely assessed cross-sectional area of the respective histological sample. Evaluation of myelin thickness was achieved by subtracting the maximal inner from the maximal outer diameters of the myelin sheaths. Every axon in the elected field was counted, regardless of its size. For the statistical evaluation, groups of sizes were deliberately chosen to achieve a histogram representing the fiber sizes. Statistical significance of the differences of (a) axon diameters/areas and (b) thickness of the myelin sheaths between wild-type animals and MAP1B −/− mice were tested using the Kolmogoroff-Smirnoff test and Student's t test. Additionally, the correlation coefficient was calculated for a possible association between axon diameters/areas versus the myelin sheaths thickness.
For analysis of retinae, wild-type mice and mice heterozygous or homozygous for the MAP1B deletion of different ages (P18, P47, and P108) were killed by cervical dislocation, the eyes were enucleated, the anterior segments removed, and the posterior eyecups immersion fixed for 15–25 min in 4% paraformaldehyde in phosphate buffer (PB, 0.1 M, pH 7.4). After fixation, the retinae were dissected from the eyecup, cryoprotected in graded sucrose solutions (10, 20, and 30%), and sectioned vertically at 12 μm on a cryostat. Retinal sections were blocked for 1 h in a solution containing 10% normal goat serum (NGS), 1% bovine serum albumin, and 0.5% Triton-X-100 in PB. MAP1B was detected using the monoclonal antibody MAP5 (Sigma-Aldrich) or the affinity purified polyclonal rabbit anti–MAP1B light chain antibody (Tögel et al. 1998). γ-Aminobutyric acid C (GABAC) receptor subunits were detected using a rabbit anti–serum (Enz et al. 1996). All primary antibodies were diluted 1:100 in 3% NGS, 1% BSA, 0.05% Triton-X-100 in PB, and applied overnight at room temperature. After washing in PB, secondary antibodies were applied for 1 h. These included: goat anti–mouse or anti–rabbit IgG conjugated to Alexa TM 488 (Molecular Probes), diluted 1:500. Control experiments with omitting primary antisera resulted in unspecific background staining. All fluorescent specimens were viewed using an Axiophot microscope (Carl Zeiss, Inc.). Micrographs were taken with a cooled CCD camera (Spot2; Diagnostic Instruments) and processed using Adobe Photoshop 5.0.1.
Behavioral Tests and Electrophysiology
F1 male mice heterozygous or homozygous for the targeted MAP1B allele as well as wild-type controls were placed into a macrolite cage and observed for 10 min for their activity, exploring behavior, and ataxia. The different patterns were quantified by normal or abnormal activity, normal or abnormal exploring, and normal gait or ataxia, as described (Thalhammer et al. 1995).
Motor nerve conduction velocities were studied in the sciatic nerve in wild-type controls and in homozygous MAP1B mutant animals. For this procedure, animals were anaesthetized by i.p. injection of 60 mg/kg sodium pentobarbital and placed on a water-circulated heating pad throughout the procedure. The nerve was stimulated with percutaneous electrodes by supramaximal stimulation at the sciatic notch (Waikar et al. 1996) and summation motor potentials were recorded with a ring electrode at the metacarpus. The ground electrode was placed between stimulation and recording sites. After the procedure, the animals recovered spontaneously.
Results
General Characteristics of MAP1B-deficient Mice
To obtain true MAP1B-deficient mice, we generated a null allele termed MAP1BΔ93 harboring a deletion of 20.5 kb of chromosomal DNA. The deletion included coding exons 3–5 and part of exon 6 (Fig. 1 A), comprising 93% of the MAP1B coding sequence (codons 96–2389 of a total of 2,464 codons) and was designed to preclude synthesis of MAP1B from regular as well as alternative transcripts (Kutschera et al. 1998). Thus, MAP1BΔ93 differed from the allele generated by Takei et al. 1997, which carried merely a truncation of exon 1 that did not prevent synthesis of MAP1B from alternative transcripts and from the allele generated by Edelmann et al. 1996 in that it could not encode a large amino-terminal fragment of the heavy chain.
Consistent with the extent of the deletion in the MAP1B gene, neither transcripts containing exon 5 sequences (not shown) nor MAP1B heavy or light chain polypeptides (Fig. 1C and Fig. D) were detectable in brains of newborn mice homozygous for MAP1BΔ93. This confirmed that MAP1BΔ93 represented a true null allele incapable of encoding MAP1B heavy or light chain from regular or alternative transcripts. Moreover, we failed to detect an amino-terminal MAP1B fragment potentially encoded by exons 1 and 2 of the targeted allele (Fig. 1 E). Analysis of the level of expression of MAP2A, MAP2B, MAP2C, tau, MAP1A (heavy and light chain), and light chain 3, a 30-kD polypeptide that forms complexes with MAP1B and MAP1A (Schoenfeld et al. 1989) did not reveal consistent alterations in MAP1B-deficient mice (Fig. 2). Apparent minor changes observed in individual experiments were not manifest when normalized with respect to the intensity of the corresponding tubulin bands. Thus, MAP1B deficiency was not compensated for by increased expression of other brain-specific MAPs.
Figure 2.

Expression of brain-specific MAPs other than MAP1B in MAP1B-deficient mice. 80 μg of total protein extracted from brains of wild-type (+/+), heterozygous (+/−) or homozygous (−/−) MAP1B mutant mice of the indicated postnatal ages were analyzed by immunoblotting using antibodies specific for the respective proteins. Expression was not determined for MAP1A heavy chain at day 1 or MAP1A light chain at day 49.
Mice heterozygous for MAP1BΔ93 were indistinguishable in appearance from their wild-type littermates. In contrast, almost all homozygous pups (94%) displayed severe reduction in body weight that was more pronounced in the first 4 wk of life, in particular in the case of females (Table ). This reflected the fact that the weakest animals died during this time (Table ), with only the stronger animals displaying less severe weight reduction surviving into adulthood. The cause of this general weakness and body weight reduction is not known at this point. Homozygous pups appeared malnourished and dehydrated and showed a delay in body hair growth and eye-lid opening. However, not even the weakest, moribund animals had obvious neurological problems or pathological alterations (apart from the developmental defects in the brain, which were also found in those homozygous mutants that survived into adulthood, see below) that could explain their condition. Adult homozygous males and females were fertile, but were poor breeders. Quantitative assessment of activity, exploring activity, and ataxia revealed that exploring activity was absent in almost all homozygous MAP1B mutants (Fig. 3), whereas general activity and gait appeared normal. All phenotypic alterations observed in MAP1B −/− mice, including the reduction in body weight and each of the defects found in the nervous system (described below), occurred in 94–100% of mice homozygous for the MAP1B deletion and were never observed in heterozygotes or wild-type controls. Only a small percentage of homozygous mutants (3/52, 6%) did not show any abnormalities.
Table 1.
Body Weight of Wild-Type and MAP1B Mutant Mice
| Age | Sex | Wild type | Heterozygotes | Homozygotes | Weight reduction | |||
|---|---|---|---|---|---|---|---|---|
| wk | g | n | g | n | g | n | % | |
| 3 | male | 11.6 ± 2.3 | 11 | 10.3 ± 1.7 | 14 | 6.9 ± 2.3 | 11 | 33 |
| 3 | female | 11.1 ± 1.4 | 9 | 11.1 ± 1.7 | 12 | 4.9 ± 0.7 | 6 | 56 |
| 8 | male | 28.1 ± 2.5 | 15 | 28.4 ± 1.7 | 24 | 20.1 ± 0.9 | 9 | 29 |
| 8 | female | 22.8 ± 2.3 | 7 | 22.9 ± 1.2 | 9 | 19.4 ± 1.6 | 6 | 15 |
Table 2.
Postnatal Lethality of MAP1B-deficient Mice
| Number of progeny | ||||||
|---|---|---|---|---|---|---|
| +/+ | +/− | −/− | ||||
| % | % | % | ||||
| Total born | 61 | 25 | 140 | 55 | 52 | 20 |
| Adult male | 27 | 68 | 18 | |||
| Adult female | 32 | 67 | 5 | |||
| Postnatal lethality | 2 | 3.3 | 5 | 3.6 | 29 | 55.8 |
Figure 3.

MAP1B-deficient mice lack exploring activity. Exploring activity of wild-type control mice (+/+) and mice homozygous (−/−) for the targeted MAP1B allele were tested for exploring activity.
Retinal Development and GABAC Receptor Clustering Are Not Impaired in MAP1B-deficient Mice
Almost all MAP1B-deficient mice (94%) displayed uni- or bilateral abnormalities of their eyes in that the eyes appeared smaller or retracted despite eyeballs of normal size. A similar phenotype was observed in more than half of homozygous MAP1B mutants in one of the previous gene-targeting studies (Takei et al. 1997). In contrast, Edelmann et al. 1996 reported gross deformation of ocular orbs in heterozygous MAP1B mutants and attributed apparent blindness of heterozygotes to disorganization of the retina. Further evidence for a role of MAP1B in retinal development came from the finding that MAP1B binds to a retina-specific GABAC receptor subunit (Hanley et al. 1999). Therefore, we reexamined retinal structure and GABAC receptor clustering in detail in the true MAP1B null mice homozygous for MAP1BΔ93.
In wild-type mice, MAP1B expression was widespread during the formation of the retinal layers, but in contrast to what had been reported for the rat (Hanley et al. 1999), it was downregulated in the adult (Fig. 4). We used heavy and light chain–specific antibodies that in rat retina produced results similar to those reported by Hanley et al. 1999, and observed in mouse retina at postnatal day 9 a distinct labeling of the inner plexiform layer (IPL) with a concentration of MAP1B in several discrete bands and staining in the outer nuclear layer (ONL; Fig. 4 A). In the adult mouse retina, only weak and diffuse label was present in the inner retina, while photoreceptors [inner segments (IS), ONL] were labeled more prominently (Fig. 4 C). The label, although diffuse, was specific, since by omitting the primary antibody it was lost (Fig. 4 B). When retinae of MAP1B −/− mice were immunostained for MAP1B, no specific fluorescence was detectable (Fig. 4 D). Despite this lack of MAP1B, the retinae of MAP1B-deficient mice did not show morphological changes (Fig. 4). The retinal layering and the cell densities appeared normal. We also immunolabeled such retinae with markers that recognize specific cell types in the mouse retina (Haverkamp, S., and H. Wässle, unpublished results). Horizontal cells immunoreactive for calbindin, amacrine cells immunoreactive for calretinin, and bipolar cells immunoreactive for protein kinase Cβ appeared unchanged (not shown).
Figure 4.
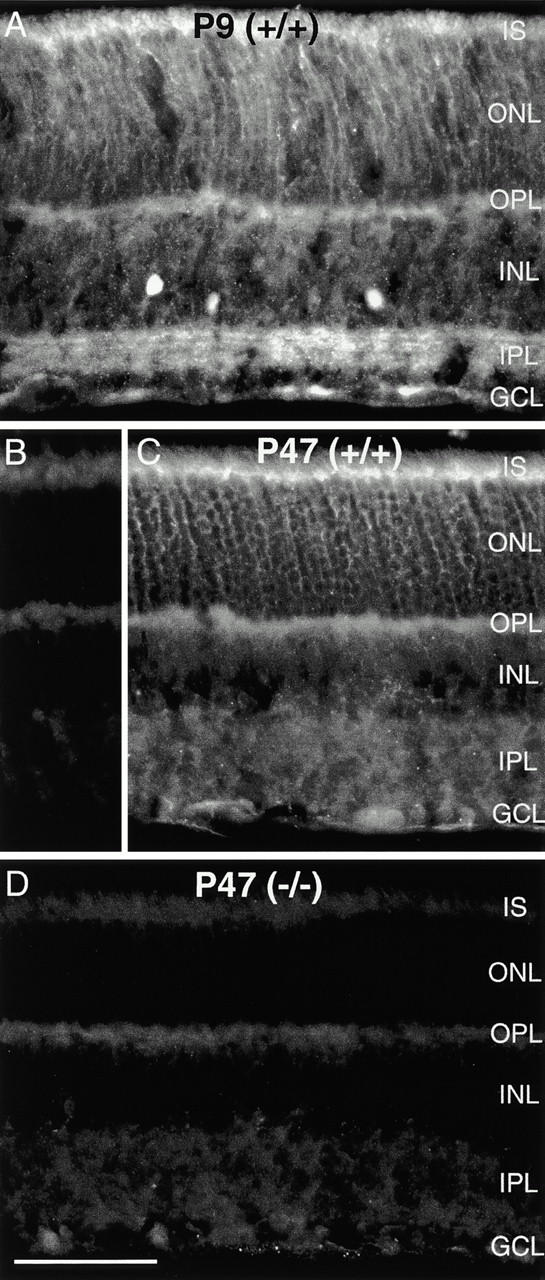
MAP1B expression in the mouse retina. (A) Fluorescence micrograph of a vertical section through a wild-type (+/+) mouse retina at postnatal day P9 that was immunostained for MAP1B. The inner plexiform layer is clearly labeled with a concentration of MAP1B in several discrete bands. MAP1B is also expressed in the outer retina. (B) Vertical section through an adult (+/+) mouse retina (postnatal day P47). In this control micrograph, the primary antibody against MAP1B was omitted and only nonspecific background label is detected. (C) Fluorescence micrograph of a vertical section through an adult (P47) wild-type (+/+) mouse retina that was immunostained for MAP1B. Diffuse, weak fluorescence is present in the inner retina, the outer retina is more intensely labeled. (D) Section through a MAP1B-deficient (−/−) mouse retina that was immunostained for MAP1B. No specific label can be detected, background intensity is identical to B. However, comparison of D and C shows that the retinal layers are unchanged in the MAP1B mutant mouse. IS, photoreceptor inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; GCL, ganglion cell layer. Bar, 50 μm.
We next analyzed the expression and distribution of GABAC receptors in wild-type and MAP1B-deficient mice. GABAC receptors show a closely similar distribution in all mammalian retinae investigated (Wässle et al. 1998). Consistent with these observations, we found that in wild-type mice GABAC receptors were expressed by rod and cone bipolar cells, including a low level of expression in their plasma membrane in the inner nuclear layer (INL, Fig. 5 A). In the outer plexiform layer (OPL), GABAC receptors were aggregated at the bipolar cell dendritic terminals. In the IPL, strong, punctate immunofluorescence was observed (Fig. 5A and Fig. C). The distribution of puncta in the IPL was not uniform, but horizontal bands of higher and lower density of puncta were present. Electron microscopy has shown that the puncta (hot spots) represent a clustering of GABAC receptors in postsynaptic densities (Enz et al. 1996; Koulen et al. 1998). This clustering was not perturbed in MAP1B-deficient mice (Fig. 5, compare A and C with B and D). Comparison of the low power micrographs (Fig. 5A and Fig. B) showed that the label in the OPL, INL, and IPL was identical between the control and the MAP1B −/− retina. This was also true for the layering of the puncta in the IPL. The high-power micrographs (Fig. 5C and Fig. D) showed the distinct clustering of the GABAC receptors indistinguishable in wild-type and MAP1B mutant mice. This was quantified by counting the number of puncta in vertical strips of 30-μm width through the IPL and calculating their densities. In the (+/+) mouse retina, the average density of GABAC immunoreactive hot spots was 34.3 ± 2.8 (n = 8) per 100 μm2. In the (−/−) mouse retina, the average density was 36.4 ± 2.8 (n = 8). These results suggest that MAP1B is not the crucial parameter in the clustering of GABAC receptors at postsynaptic densities. We also found no change in MAP1B-deficient mice in the clustering of GABAA receptor subunits α1, α2, α3, and γ2, the glycine receptors, the glutamate receptor subunits GluR1, 4, and 6/7, the N-methyl-d-arginine receptor subunits NR1C2′ and NR2B, and the postsynaptic density protein PSD-95 (not shown).
Figure 5.

GABAC receptor clustering in MAP1B-deficient retina. (A) Section through a (+/+) mouse retina that was immunolabeled for the GABAC receptor subunits. (B) GABAC receptor immunolabeling in a(−/−) mouse retina. (C) High power micrograph of a (+/+) mouse retina showing the GABAC receptor synaptic clusters in the IPL. (D) GABAC receptor synaptic clusters in the IPL of a (−/−) mouse retina. IS, photoreceptor inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bar: 55 μm (A and B) and 20 μm (C and D).
Agenesis of the Corpus Callosum in MAP1B-deficient Mice
The neuropathological hallmark of MAP1B-deficient animals was agenesis of the corpus callosum. 80% of the animals with homozygous deletion of MAP1B (n = 15) displayed total agenesis of the corpus callosum (Fig. 6 and Fig. 7). In the parietal portions of the brain, the corpus callosum was replaced by a fine fibrous membrane in which the choroide plexus of the third ventricle was inserted. This membrane was frequently lacking in more posterior portions of the brain, where the third ventricle was found in direct continuity with the interhemispheric subarachnoid space. In most animals, a longitudinal bundle of myelinated fibers was found (Fig. 6b, Fig. d, Fig. f, and Fig. h), resembling the Probst bundle present in agenesis of the corpus callosum in humans (Probst 1901). It was located at the most medial parts of the hemispheric white matter, traversing through the entire hemisphere of the brain from the frontal to the occipital lobe. The hemispheric cortex was normally developed, although the gyrus cinguli was in part rotated ventrally. In addition, prominent aberrant bundles of myelinated axons were present in the deeper layers of the parietal cortex (Fig. 6d and Fig. f). No astrocytic scaring was found in comparison with normal wild-type animals (Fig. 6i and Fig. j). The hippocampi showed a regular cortical architecture, yet they were dislocated laterally and the hippocampal commissure was absent. In 20% of the homozygous animals, the developmental defect described above was incomplete. This was either reflected by an unusually thin corpus callosum throughout the whole brain or a lack of the corpus callosum with similar abnormalities as described above, restricted to the posterior portions of the forebrain.
Figure 6.
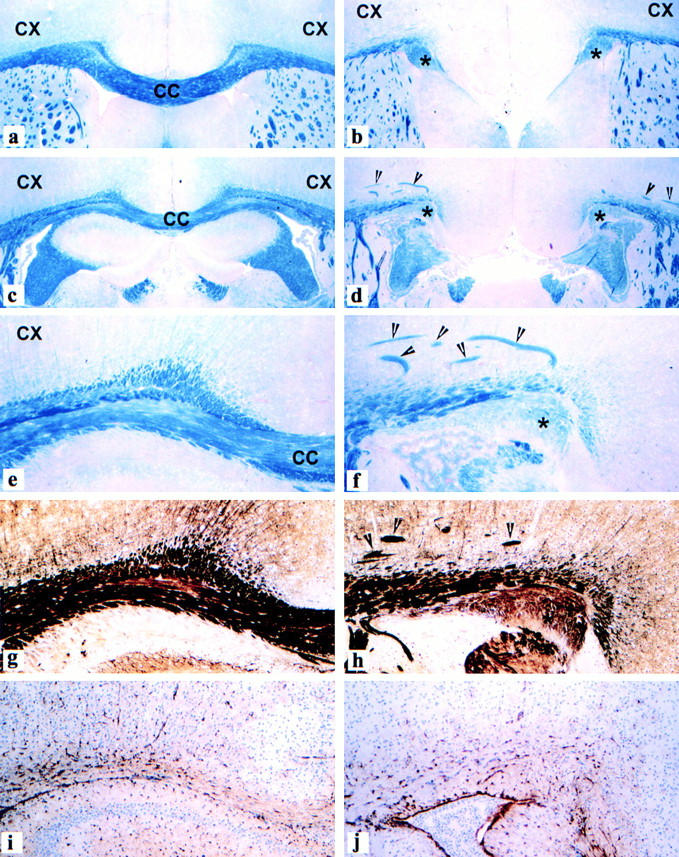
Agenesis of the corpus callosum and formation of Probst bundles in the brain of MAP1B-deficient mice. (a, c, e, g, and i) Frontal sections through the cerebral white matter and corpus callosum (CC) of a normal wild-type mouse brain, showing normal architecture of myelinated fiber tracts including the corpus callosum (a, c, and e, blue), axons (g, dark brown), and astrocytes (i, brown). (b, d, f, h, and j) Corresponding brain region of a homozygous MAP1B-deficient mouse. The corpus callosum is absent. The cerebral white matter ends medially in a thick bundle of myelinated fibers (*Probst bundle). Within the deeper layers of the cortex, thick bundles of myelinated axons are visible (arrowheads). Despite the massive structural changes in the brain tissue, there is no increase in reactive astrocytes. CX, cerebral cortex. (a–d) Luxol fast blue myelin stain; myelinated fiber tracts such as the corpus callosum are stained blue, areas of grey matter are stained red, 40×. (e and f) Luxol fast blue myelin stain, 120×. (g and h) Bielschowski silver impregnation; axons within fiber tracts are stained black, areas of grey matter reveal light brown staining, 120×. (i and j) Immunocytochemistry for glial fibrillary acidic protein; astrocytes containing glial fibrillary acidic protein are stained brown. Cellular nuclei of the tissue are counterstained with hematoxylin blue, 120×.
Figure 7.
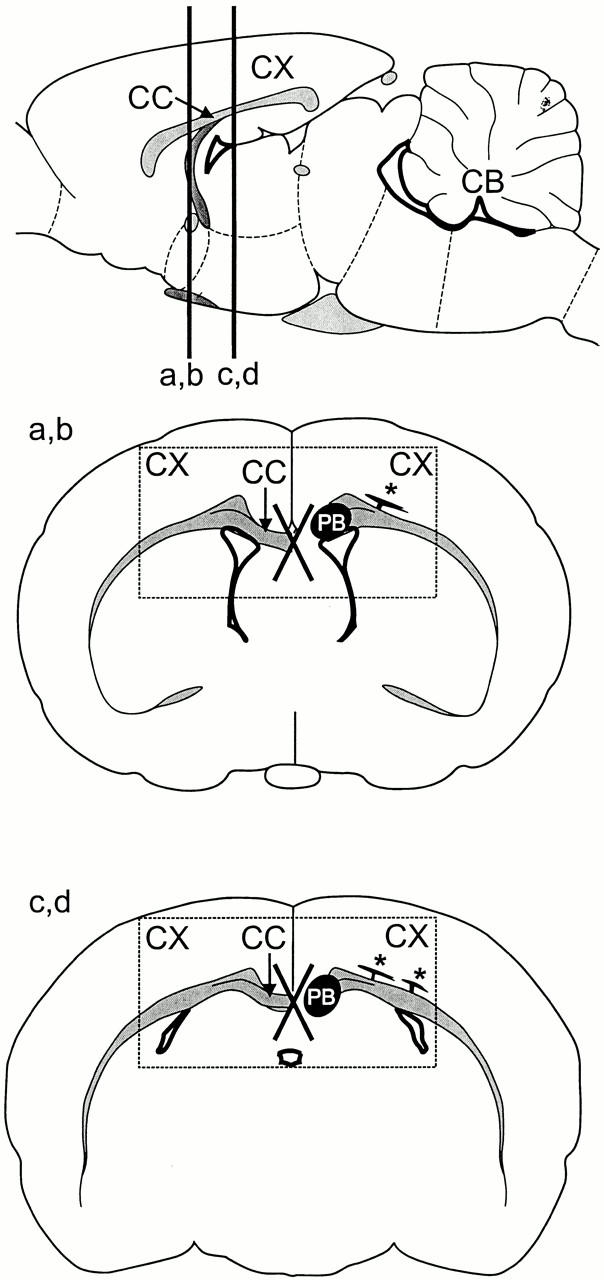
Schematic representation of brain areas shown in Fig. 6. (Top) Side view of the rodent brain (sagittal section). The locations of the frontal sections shown in Fig. 6, a and b and c and d, are indicated by vertical lines labeled a,b and c,d, respectively. CB, cerebellum. (Middle and bottom) Schematic representation of the frontal sections shown in Fig. 6, a and b (a,b) and c and d (c,d). The left side of the brain in each schematic shows the normal situation, the right side the changes in MAP1B-deficient animals. In the normal brain, nerve fibers insert into the corpus callosum (CC) and cross the midline. In MAP1B-deficient animals, these fibers do not cross the midline (indicated by the X), but form a thick bundle of fibers that runs in the longitudinal direction of the brain (PB, Probst bundle). In addition, thick aberrant fiber bundles leave the centrum semiovale and enter the basal layers of the cortex, where they turn by 90° and further traverse in parallel to the cortical surface (*). The rectangles in the middle and bottom panels delineate the areas shown in Fig. 6, a and b and c and d, respectively. CX, cerebral cortex.
The pathology of MAP1B-deficient animals was studied on step serial sections of 0.1-mm distance covering the whole brain and spinal cord. No other malformations were found. In particular, other commissures, such as the commissura anterior, the commissura posterior, the commissura habenularum, and the brain stem midline structures, revealed no defects. No developmental abnormalities were found in mice heterozygous for the MAP1B mutation or in wild-type controls.
Peripheral Nerves in MAP1B-deficient Mice Have Axonal and Myelinization Defects Resulting in Reduced Motor Nerve Conduction Velocity
Histological and histomorphometrical assessment of the sciatic nerves revealed a marked predominance of small-caliber myelinated axons in MAP1B-deficient mice (Fig. 8 A). With the exception of axons in the 70- and 80-μm2 classes, numbers of axons in intermediate and large size classes were reduced compared with wild-type controls, resulting in a left shift of the size distribution histogram. In addition, the myelin sheaths of axons of a given diameter class had reduced thickness in MAP1B-deficient mice (Fig. 8 B). Concomitantly, the proportion of endoneural connective tissue was increased, probably due to reduced axon diameters, since we found no evidence for ongoing myelin degeneration or myelinophagocytosis. The overall number of axons appeared unchanged (2.93 ± 0.31 × 104, n = 3 in wild-type controls, and 3.05 ± 0.68 × 104, n = 4 in homozygous mutants). Consistent with this observation, motor nerve conduction velocity was reduced to 19.82 ± 2.93 (mean ± SD, n = 8) in homozygous mutants compared with 30.60 ± 6.71 (mean ± SD, n = 16) in wild-type controls. None of the conduction velocities of the homozygous animals was within 1 SD of the values observed in wild-type mice. The difference in conduction velocities between controls and homozygotes was statistically significant (P < 0.05) when compared with one way analysis of variance.
Figure 8.
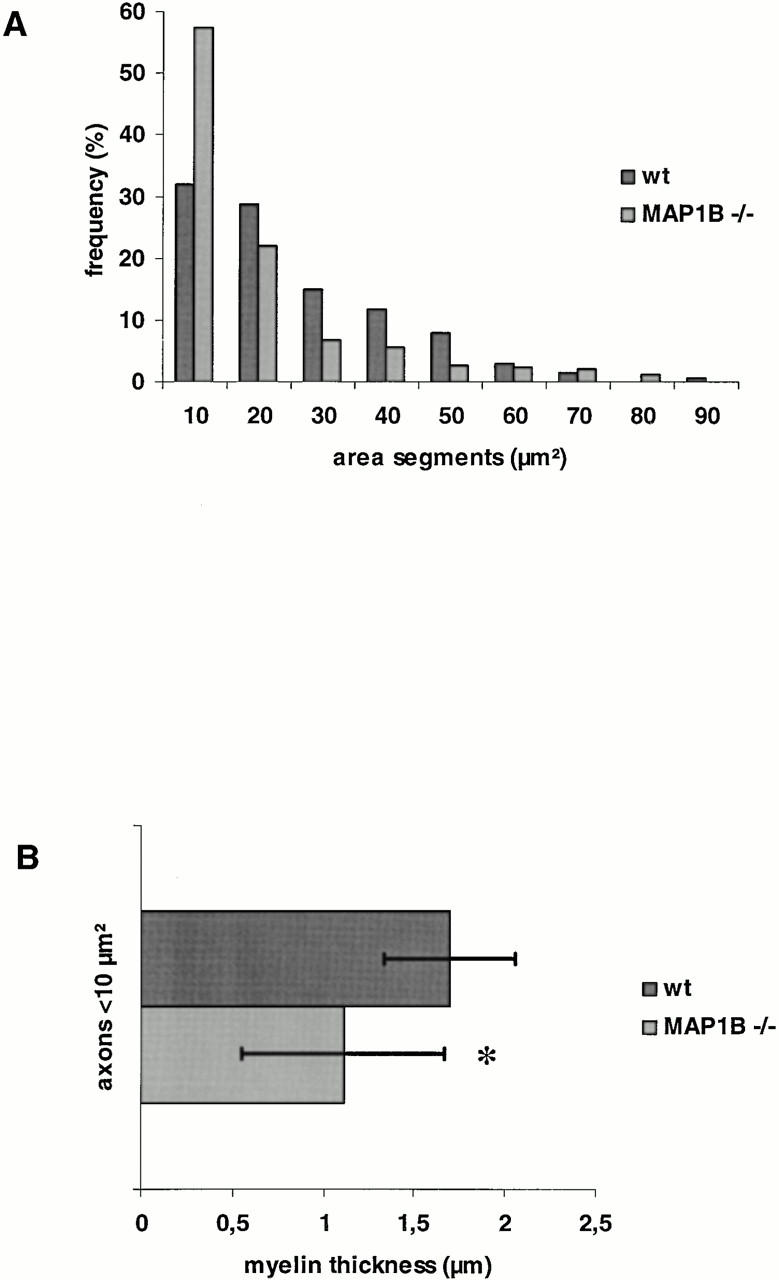
Predominance of small myelinated nerve fibers (A) and reduced thickness of myelin sheaths (B) in the sciatic nerve of MAP1B-deficient mice. Sciatic nerve preparations from three wild-type and four MAP1B-deficient mice were used to obtain the data. (A) The size distribution of axon areas differed significantly (Kolmogoroff-Smirnoff test, P < 0.001) between MAP1B-deficient (n = 465) and wild-type (n = 206) sciatic axons. (B) Myelin sheath thickness was assessed from axons with areas <10 μm (MAP1B −/−, n = 265, vs. wild-type, n = 66). *The mean values of the myelin sheath thickness were significantly smaller (t test, P < 0.001) in MAP1B −/− mice (1.11 ± 0.36 μm) compared with wild-type animals (1.70 ± 0.57 μm).
Discussion
Mice Homozygous for MAP1BΔ93 Are the First True MAP1B-deficient Mice
By deleting 93% of the coding capacity of the MAP1B gene, we have generated a true null allele, MAP1BΔ93, and, in contrast to the two previous MAP1B gene-targeting studies (Edelmann et al. 1996; Takei et al. 1997), were able to assess for the first time the effects of MAP1B deficiency on the development of the murine nervous system. The previous MAP1B targeting studies produced inconclusive and contradictory results. The targeting strategy employed by Takei et al. 1997 reduced MAP1B to ∼5% of its normal level, but did not result in complete ablation of MAP1B expression. Not surprisingly, homozygous mutants merely displayed a transient retardation in development and did not reveal the effects of true MAP1B deficiency, a caveat also limiting the significance of their recent results obtained in MAP1B/tau double knockout mice in which they used the same incompletely inactivated MAP1B allele (Takei et al. 2000).
Contradictory to the results obtained by Takei et al. 1997, Edelmann et al. 1996 reported that mice homozygous for a MAP1B allele termed MAP1B571 (which is interrupted at codon 571 by a stop codon) died in early embryonic development and 20% of mice heterozygous for MAP1B571 (which still expressed MAP1B from the intact allele) displayed ataxia and spastic tremor and abnormalities in the cerebellum. At present, it is not clear why the phenotype of MAP1B571 was so severe. One possibility is that the severity is influenced by the genetic background on which this mutation was introduced. In contrast to the mice generated in the present study, one component of the background of MAP1B571 mice was derived from 129P2/Ola, a strain displaying axonal guidance problems and defects in brain development in wild-type mice (Wahlsten 1982). It is conceivable that the additional reduction or ablation of MAP1B expression on this background lead to severe developmental problems. A second likely factor that might limit analysis of MAP1B function in knockout mice of Edelmann et al. 1996 is the potential expression of the 571 amino acid amino-terminal MAP1B fragment encoded by the targeted allele. This fragment might have dominant-negative activity (Tögel et al. 1998), resulting in the dominant phenotypes observed in mice heterozygous for MAP1B571.
Here we generated MAP1BΔ93, an allele carrying a deletion of most of the MAP1B gene. This deletion prevented synthesis of MAP1B from regular as well as alternative transcripts, resulted in complete ablation of MAP1B heavy chain and light chain expression, prevented the expression of amino-terminal MAP1B fragments, did not lead to dominant alterations in heterozygous mutants, and was analyzed on a genetic background permissive to complete MAP1B deficiency.
MAP1B Is Required for Axon Guidance
By generating true MAP1B-deficient mice, we were able to demonstrate that MAP1B was required for axon guidance in a subset of neurons during brain development. Histological examination of homozygous MAP1B mutant mice revealed a specific developmental brain defect, agenesis of the corpus callosum, and the formation of Probst bundles (Probst 1901). In addition, we found prominent aberrant growth of fibers in thick bundles, located in the lower layers of the cortex. This may suggest that callosal fibers, which are unable to cross the midline, still are at least in part targeted to cortical regions, albeit on the ipsilateral side. Agenesis of the corpus callosum has been observed in some inbred strains of mice and is also influenced by rearing conditions (Wahlsten 1982). However, in the present study, agenesis of the corpus callosum and the formation of Probst bundles strictly correlated with the MAP1B genotype and was observed in all mice homozygous for the MAP1B deletion, but never in heterozygous mutants or wild-type controls, demonstrating that MAP1B is essential for corpus callosum formation.
Despite the fact that MAP1B is expressed in axons, dendrites, and growth cones throughout the central nervous system during development, the axonal guidance defect is limited to the corpus callosum and the hippocampal commissure. One possible explanation for this specificity is that redundant backup mechanisms exist for the guidance of most axons, whereas corpus callosum formation is critically dependent on all components of a single mechanism. MAP1B might be a component of a signal transduction pathway guiding axons across an environment determined by midline glia cells. These cells express laminin on their cell surface (Liesi and Silver 1988) and it has been shown that MAP1B is necessary for laminin-enhanced axonal growth in vitro (DiTella et al. 1996). In addition, callosal axon guidance defects are observed in mice lacking the cdk5 activator p35 (Kwon et al. 1999), a protein implicated in regulation of laminin-induced MAP1B phosphorylation (Paglini et al. 1998). Thus, MAP1B might be involved in laminin/integrin-mediated cell adhesion and/or signaling and this pathway could be crucial for corpus callosum formation. Likewise, MAP1B, capable of binding to actin fibers (Pedrotti and Islam 1996; Tögel et al. 1998), might be involved in the regulation of actin polymerization in the growth cone by the netrin/MENA pathway, which is also involved in corpus callosum formation (Serafini et al. 1996; Fazeli et al. 1997; Lanier et al. 1999). Involvement of MAP1B in laminin or netrin signaling might not be mutually exclusive, since laminin and netrin share structural homologies (Serafini et al. 1996).
An alternative explanation for the selective defect in MAP1B-deficient brains might be that midline crossing of callosal fibers is limited to a temporal window. If callosal axons were to be slowed down in their progress towards the midline as a result of MAP1B deficiency, they might miss the permissive window and, instead of crossing, extend parallel to the midline or turn back. In this context, it is of interest that a Drosophila MAP termed Futsch is necessary for axonal growth and extension (Hummel et al. 2000). Futsch is quite distinct from any of the vertebrate MAPs, but shares two stretches of sequence homology with MAP1B and MAP1A.
MAP1B Is Required for Development and Function of the Peripheral Nervous System
In contrast to the previous MAP1B targeting studies, we were able to demonstrate that MAP1B deficiency lead to permanent defects in the peripheral nervous system. Morphometric analysis revealed that the axons constituting the adult sciatic nerve of MAP1B-deficient mice were of decreased diameters and that the thickness of myelin sheaths of the remaining axons was reduced. Electrophysiological measurements in the sciatic nerve showed a reduction in conductance velocities in MAP1B-deficient mice by ∼35%, consistent with the observed alterations in axon diameter and myelin sheaths. Thus, our findings extend observations of Takei et al. 1997, who reported that reduction of MAP1B expression leads to a delay in myelination in the optic nerve. The reduction in numbers of large myelinated axons observed in our mice is likely to be due to the failure of most MAP1B-deficient axons to grow to larger diameters and not to a selective loss of large-caliber axons in the adult, since we were unable to observe any signs of axon-degeneration and the numbers of axons per sciatic nerve did not significantly differ in wild-type and homozygous mutant mice. The reduction in the thickness of myelin sheaths could possibly be due to the lack of MAP1B in Schwann cells and/or the corresponding axons during the process of myelination. Both Schwann cells and axons contain MAP1B (Fawcett et al. 1994; Bush et al. 1996b; Ramón-Cueto and Avila 1997).
MAP1B Is Not Essential for Retina Development and GABAC Receptor Clustering
The recent finding that MAP1B interacts with the cytoplasmic domain of the retina-specific GABAC receptor subunit ρ1 has lead to the hypothesis that MAP1B belongs to a family of synapse-associated proteins that are involved in clustering ion channels at postsynaptic sites providing a link to the cytoskeleton and possibly contributing to the regulation of synaptic efficacy by electrical activity (Hanley et al. 1999). In contrast to previous MAP1B targeting studies, the generation of a true null allele of MAP1B allowed us to address the question of whether MAP1B was indeed essential for retina development and GABAC receptor clustering. If this were the case, one would expect an absence of GABAC immunoreactive puncta, or at least a reduction of their density, in the MAP1B-deficient mouse retina. However, we found no difference in the pattern of GABAC staining between MAP1B-deficient and wild-type mice. Whereas our results do not rule out that MAP1B participates in receptor clustering and linkage to the cytoskeleton, they do demonstrate that MAP1B does not play an essential role in GABAC receptor clustering.
Acknowledgments
We are grateful to J.A. Hammarback for providing the LC3 antibody and to M. Kraus for the neo cassette neoflox-8, and to P. Holzfeind and T. Raffelsberger for their help with statistical analysis.
This research was supported by a grant from the Austrian Science Fund (Project No. F607).
Footnotes
Abbreviations used in this paper: GABA, γ-aminobutyric acid; IPL, inner plexiform layer; MAP, microtubule-associated protein.
References
- Black M.M., Slaughter T., Fischer I. Microtubule-associated protein 1b (MAP1b) is concentrated in the distal region of growing axons. J. Neurosci. 1994;14:857–870. doi: 10.1523/JNEUROSCI.14-02-00857.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugg B., Reddy D., Matus A. Attenuation of microtubule-associated protein 1B expression by antisense oligonucleotides inhibits initiation of neurite outgrowth. Neuroscience. 1993;52:489–496. doi: 10.1016/0306-4522(93)90401-z. [DOI] [PubMed] [Google Scholar]
- Bush M.S., Goold R.G., Moya F., Gordon-Weeks P.R. An analysis of an axonal gradient of phosphorylated MAP 1B in cultured rat sensory neurons Eur. J. Neurosci. 8 1996. 235 248a [DOI] [PubMed] [Google Scholar]
- Bush M.S., Tonge D.A., Woolf C., Gordon-Weeks P.R. Expression of a developmentally regulated, phosphorylated isoform of microtubule-associated protein 1B in regenerating axons of the sciatic nerve Neuroscience. 73 1996. 553 563b [DOI] [PubMed] [Google Scholar]
- DiTella M.C., Feiguin F., Carri N., Kosik K.S., Cáceres A. MAP-1B/TAU functional redundancy during laminin-enhanced axonal growth. J. Cell Sci. 1996;109:467–477. doi: 10.1242/jcs.109.2.467. [DOI] [PubMed] [Google Scholar]
- Edelmann W., Zervas M., Costello P., Roback L., Fischer I., Hammarback J.A., Cowan N., Davies P., Wainer B., Kucherlapati R. Neuronal abnormalities in microtubule-associated protein 1B mutant mice. Proc. Natl. Acad. Sci. USA. 1996;93:1270–1275. doi: 10.1073/pnas.93.3.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enz R., Brandstätter J.H., Wässle H., Bormann J. Immunocytochemical localization of the GABAC receptor rho subunits in the mammalian retina. J. Neurosci. 1996;16:4479–4490. doi: 10.1523/JNEUROSCI.16-14-04479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett J.W., Mathews G., Housden E., Goedert M., Matus A. Regenerating sciatic nerve axons contain the adult rather than the embryonic pattern of microtubule associated proteins. Neuroscience. 1994;61:789–804. doi: 10.1016/0306-4522(94)90402-2. [DOI] [PubMed] [Google Scholar]
- Fazeli A., Dickinson S.L., Hermiston M.L., Tighe R.V., Steen R.G., Small C.G., Stoeckli E.T., Keino-Masu K., Masu M., Rayburn H. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature. 1997;386:796–804. doi: 10.1038/386796a0. [DOI] [PubMed] [Google Scholar]
- García-Pérez J., Avila J., Díaz-Nido J. Implication of cyclin-dependent kinases and glycogen synthase kinase 3 in the phosphorylation of microtubule-associated protein 1B in developing neuronal cells. J. Neurosci. Res. 1998;52:445–452. doi: 10.1002/(SICI)1097-4547(19980515)52:4<445::AID-JNR8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Hammarback J.A., Obar R.A., Hughes S.M., Vallee R.B. MAP1B is encoded as a polyprotein that is processed to form a complex N-terminal microtubule-binding domain. Neuron. 1991;7:129–139. doi: 10.1016/0896-6273(91)90081-a. [DOI] [PubMed] [Google Scholar]
- Hanley J.G., Koulen P., Bedford F., Gordon-Weeks P.R., Moss S.J. The protein MAP-1B links GABAC receptors to the cytoskeleton at retinal synapses. Nature. 1999;397:66–69. doi: 10.1038/16258. [DOI] [PubMed] [Google Scholar]
- Hogan B., Beddington R., Constantini F., Lacy E. Manipulating the mouse embryo. A laboratory manual. Cold Spring Harbor Laboratory Press, ; Cold Spring Harbor, NY: 1994. [Google Scholar]
- Hummel T., Krukkert K., Roos J., Davis G., Klämbt C. Drosophila Futsch/22C10 is a MAP1B-like protein required for dendritic and axonal development. Neuron. 2000;26:357–370. doi: 10.1016/s0896-6273(00)81169-1. [DOI] [PubMed] [Google Scholar]
- Koulen P., Brandstätter J.H., Enz R., Bormann J., Wässle H. Synaptic clustering of GABA(C) receptor rho-subunits in the rat retina. Eur. J. Neurosci. 1998;10:115–127. doi: 10.1046/j.1460-9568.1998.00005.x. [DOI] [PubMed] [Google Scholar]
- Kutschera W., Zauner W., Wiche G., Propst F. The mouse and rat MAP1B genesgenomic organization and alternative transcription. Genomics. 1998;49:430–436. doi: 10.1006/geno.1998.5294. [DOI] [PubMed] [Google Scholar]
- Kwon Y.T., Tsai L.-H., Crandall J.E. Callosal axon guidance defects in p35−/− mice. J. Comp. Neurol. 1999;415:218–229. doi: 10.1002/(sici)1096-9861(19991213)415:2<218::aid-cne6>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Lanier L.M., Gates M.A., Witke W., Menzies A.S., Wehman A.M., Macklis J.D., Kwiatkowski D., Soriano P., Gertler F.B. Mena is required for neurulation and commissure formation. Neuron. 1999;22:313–325. doi: 10.1016/s0896-6273(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Lien L.L., Feener C.A., Fischbach N., Kunkel L.M. Cloning of human microtubule-associated protein 1B and the identification of a related gene on chromosome 15. Genomics. 1994;22:273–280. doi: 10.1006/geno.1994.1384. [DOI] [PubMed] [Google Scholar]
- Liesi P., Silver J. Is astrocyte laminin involved in axon guidance in the mammalian CNS? Dev. Biol. 1988;130:774–785. doi: 10.1016/0012-1606(88)90366-1. [DOI] [PubMed] [Google Scholar]
- Lucas F.R., Goold R.G., Gordon-Weeks P.R., Salinas P.C. Inhibition of GSK-3β leading to the loss of phosphorylated MAP-1B is an early event in axonal remodelling induced by WNT-7a or lithium. J. Cell Sci. 1998;111:1351–1361. doi: 10.1242/jcs.111.10.1351. [DOI] [PubMed] [Google Scholar]
- Paglini G., Pigino G., Kunda P., Morfini G., Maccioni R., Quiroga S., Ferreira A., Caceres A. Evidence for the participation of the neuron-specific CDK5 activator P35 during laminin-enhanced axonal growth. J. Neurosci. 1998;18:9858–9869. doi: 10.1523/JNEUROSCI.18-23-09858.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrotti B., Islam K. Dephosphorylated but not phosphorylated microtubule associated protein MAP1B binds to microfilaments. FEBS Lett. 1996;388:131–133. doi: 10.1016/0014-5793(96)00520-0. [DOI] [PubMed] [Google Scholar]
- Probst M. Über den Bau des balkenlosen Großhirns, sowie über Mikrogyrie und Heterotypie der grauen Substanz. Arch. Psychiatr. 1901;34:709–786. [Google Scholar]
- Ramón-Cueto A., Avila J. Differential expression of microtubule-associated protein 1B phosphorylated isoforms in the adult rat nervous system. Neuroscience. 1997;77:485–501. doi: 10.1016/s0306-4522(96)00437-x. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Molecular cloninga laboratory manual. Cold Spring Harbor Laboratory Press, ; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Schoenfeld T.A., McKerracher L., Obar R., Vallee R.B. MAP 1A and MAP 1B are structurally related microtubule associated proteins with distinct developmental patterns in the CNS. J. Neurosci. 1989;9:1712–1730. doi: 10.1523/JNEUROSCI.09-05-01712.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini T., Colamarino S.A., Leonardo E.D., Wang H., Beddington R., Skarnes W.C., Tessier-Lavigne M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 1996;87:1001–1014. doi: 10.1016/s0092-8674(00)81795-x. [DOI] [PubMed] [Google Scholar]
- Simpson E.M., Linder C.C., Sargent E.E., Davisson M.T., Mobraaten L.E., Sharp J.J. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat. Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- Takei Y., Kondo S., Harada A., Inomata S., Noda T., Hirokawa N. Delayed development of nervous system in mice homozygous for disrupted microtubule-associated protein 1B (MAP1B) gene. J. Cell Biol. 1997;137:1615–1626. doi: 10.1083/jcb.137.7.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y., Teng J., Harada A., Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalhammer J.G., Vladimirova M., Bershadsky B., Strichartz G.R. Neurologic evaluation of the rat during sciatic nerve block with lidocaine. Anesthesiology. 1995;82:1013–1025. doi: 10.1097/00000542-199504000-00026. [DOI] [PubMed] [Google Scholar]
- Threadgill D.W., Yee D., Matin A., Nadeau J.H., Magnuson T. Genealogy of the 129 inbred strains129/SvJ is a contaminated inbred strain. Mamm. Genome. 1997;8:390–393. doi: 10.1007/s003359900453. [DOI] [PubMed] [Google Scholar]
- Tögel M., Eichinger R., Wiche G., Propst F. A 45-amino acid residue domain necessary and sufficient for proteolytic cleavage of the MAP1B polyprotein precursor. FEBS Lett. 1999;451:15–18. doi: 10.1016/s0014-5793(99)00523-2. [DOI] [PubMed] [Google Scholar]
- Tögel M., Wiche G., Propst F. Novel features of the light chain of microtubule-associated protein MAP1Bmicrotubule stabilization, self interaction, actin filament binding, and regulation by the heavy chain. J. Cell Biol. 1998;143:695–707. doi: 10.1083/jcb.143.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlsten D. Deficiency of corpus callosum varies with strain and supplier of the mice. Brain Res. 1982;239:329–347. doi: 10.1016/0006-8993(82)90513-3. [DOI] [PubMed] [Google Scholar]
- Waikar S.S., Thalhammer J.G., Raymond S.A., Huang J.H., Chang D.S., Strichartz G.R. Mechanoreceptive afferents exhibit functionally-specific activity dependent changes in conduction velocity. Brain Res. 1996;721:91–100. doi: 10.1016/0006-8993(96)00165-5. [DOI] [PubMed] [Google Scholar]
- Wässle H., Koulen P., Brandstätter J.H., Fletcher E.L., Becker C.M. Glycine and GABA receptors in the mammalian retina. Vision Res. 1998;38:1411–1430. doi: 10.1016/s0042-6989(97)00300-3. [DOI] [PubMed] [Google Scholar]