

Abstract
Peptide growth factors control diverse cellular functions by regulating distinct signal transduction pathways. In cultured myoblasts, insulin-like growth factors (IGFs) stimulate differentiation and promote hypertrophy. IGFs also maintain muscle cell viability. We previously described C2 skeletal muscle lines lacking expression of IGF-II. These cells did not differentiate, but underwent progressive apoptotic death when incubated in differentiation medium. Viability could be sustained and differentiation enabled by IGF analogues that activated the IGF-I receptor; survival was dependent on stimulation of phosphatidylinositol 3-kinase (PI3-kinase). We now find that IGF action promotes myoblast survival through two distinguishable PI3-kinase–regulated pathways that culminate in expression of the cyclin-dependent kinase inhibitor, p21. Incubation with IGF-I or transfection with active PI3-kinase led to rapid induction of MyoD and p21, and forced expression of either protein maintained viability in the absence of growth factors. Ectopic expression of MyoD induced p21, and inhibition of p21 blocked MyoD-mediated survival, thus defining one PI3-kinase–dependent pathway as leading first to MyoD, and then to p21 and survival. Unexpectedly, loss of MyoD expression did not impede IGF-mediated survival, revealing a second pathway involving activation by PI3-kinase of Akt, and subsequent induction of p21. Since inhibition of p21 caused death even in the presence of IGF-I, these results establish a central role for p21 as a survival factor for muscle cells. Our observations also define a MyoD-independent pathway for regulating p21 in muscle, and demonstrate that distinct mechanisms help ensure appropriate expression of this key protein during differentiation.
Keywords: insulin-like growth factors, p21, MyoD, phosphatidyl inositol 3-kinase, Akt
Introduction
The insulin-like growth factors (IGFs), IGF-I and IGF–II, comprise with insulin a structurally related family of peptides of fundamental importance for normal somatic growth, for intermediary metabolism, and for the survival, proliferation, and terminal differentiation of many different cell types (Jones and Clemmons 1995; Stewart and Rotwein 1996a; Baserga et al. 1997). The biological effects of these proteins are mediated by a pair of related receptors. Both the IGF-I and insulin receptors are heterotetrameric, membrane-spanning, tyrosine protein kinases that activate a number of shared intracellular signal transduction pathways upon ligand binding (LeRoith et al. 1995; Virkamaki et al. 1999). Insulin functions primarily as a hormone of intermediary metabolism, being synthesized in the beta cells of the endocrine pancreas, and reaching its sites of action in liver, muscle, fat, and other cell types through the circulation (Virkamaki et al. 1999). By contrast, IGFs function primarily as growth, survival, and differentiation factors, and reach target tissues through the circulation, or through local synthesis near sites of action (Jones and Clemmons 1995; Stewart and Rotwein 1996a).
IGF action plays key roles in the formation and maintenance of skeletal muscle. Mice engineered to lack the IGF-I receptor, or deficient in IGF-I and IGF-II, exhibit marked muscle hypoplasia and die in the neonatal period because of inadequate muscle mass to inflate their lungs (Liu et al. 1993; Powell-Braxton et al. 1993). Conversely, mice with enhanced expression of IGF-I in muscle develop enlarged myofibers (Coleman et al. 1995; Barton-Davis et al. 1998). In cultured skeletal muscle, activation of the IGF-I receptor stimulates terminal differentiation through an autocrine pathway dependent on expression of IGF-II (Florini et al. 1991; Montarras et al. 1996; Tollefsen et al. 1989a,Tollefsen et al. 1989b). Endogenously produced IGF-II also plays an important role in maintaining cell survival during the transition from proliferating to terminally differentiating myoblasts (Stewart and Rotwein 1996b; Lawlor et al. 2000). The signal transduction pathways and mechanisms involved in IGF-mediated muscle cell survival and differentiation have not been completely elucidated. Recent studies have indicated that two classes of intracellular signaling molecules, phosphatidylinositol 3-kinase (PI3-kinase) and mitogen-activated protein kinases, are involved in muscle differentiation (Kaliman et al. 1996, Kaliman et al. 1998; Bennett and Tonks 1997; Coolican et al. 1997; Sarbassov et al. 1997; Gredinger et al. 1998; Jiang et al. 1998; Sarbassov and Peterson 1998; Cuenda and Cohen 1999; Musaro and Rosenthal 1999; Rommel et al. 1999; Zetser et al. 1999; Tamir and Bengal 2000; Wu et al. 2000), with the PI3-kinase pathway being considered more critical. At present, the mechanisms have not been established by which these signaling molecules or other pathways activated by the IGF-I receptor might collaborate with myogenic regulatory factors to regulate muscle cell viability or differentiation.
The muscle-specific basic helix-loop-helix (bHLH) transcription factors, MyoD, myogenin, MRF4, and Myf5, were initially identified as master regulators of cell fate because of their ability to confer a skeletal muscle phenotype on nonmuscle cells (Weintraub 1993; Olson and Klein 1994). These proteins function by activating genes that are required for muscle determination and/or differentiation through the formation of heterodimers with ubiquitous bHLH proteins, and subsequent binding to specific sequences termed E boxes in the promoter-regulatory regions of muscle-restricted target genes (Weintraub 1993; Olson and Klein 1994). One of the targets of MyoD is the gene encoding the cyclin-dependent protein kinase inhibitor, p21, also known as Waf1 and Cip1 (Ball 1997; El-Deiry 1998). This protein, and related molecules, p27/Kip1 and p57/Kip2 (Ball 1997), act to block progression through the cell cycle by reversibly inhibiting complexes of several different cyclins and cyclin-dependent kinases (Elledge 1996). In cultured muscle cells, p21 expression is induced as an early event during differentiation (Guo et al. 1995; Halevy et al. 1995; Parker et al. 1995; Mal et al. 2000). The increase in p21 is temporally associated with an ongoing decline in cyclin-dependent kinase activity as differentiation proceeds (Guo et al. 1995; Mal et al. 2000), and p21 has been found to become part of a complex containing cyclin E and cdk2 in differentiating C2 muscle cells (Mal et al. 2000). In addition, induction of p21 has been shown to correlate with development of an apoptosis-resistant phenotype during differentiation (Wang and Walsh 1996). The significance of p21 action in skeletal muscle is underscored by observations that high levels of p21 mRNA are detected in early muscle fibers in the mouse embryo (Parker et al. 1995), and that mice deficient in both p21 and p57 have defective muscle formation and exhibit increased rates of myoblast apoptosis (Zhang et al. 1999). It generally has been accepted that MyoD is a key transcription factor regulating p21 gene expression during muscle differentiation. MyoD has been found to enhance activity of the p21 promoter in transient transfection experiments (Halevy et al. 1995), and to stimulate p21 mRNA and protein accumulation in muscle cells and fibroblasts (Guo et al. 1995; Halevy et al. 1995; Parker et al. 1995), including cells lacking p53, the tumor suppressor protein that also can activate the p21 gene (Halevy et al. 1995).
In this manuscript, we address the question of how IGF action promotes muscle cell survival through study of C2 myoblasts and a derived cell line that lacks expression of IGF-II (Stewart and Rotwein 1996b). These cells undergo rapid apoptotic death when incubated in low serum differentiation medium (Stewart and Rotwein 1996b; Lawlor et al. 2000; Lawlor and Rotwein 2000). Addition of IGF-I or analogues that activate the IGF-I receptor maintain cell viability, but this is reversed by inhibitors of PI3-kinase (Lawlor et al. 2000). We now find that IGF action promotes myoblast survival by two different PI3-kinase–dependent pathways that converge on p21. Incubation with IGF-I, or transfection with active PI3-kinase, leads to rapid induction of MyoD and p21, and ectopic expression of either protein sustains muscle cell survival in the absence of growth factors. Forced expression of MyoD induces p21, and inhibition of p21 expression blocks MyoD-mediated survival, thus defining one pathway as leading through PI3-kinase to MyoD, and then to p21 and survival. The second IGF-stimulated pathway involves activation of the serine-threonine kinase, Akt, a protein that has been shown to play a key role in the survival of many different cell types (Datta et al. 1999). We find that Akt induces p21 in myoblasts by a mechanism that does not require MyoD. Since inhibition of p21 causes apoptotic death even in the presence of IGF-I, our results establish a central role for p21 as a survival factor for muscle cells, and demonstrate that distinct but integrated mechanisms help ensure the appropriate expression of this key protein during myogenic differentiation.
Materials and Methods
Materials
Fetal calf serum, newborn calf serum, horse serum, Dulbecco's modified Eagle's medium, phosphate buffered saline, G418, PDGF-BB, and TRIzol were purchased from Life Technologies. The long-lasting IGF-I analogue, R3IGF-I, was from Gropep. Effectene was purchased from QIAGEN. Restriction enzymes, ligases, and polymerases were from New England Biolabs. The pEGFP-N3 plasmid was purchased from CLONTECH Laboratories, Inc. Protease inhibitor tablets were from Roche Molecular Biochemicals. The BCA protein assay kit was from Pierce Chemical Co. Antibodies to p21 and CDK-4 used for immunoblotting were purchased from Santa Cruz Biotechnology, Inc. The antibody to MyoD was a gift from Dr. Peter J. Houghton (St. Jude Children's Research Hospital, Memphis, TN). Secondary antibodies were from Sigma-Aldrich. Antibodies to MyoD and p21 used for immunocytochemistry were obtained from Santa Cruz Biotechnology, Inc. and PharMingen, respectively. The antibody to influenza hemagglutinin (HA) was from BabCO, and the anti–myc antibody (clone 9E-10) was from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Secondary antibodies conjugated to fluorophores were purchased from Molecular Probes. Nitrocellulose was from Schleicher & Schuell, and ECL reagents from Amersham-Pharmacia Biotech. X-ray film was purchased from Eastman Kodak Co. All other chemicals were reagent grade.
Cell Culture
C2 myoblasts stably transfected with the coding region of a mouse IGF-II cDNA in the antisense orientation (C2AS12 cells; Stewart and Rotwein 1996b; Lawlor et al. 2000) were grown until >95% confluent on gelatin-coated tissue culture dishes in DMEM supplemented with 10% heat-inactivated FCS, 10% heat-inactivated newborn calf serum, 2 mM l-glutamine, and 400 μg/ml G418 (active drug) (growth medium). C2 myoblasts (Yaffe and Saxel 1977) were grown until >95% confluent on gelatin-coated plates in growth medium minus G418. For both cell lines, differentiation was initiated after washing with PBS by incubating in differentiation medium (DM) containing DMEM plus 2% horse serum, or in DM supplemented with 0.4 nM PDGF-BB or 2 nM R3IGF-I. At different intervals, adherent cells were trypsinized and counted by hemocytometer or by Coulter particle counter. Cos7 cells were grown in DMEM supplemented with 10% heat-inactivated FCS and 2 mM l-glutamine.
RNA Isolation and Ribonuclease Protection Assays
Total RNA was isolated from cells using TRIzol and quantitated by spectrophotometry. RNA integrity was assessed by electrophoresis though 1% agarose-formaldehyde gels after staining with ethidium bromide. Solution hybridization ribonuclease protection assays were performed as described (Stewart et al. 1996), using single stranded [α-32P]CTP-labeled antisense riboprobes synthesized from linearized plasmid templates. Results were quantitated with a PhosphorImager (GS 525; Bio-Rad Laboratories).
Protein Isolation and Immunoblotting
Protein extracts were isolated after washing cells twice with cold PBS by incubating for 30 min at 4°C in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.1% SDS, 1.0% NP-40, and 1.0% deoxycholate) containing protease inhibitors, 1 μM okadaic acid, and 1 μM sodium orthovanadate. After removal of insoluble material by centrifugation at 14,000 rpm for 10 min at 4°C, protein concentration was determined by BCA assay. Protein extracts (60 μg) were separated by SDS-polyacrylamide gel electrophoresis under denaturing and reducing conditions before transfer to 0.2 μM nitrocellulose membranes at 18 V for 45 min using a semidry blotter. Membranes were blocked for 2 h at 25°C in TBST (Tris buffered saline plus 0.1% Tween-20) containing 5% nonfat dry milk (blocking buffer) before being incubated with primary antibody (anti–MyoD undiluted supernatant, anti–p21 1:75, and anti–CDK4 1:500 in blocking buffer). After incubation with horseradish peroxidase–conjugated secondary antibodies (1:2,000 in blocking buffer), proteins were detected by ECL, followed by exposure to x-ray film. Results were quantitated by densitometry (GS 700; Bio-Rad Laboratories).
Immunocytochemistry
Cells were washed twice with PBS before fixation in 1% paraformaldehyde for 10 min. Cells were then incubated for 5 min in PBS containing 0.2% Triton X-100 (PBST), washed twice with PBST, and incubated with primary antibodies: polyclonal rabbit anti–MyoD (1:500), polyclonal rabbit anti–p21 IgG (1:2,000), monoclonal anti–HA IgG (1:100), or monoclonal anti–myc (1:500) in PBST plus 3% bovine serum albumin for 3–16 h at 4°C. Cells were washed three times with PBST before incubation with polyclonal secondary antibodies, either Alexa 594-tagged goat anti–rabbit IgG (1:2,000), fluorescein-conjugated goat anti–rabbit IgG, or fluorescein-conjugated goat anti–mouse IgG (each 1:1,000), for 45 min in the dark. Images were captured with a fluorescence microscope (Eclipse TE 300; Nikon) and an CCD camera (Optronics) using Scion Image 1.62 software. Images were save in Photoshop 5.5 (Adobe Systems).
Construction of Bicistronic Expression Plasmids
The internal ribosome entry site (IRES) from mouse encephalomyocarditis virus (Ghattas et al. 1991) was subcloned into the polylinker of pEGFP-N3 to generate pIRES-EGFP. An XhoI–BamHI DNA fragment containing the coding region of murine MyoD in the sense orientation was excised from pBluescript and subcloned 5′ to the IRES to generate MyoD-IRES-EGFP. A BamHI–SalI DNA fragment containing the mouse MyoD coding region in the antisense orientation was used to generate MyoDAS-IRES-EGFP. The p21-IRES-EGFP, p21AS-IRES-EGFP, PI3-kinase, and iAkt plasmids have been described (Lawlor et al. 2000; Lawlor and Rotwein 2000).
Transfections
Myoblasts were plated at 13,000 cells/cm2 onto 12-well tissue culture dishes and incubated for 24 h in growth medium. DNA-mediated gene transfer was performed using Effectene, following the protocol of the manufacturer. A total of 1 μg of DNA of sense plasmids was used per well. For antisense plasmids [MyoDAS-IRES-EGFP and p21AS-IRES-EGFP DNA], or control [EGFP], 2 μg of DNA were used, and for cotransfections a total of 2 μg of DNA was added to cells. After incubation with DNA for 16–18 h, fresh growth medium was added to the cells. When cells reached confluent density at ∼48 h after transfection, they were incubated in DM without or with growth factors for an additional 24 or 48 h. Transfection efficiencies ranged from 12–20% (C2AS12 cells) to 19–23% (C2 myoblasts). Cos 7 cells were grown in six-well dishes and were transfected with 2 μg of DNA using the calcium phosphate precipitation method (Stewart and Rotwein 1996b). Expression of p21 and MyoD was analyzed by immunoblotting.
Survival Assays of Transfected Cells
Myoblasts were transfected as described above. When cells reached confluent density at ∼48 h after transfection, two wells were harvested and both total cell number [T0(total)] and transfection efficiency were assessed. The latter value was determined by averaging the fraction of cells expressing EGFP in 20 hemocytometer fields at a magnification of 200× [T0(transfected)]. The remaining wells were incubated in DM without or with growth factors for 24 or 48 h. At each interval, cells were harvested and total and transfected cells were counted. Survival of transfected cells at 24 h was determined by the following formula: % survival = [T24(transfected)/T0(transfected) × (T24(total)/T0(total)] × 100. Survival at 48 h was assessed similarly.
Statistical Analysis
Results are presented as the mean ± SEM. Statistical significance was determined using independent Student's t test for paired samples. Results were considered statistically significant when P < 0.05.
Results
Prevention of Myoblast Death by IGF-I
We have shown previously that signaling through the IGF-I receptor by endogenously produced IGF-II is essential for myoblast survival as cells begin to differentiate (Stewart and Rotwein 1996b; Lawlor et al. 2000; Lawlor and Rotwein 2000). As seen in Fig. 1, C2 myoblasts engineered to lack IGF-II expression (C2AS12 cells) underwent rapid death when incubated in DM. Only 48 ± 1% of cells remained alive after 24 h, and only 34 ± 1% survived after 48 h. Viability continued to decline progressively with longer incubations in DM (Lawlor and Rotwein 2000). Addition of IGF-I caused complete survival (102 ± 3% at 24 h, 98 ± 4% at 48 h). Other growth factors such as PDGF-BB also maintain viability of C2AS12 cells (Lawlor et al. 2000, and data not shown). Nontransfected C2 myoblasts also underwent apoptotic death when incubated in DM, with ∼30% of cells dying at 24 h (see Fig. 7; and Lawlor and Rotwein 2000). No further death occurred upon longer incubation in DM, with survival correlating with expression of IGF-II (Tollefsen et al. 1989b; Lawlor and Rotwein 2000).
Figure 1.

IGF-I treatment maintains myoblast survival. Results are shown for cell counts (expressed as percent at time 0, % T0) of C2AS12 myoblasts after a 24 or 48 h incubation in DM with or without R3IGF-I (2 nM). Asterisks indicate that significantly fewer cells survived (*P < 0.005, **P < 0.001) in untreated than in growth factor–treated cells (mean ± SEM of three experiments, each performed in duplicate).
Figure 7.

Inhibition of MyoD does not block survival of C2 myoblasts. C2 myoblasts were transiently transfected with MyoDAS-IRES-EGFP or p21AS-IRES-EGFP expression plasmids, as described in Materials and Methods. Cell counts of transfected cells were performed after a 24 or 48 h incubation in DM. Results are expressed as the mean ± SEM of three independent experiments, each performed in duplicate. The asterisks denote that viability was significantly less in myoblasts transfected with p21AS than in cells transfected with MyoDAS or untransfected cells (*P < 0.003, **P < 0.00002).
Stimulation of MyoD and p21 mRNA and Protein Expression after IGF-I Treatment
The results in Fig. 1 prompted investigation of mechanisms of IGF-mediated muscle cell survival. The cyclin-dependent kinase inhibitor, p21, has been found to function as a survival factor for muscle and other cell types (Poluha et al. 1996; Wang and Walsh 1996; Levkau et al. 1998), and MyoD has been shown to induce p21 expression (Guo et al. 1995; Halevy et al. 1995; Parker et al. 1995). We thus asked whether IGF treatment stimulated expression of MyoD or p21 in C2AS12 cells. Results of time course studies are shown in Fig. 2. Treatment with IGF-I caused a sustained increase in the abundance of both MyoD and p21 mRNAs beginning at 8 h, the earliest time point examined, and lasting up to 48 h. Peak values of 7- and 10-fold above baseline were seen at 24 h for MyoD and p21, respectively. By contrast, PDGF-BB had a minimal effect. Similar results were seen when MyoD and p21 protein expression was examined. A progressive increase was seen in the abundance of both proteins after incubation with IGF-I, reaching fourfold above T0 for MyoD and sevenfold for p21 (mean ± SEM of three experiments). PDGF caused at best a transient rise in MyoD that returned to baseline levels or below within 8 h (Fig. 2 B). Additionally, as shown by immunocytochemistry, MyoD and p21 could be detected in nearly all myoblasts after a 24-h incubation with IGF-I, while few cells expressed either protein after treatment with PDGF (Fig. 2 C). Thus, IGF-I caused a progressive and sustained induction of MyoD and p21 mRNA and protein expression in C2AS12 cells.
Figure 2.


Treatment with IGF-I stimulates expression of MyoD and p21. (A) Induction of MyoD and p21 mRNAs by IGF-I. The autoradiographs show results of representative ribonuclease protection assays performed using total RNA isolated from C2AS12 cells incubated with either R3IGF-I (2 nM) or PDGF-BB (0.4 nM) for the indicated times (top, MyoD; middle, p21). (Bottom) Photograph of an ethidium bromide–stained gel of the RNA used in these studies. Similar results were seen in three independent experiments. (B) Induction of MyoD and p21 proteins by IGF-I. Representative immunoblots are shown using whole-cell protein extracts from C2AS12 cells incubated with either R3IGF-I (2 nM) or PDGF-BB (0.4 nM) for the indicated times, and antibodies to MyoD (top), p21 (middle), and CDK-4 (bottom). Similar results were seen in three independent experiments. (C) Representative fluorescence micrographs of C2AS12 myoblasts treated with either R3IGF-I (2 nM) or PDGF-BB (0.4 nM) for 24 h, as described in Materials and Methods, and immunostained with antibodies to MyoD (top left), or p21 (top right), and incubated with Hoechst nuclear dye (bottom).
Forced Expression of MyoD or p21 Promotes Myoblast Survival
We next asked whether MyoD or p21 could maintain muscle cell survival in the absence of growth-factor treatment. We generated bicistronic expression plasmids using cDNAs for mouse MyoD or mouse p21, followed by a cassette containing an IRES derived from murine encephalomyocarditis virus and the marker protein EGFP. C2AS12 myoblasts were transiently transfected with these recombinant plasmids or with a control encoding EGFP alone, and cell survival was measured by cell counting after 24 h in DM. As depicted in Fig. 3, MyoD and p21 each promoted a significant increase in myoblast viability over results obtained with EGFP (MyoD: 84 ± 5% survival; p21: 98 ± 4%; EGFP: 52 ± 5%; P < 0.001 compared with EGFP). The latter value is similar to the 48 ± 1% survival seen in nontransfected myoblasts (Fig. 1). Incubation with IGF-I maintained complete cell viability, indicating that neither the recombinant plasmids nor the process of transfection were toxic. Comparable results were obtained after 48 h in DM (data not shown).
Figure 3.
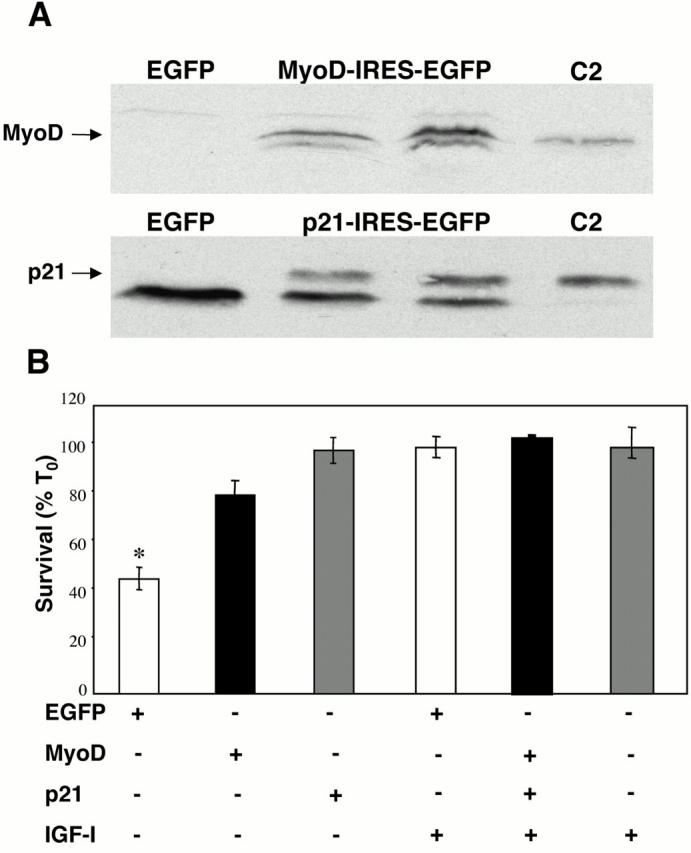
MyoD and p21 promote myoblast survival. (A) Expression of MyoD and p21 in transfected Cos7 cells. Cos7 cells were transiently transfected with expression plasmids encoding MyoD-IRES-EGFP (MyoD), p21-IRES-EGFP (p21), or EGFP alone, as outlined in Materials and Methods. Whole-cell protein extracts were isolated 48 h after transfection and used for immunoblotting with antibodies to MyoD (top) or p21 (bottom). Whole-cell extracts from differentiating C2 myoblasts (C2) were used as a positive control. (B) MyoD and p21 promote muscle cell survival. C2AS12 myoblasts were transfected with plasmids encoding either MyoD-IRES-EGFP (MyoD), p21-IRES-EGFP (p21), or EGFP alone. Counts of transfected cells were performed after a 24-h incubation in DM without or with R3IGF-I (2 nM). Results are presented as percent survival compared with T0 (mean ± SEM of four experiments, each performed in duplicate). *Survival was significantly lower in myoblasts transfected with EGFP than in cells transfected with MyoD or p21, or treated with IGF-I (P < 0.001).
p21 Is Required for MyoD-stimulated Muscle Cell Survival
Based on published studies showing that MyoD stimulated p21 expression in muscle and fibroblasts (Guo et al. 1995; Halevy et al. 1995; Parker et al. 1995), we anticipated that forced expression of MyoD would promote accumulation of endogenous p21 in our cell line. C2AS12 myoblasts were transiently transfected with the MyoD expression plasmid and cells were immunostained for p21 after incubation for 24 h in DM. As shown in Fig. 4 A and graphed in B, MyoD induced p21 protein expression in the majority of transfected cells (75 ± 5%), compared with only 25 ± 2% of cells transfected with EGFP (P < 0.005), a value similar to that seen in nontransfected myoblasts (data not shown). To determine whether p21 was necessary for MyoD-mediated muscle cell survival, we cotransfected C2AS12 myoblasts with MyoD and either a p21 antisense plasmid (p21AS) or a plasmid encoding EGFP. We have shown recently that this p21AS cDNA blocked IGF-induced p21 protein expression in C2AS12 myoblasts (Lawlor and Rotwein 2000). Cell survival was assessed after incubation for 24 h in DM. As seen in Fig. 4 C, MyoD was unable to maintain muscle cell viability in the presence of the p21AS plasmid (30 ± 4% survival), but could sustain viability when cotransfected with EGFP (73 ± 3%, P < 0.001). These results indicate that MyoD promotes muscle cell survival by inducing p21.
Figure 4.
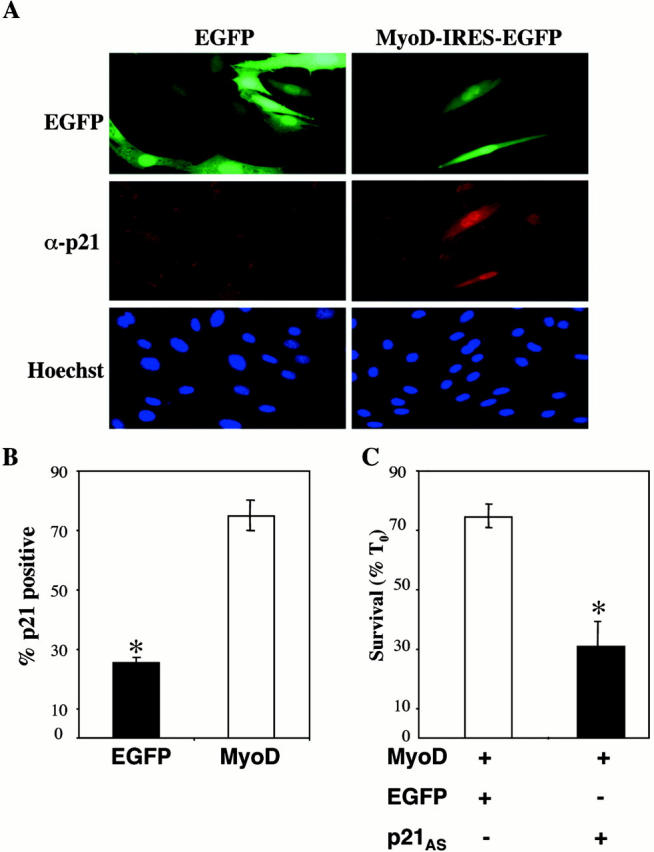
p21 is required for MyoD-stimulated muscle cell survival. (A) MyoD induces p21 protein expression. C2AS12 cells were transfected with either EGFP or MyoD-IRES-EGFP expression plasmids, and following a 24-h incubation in DM, fixed, and stained with an antibody to p21. (Top) Expression of EGFP, (middle) immunostaining for p21, and (bottom) nuclear staining with Hoechst dye. (B) The graph records the percent of transfected cells expressing p21. Significantly fewer cells are positive for p21 after transfection with EGFP than with MyoD-IRES-EGFP (*P < 0.005; mean ± SEM of three experiments, 100 transfected cells counted per experiment). (C) Inhibition of p21 prevents MyoD-stimulated muscle cell survival. C2AS12 cells were cotransfected with MyoD-IRES-EGFP (MyoD) and either EGFP or p21AS-IRES-EGFP (p21AS), as outlined in Materials and Methods. Cell counts of transfected myoblasts were performed after a 24-h incubation in DM. Results are presented as percent survival compared with T0 (mean ± SEM of three experiments, each performed in duplicate). *Survival was significantly less in myoblasts cotransfected with p21AS-IRES-EGFP than with EGFP (P < 0.014).
MyoD Is Not Required for IGF-mediated Myoblast Survival
We have demonstrated that p21 is necessary for IGF-mediated survival of C2AS12 cells and wild-type C2 and L6 myoblasts (Lawlor and Rotwein 2000). Since both MyoD and p21 are induced by IGF-I, and MyoD stimulates expression of p21, we next asked if MyoD was a necessary component of an IGF-stimulated muscle cell survival pathway. We first tested the effectiveness of a MyoD antisense cDNA (MyoDAS) to prevent IGF-stimulated MyoD protein expression. C2AS12 cells were transiently transfected with an expression plasmid containing a mouse MyoD cDNA in the antisense orientation (MyoDAS-IRES-EGFP), and were immunostained for MyoD after a 24-h incubation in DM plus IGF-I. As seen in Fig. 5, the MyoDAS plasmid caused a marked reduction in IGF-stimulated expression of MyoD. MyoD was detected in only 34 ± 2% of transfected cells, compared with 78 ± 5% of myoblasts transfected with EGFP (P < 0.014).
Figure 5.

Forced expression of a MyoD antisense plasmid blocks IGF-I–stimulated MyoD expression. (A) C2AS12 cells were transiently transfected with the MyoDAS-IRES-EGFP expression plasmid, as described in Materials and Methods, and incubated in DM plus R3IGF-I (2 nM) for 24 h, followed by fixation and staining for MyoD. (Left) Expression of EGFP, (center) immunostaining for MyoD, and (right) merged image. (B) The graph indicates the percentage of transfected cells expressing MyoD, as assessed by immunocytochemistry following IGF-I treatment. Significantly fewer cells are positive for MyoD after transfection with MyoDAS-IRES-EGFP than with EGFP (*P < 0.01; mean ± SEM of three experiments, counting 100 cells per experiment).
The MyoDAS plasmid did not reduce IGF-mediated muscle cell viability. As shown in Fig. 6 A, IGF-I maintained complete survival of myoblasts transfected with the MyoDAS cDNA. In the absence of growth factor treatment, 49 ± 5% of transfected cells remained alive after a 24-h incubation in DM, a level similar to that seen in nontransfected myoblasts (48 ± 1%, see Fig. 1), and contrasting with the 82 ± 4% viability of cells transfected with the MyoD sense vector (P < 0.016). In contrast with our recent observations that a p21AS plasmid inhibited IGF-mediated muscle cell survival (Lawlor and Rotwein 2000), forced expression of the MyoDAS plasmid did not attenuate the ability of IGF-I to promote complete myoblast viability (100 ± 3%). These results indicate that while MyoD can sustain muscle cell viability through p21, it is not necessary for IGF-regulated myoblast survival. Based on these observations, we next asked if IGF-I could stimulate p21 expression in cells transfected with the MyoDAS plasmid. As shown by immunocytochemistry in Fig. 6 B, incubation with IGF-I equivalently induced p21 in myoblasts transfected with MyoDAS or EGFP (84% for MyoDAS vs. 78% for EGFP). Thus, MyoD does not appear to be a critical component of a survival pathway involving IGF-I and p21.
Figure 6.
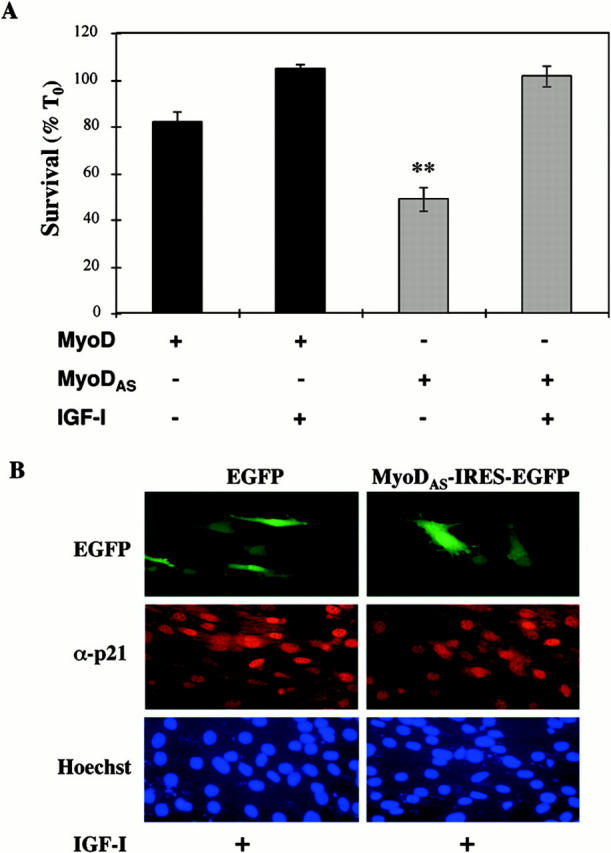
MyoD is not required for IGF-mediated survival of C2AS12 myoblasts. (A) C2AS12 cells were transiently transfected with MyoD-IRES-EGFP or MyoDAS-IRES-EGFP expression plasmids, as described in Materials and Methods. Cell counts of transfected myoblasts were performed after a 24-h incubation in DM or in DM supplemented with R3IGF-I (2 nM). Results are expressed as the mean ± SEM of three independent experiments, each performed in duplicate. **Survival was significantly less in myoblasts transfected with MyoDAS than in cells transfected with MyoD or treated with IGF-I (P < 0.001). (B) Inhibition of MyoD expression does not block IGF-mediated induction of p21. Representative fluorescence micrographs of C2AS12 myoblasts transiently transfected with expression plasmids for MyoDAS-IRES-EGFP or EGFP alone, and treated with IGF-I (2 nM) in DM for 24 h. (Top) Expression of EGFP, (middle) immunostaining for p21, and (bottom) nuclei stained with Hoechst dye. As determined by cell counting, ∼80% of cells transfected with either plasmid expressed p21 after incubation with IGF-I.
A MyoD Antisense cDNA Does Not Inhibit Survival of C2 Myoblasts
Parental C2 myoblasts, which produce IGF-II during differentiation (Tollefsen et al. 1989b), undergo limited apoptotic death during incubation in DM, with ∼70% viability over the first 24 h, and little cell death subsequently (Lawlor and Rotwein 2000). We next asked if MyoD was involved in the survival of these cells. C2 myoblasts were transfected with the MyoDAS plasmid or with a p21AS expression construct that we have shown diminishes C2 cell survival (Lawlor and Rotwein 2000). Fig. 7 demonstrates that forced expression of MyoDAS had little effect on C2 muscle cell survival beyond what was seen in the nontransfected population (24 h viability: 72 ± 3% vs. 73 ± 3%; 48 h viability: 70 ± 2% vs. 69 ± 3%, respectively, P = NS). By contrast, forced expression of p21AS resulted in a dramatic decrease in viability, with 42 ± 4% of transfected cells remaining alive after a 24-h incubation in DM, and only 17 ± 1% by 48 h. Taken together with results shown in Fig. 6, these observations indicate that induction of p21 expression by MyoD is not a critical pathway for IGF-mediated muscle cell survival.
Differential Expression of MyoD and p21 by IGF-activated Signaling Pathways
The results described above suggest that IGF-I induces the expression of MyoD and p21 by distinct mechanisms. In recent studies, we demonstrated that IGF-I treatment resulted in the sustained stimulation of both PI3-kinase and Akt kinase activities in muscle cells, and showed that a constitutively active PI3-kinase or an inducible Akt could maintain myoblast survival in the absence of growth factors (Lawlor et al. 2000). Based on these observations and on the results in Fig. 2, we next asked if either signaling molecule was involved in IGF-mediated stimulation of MyoD or p21. Transient transfection of an active PI3-kinase (p110*) into C2AS12 myoblasts led to the induction of MyoD and p21 in the absence of IGF-I treatment, as determined by immunocytochemistry after a 24-h incubation in DM (Fig. 8). Over 80% of cells positive for p110* also expressed both MyoD and p21. By contrast, in cells transfected with an inactive PI3-kinase (p110Δkin), the level of expression of MyoD or p21 (31–36%) did not exceed values detected in nontransfected myoblasts (data not shown). Thus, PI3-kinase can coordinately stimulate the expression of MyoD and p21 in muscle cells.
Figure 8.

Induction of MyoD and p21 after transfection of active PI3-kinase. (A) Induction of MyoD by PI3-kinase. Pictured are representative fluorescence micrographs of C2AS12 muscle cells transiently transfected with plasmids encoding active (p110*) or inert (p110Δkin) PI3-kinase, as described in Materials and Methods. After a 24-h incubation in DM, cells were fixed and immunostained for expression of either PI-3 kinase using an antibody directed against the c-myc epitope tag (top) or for MyoD (bottom). The graph shows that the percent of transfected cells expressing MyoD was significantly less after transfection with p110Δkin than with p110* (mean ± SEM of three experiments, counting 100 cells per experiment, *P < 0.01). (B) Induction of p21 by PI3-kinase. Illustrated are representative fluorescence micrographs of C2AS12 muscle cells transiently transfected with the p110* or p110Δkin expression plasmids, as described in A. After a 24-h incubation in DM cells were immunostained for expression of either protein (top), and for p21 (bottom). The graph shows that the percentage of transfected cells expressing p21 was significantly smaller after transfection with p110Δkin than with p110* (mean ± SEM of three experiments, counting 100 cells per experiment, **P < 0.001).
We next examined the ability of Akt to stimulate MyoD and p21 protein accumulation. Myoblasts were transfected with an expression plasmid encoding hydroxytamoxifen (HT)-inducible HA-tagged Akt (iAkt), and MyoD and p21 were detected by immunocytochemistry after incubation for 24 h in DM without or with HT. Upon transfection with the iAkt plasmid, Akt protein is expressed, but its enzymatic activity requires induction by HT (Lawlor et al. 2000). As shown in Fig. 9 A, HT treatment had little effect on the low level of MyoD protein accumulation observed (36 ± 2% vs. 33 ± 2% without HT, P = NS). By contrast, addition of HT caused a significant increase in p21 protein expression (78 ± 4% vs. 24 ± 4%, P < 0.01), as demonstrated previously (Lawlor and Rotwein 2000). Based on these observations, and on the results in Fig. 6, we conclude that IGF-activated signal transduction pathways control muscle cell survival by at least two mechanisms, one involving induction of MyoD mRNA and protein through PI3-kinase, with the subsequent stimulation of p21 expression, and the other through induction of p21 by Akt.
Figure 9.

Induction of p21 but not MyoD after activation of transfected Akt. (A) Akt does not induce MyoD. Pictured are representative fluorescence micrographs of C2AS12 cells transiently transfected with hydroxytamoxifen-inducible HA-tagged Akt (iAkt), as described in Materials and Methods. After a 24-h incubation with DM in the absence or presence of hydroxytamoxifen, cells were fixed and immunostained using antibodies to the HA tag (top) and MyoD (bottom). The graph shows that the fraction of transfected cells expressing MyoD was not significantly different in cells incubated without or with HT (mean ± SEM of three experiments, counting 100 cells per experiment). (B) Akt induces p21. Illustrated are representative fluorescence micrographs of C2AS12 cells transiently transfected with iAkt, as described in Materials and Methods. After a 24-h incubation in DM without or with HT, the cells were immunostained using antibodies to the HA tag (top) and to p21 (bottom). The graph shows that the percentage of transfected cells expressing p21 was significantly greater after incubation with HT (mean ± SEM of three experiments, counting 100 cells per experiment, *P < 0.01).
Discussion
Muscle differentiation is a multi-step process involving permanent withdrawal from the cell cycle, expression of muscle-specific genes and proteins, and fusion of myoblasts into multinucleated myotubes (Lassar et al. 1994; Arnold and Winter 1998). In this paper, we focus on steps involved in control of myoblast survival during early phases of differentiation. We show that the cyclin-dependent kinase inhibitor, p21, plays a key role in maintaining muscle cell viability and that its expression is regulated by two interdependent, yet distinguishable pathways. One pathway involves stimulation of MyoD expression by PI3-kinase, and subsequent induction of p21 by MyoD; the other involves activation of Akt by PI3-kinase, followed by stimulation of p21 expression. Both pathways collaborate to maintain survival and are connected through PI3-kinase, which itself is required for IGF-regulated muscle cell viability (Lawlor et al. 2000). The pathways diverge distal to PI3-kinase, as Akt induces expression of p21 but not MyoD, and inhibition of MyoD does not prevent IGF-stimulated production of p21 or IGF-mediated myoblast survival. A diagram outlining the interrelationships between these pathways is presented in Fig. 10.
Figure 10.

Model for IGF-regulated muscle cell survival. Ligand-induced activation of the IGF-I receptor (IGF-IR) leads to stimulation of PI3-kinase enzymatic activity through the adapter molecules, IRS-1 and -2. PI3-kinase subsequently activates Akt, which stimulates expression of p21 mRNA and protein, which then promotes muscle cell survival. PI3-kinase through unidentified steps also stimulates expression of MyoD, which then induces p21. MyoD plays a secondary role in the induction of p21 and in IGF-mediated cell survival.
It has been established in cultured muscle cells that p21 mRNA and protein expression increase dramatically with the onset of differentiation (Guo et al. 1995; Halevy et al. 1995; Parker et al. 1995), and that p21 contributes to the cell cycle arrest and resistance to apoptosis that occur early in differentiation (Andres and Walsh 1996; Wang and Walsh 1996). Although p21 null mice do not demonstrate abnormalities in skeletal muscle (Deng et al. 1995), mice deficient in both p21 and the related cyclin-dependent kinase inhibitor, p57, display marked defects in muscle differentiation and exhibit increased myoblast apoptosis during embryonic development (Zhang et al. 1999). These results confirm observations made with cultured muscle cells, but illustrate the complexity and functional redundancy within this family of cyclin-dependent kinase inhibitors in vivo. Little p57 can be detected in C2 myoblasts (Zhang et al. 1999), thus explaining why abrogation of p21 expression enhances cell death. A major function for p21 (and presumably p57) in muscle cells is to promote arrest in the G1 phase of the cell cycle by inhibiting activity of cyclin-dependent kinases, which otherwise would phosphorylate and inactivate the retinoblastoma protein, pRb (Elledge 1996; Jacks and Weinberg 1998). Active pRb in turn restrains E2F transcription factors, thus preventing induction of genes involved in cell-cycle progression (Weinberg 1995). It remains to be established whether this is also the mechanism by which p21 sustains myoblast survival. Of interest, transgenic mice expressing low levels of pRb in an otherwise pRb null background have muscle defects similar to those of p21- and p57-deficient mice (Zackenhaus et al. 1996).
It has been shown by several investigators that MyoD induces p21 gene and protein expression in differentiating myoblasts (Guo et al. 1995; Halevy et al. 1995; Parker et al. 1995), in fibroblasts from intact and p53-deficient mice, and in other cell types by transcriptional pathways (Halevy et al. 1995) that are not dependent on ongoing protein synthesis (Otten et al. 1997), implying that MyoD transactivates the p21 gene. Our results now define a MyoD-independent mechanism for controlling p21 expression in muscle cells through an IGF-I–activated signal transduction pathway involving PI3-kinase and Akt. Previously published experiments from others also may be interpreted to support the idea that MyoD and p53 are not the only factors regulating p21 expression in muscle. During embryonic development in mice, p21 mRNA is detected in somites destined to form muscle at day 8.5 post coital, a full 2 d before MyoD gene expression is measurable (Parker et al. 1995). In addition, p21 mRNA is equivalently expressed in presumptive muscle in developing wild-type mice and in mice lacking both MyoD and myogenin (Parker et al. 1995). It has not been established if other myogenic basic helix-loop-helix transcription factors are responsible for induction of p21 under these circumstances or whether additional pathways are involved, such as those activated by the IGF-I receptor. Since IGF-II is strongly expressed in the somites at day 8.5 post coital and throughout muscle development in mice (Lee et al. 1990), it is conceivable that IGF-mediated mechanisms contribute to MyoD-independent regulation of p21 expression during embryonic development.
IGF-I receptor null mice also display muscle hypoplasia (Liu et al. 1993), but it is not known whether the underlying defect results from decreased proliferation of muscle precursor cells or from increased apoptosis. In this report, we define two IGF-I receptor-activated survival pathways in cultured myoblasts that require PI3-kinase. A role for PI3-kinase in muscle differentiation had been established previously. Inhibition of kinase activity had been shown to blunt differentiation (Kaliman et al. 1996, Kaliman et al. 1998; Coolican et al. 1997; Jiang et al. 1998, Jiang et al. 1999), and forced expression of active enzyme had been found to enhance differentiation (Jiang et al. 1998), although in no studies have the downstream signaling pathways been delineated. We now demonstrate that activation of PI3-kinase leads to induction of MyoD expression by a mechanism that does not appear to require Akt. Other downstream effectors of PI3-kinase have been identified in several cell types, including p70 S6 kinase, and atypical protein kinases C, δ, and ζ (Chou et al. 1998; Le Good et al. 1998; Pullen et al. 1998). We have been unable to establish in preliminary experiments a role for either p70 S6 kinase or protein kinase C ζ in myoblast survival (data not shown). It also has been found recently that PI3-kinase can induce the phosphorylation and enhance the activity of the MEF2 muscle transcription factors (Tamir and Bengal 2000), although this has not been shown to be a direct effect, and it is not known if MEF2 proteins contribute to muscle cell survival. Additional signaling molecules potentially involved in regulating myoblast differentiation and downstream of PI3-kinase include members of the Rho family of small GTPases, RhoA, Rac, and Cdc42 (Nobes and Hall 1994). Inhibition of activity of these proteins, either through use of dominant interfering mutants or overexpression of the GDP dissociation inhibitor, RalGDI, can block expression of muscle-specific genes and prevent differentiation (Carnac et al. 1998; Ramocki et al. 1998; Takano et al. 1998; Wei et al. 1998). To date, the Rho family has not been linked to muscle cell survival pathways controlled by the IGF-I receptor.
The signaling pathways and mechanisms by which Akt enhances p21 gene expression are similarly unknown. In some cell types, Akt has been shown to activate the transcription factor NF-κB by stimulating the kinases that phosphorylate its inhibitor, IκB, and target it for destruction (Kane et al. 1999; Romashkova and Makarov 1999; Sizemore et al. 1999). Although NF-κB is expressed in C2 myoblasts (Kaliman et al. 1999), we have found that it is not induced by IGF-I in these cells (Lawlor and Rotwein 2000), thus making it unlikely to be involved in Akt-mediated gene activation. Members of the p38 family of mitogen-activated protein (MAP) kinases have been demonstrated to phosphorylate and activate MEF2 proteins (Yang et al. 1999; Zhao et al. 1999), but p38 MAP kinases are not activated by Akt and have not been found to stimulate p21 gene expression or promote myoblast survival.
In summary, we have identified two distinct IGF-I receptor and PI3-kinase–activated signal transduction pathways that contribute to the maintenance of myoblast viability through induction of p21. This dual mechanism of p21 regulation may function to ensure the appropriate expression of this critical survival factor. It remains to be determined whether similarly multilayered interdependent pathways control p21 gene expression in vivo, where they potentially may act to modulate myoblast viability and muscle mass during embryonic and adult life.
Acknowledgments
We thank the following individuals for plasmids: Dr. Richard Roth (Stanford University, Stanford, CA) for iAkt, Dr. Bert Vogelstein (Johns Hopkins University School of Medicine, Baltimore, MD) for the mouse p21 cDNA, Dr. Anke Klippel (Chiron Corp., Emeryville, CA) for constitutively active and inert PI3-kinases, Dr. Andrew Lassar (Harvard University, Cambridge, MA) for mouse MyoD, and Dr. Alex Toker (Boston Biomedical Research Institute, Boston, MA) for protein kinase C ζ. We also thank Dr. Peter J. Houghton (St. Jude Children's Research Hospital, Memphis, TN) for the antibody to MyoD. We appreciate the technical assistance of Barb Rainish and Daniel Everding.
This study was supported by research grant 5RO1-DK42748 from the National Institutes of Health to P. Rotwein.
Footnotes
Abbreviations used in this paper: DM, differentiation medium; HA, hemagglutinin; HT, hydroxytamoxifen; IGF, insulin-like growth factor; IRES, internal ribosome entry site; PI3-kinase, phosphatidylinositol 3-kinase.
References
- Andres V., Walsh K. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J. Cell Biol. 1996;132:657–666. doi: 10.1083/jcb.132.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold H.H., Winter B. Muscle differentiationmore complexity to the network of myogenic regulators. Curr. Opin. Genet. Dev. 1998;8:539–544. doi: 10.1016/s0959-437x(98)80008-7. [DOI] [PubMed] [Google Scholar]
- Ball K.L. p21structure and functions associated with cyclin-CDK binding. Prog. Cell Cycle Res. 1997;3:125–134. doi: 10.1007/978-1-4615-5371-7_10. [DOI] [PubMed] [Google Scholar]
- Barton-Davis E.R., Shoturma D.I., Musaro A., Rosenthal N., Sweeney H.L., Kurabayashi M., Dutta S., Kedes L. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc. Natl. Acad. Sci. USA. 1998;95:5603–15607. doi: 10.1073/pnas.95.26.15603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baserga R., Resnicoff M., D'Ambrosio C., Valentinis B. The role of the IGF-I receptor in apoptosis. Vitam. Horm. 1997;53:65–98. doi: 10.1016/s0083-6729(08)60704-9. [DOI] [PubMed] [Google Scholar]
- Bennett A.M., Tonks N.K. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997;278:1288–1291. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- Carnac G., Primig M., Kitzmann M., Chafey P., Tuil D., Lamb N., Fernandez A. Rhoa GTPase and serum response factor control selectively the expression of MyoD without affecting Myf5 in mouse myoblasts. Mol. Biol. Cell. 1998;9:1891–1902. doi: 10.1091/mbc.9.7.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou M.M., Hou W., Johnson J., Graham L.K., Lee M.H., Chen C.S., Newton A.C., Schaffhausen B.S., Toker A. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr. Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- Coleman M.E., DeMayo F., Yin K.C., Lee H.M., Geske R., Montgomery C., Schwartz R.J. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J. Biol. Chem. 1995;270:12109–12116. doi: 10.1074/jbc.270.20.12109. [DOI] [PubMed] [Google Scholar]
- Coolican S.A., Samuel D.S., Ewton D.Z., McWade F.J., Florini J.R. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J. Biol. Chem. 1997;272:6653–6662. doi: 10.1074/jbc.272.10.6653. [DOI] [PubMed] [Google Scholar]
- Cuenda A., Cohen P. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J. Biol. Chem. 1999;274:4341–4346. doi: 10.1074/jbc.274.7.4341. [DOI] [PubMed] [Google Scholar]
- Datta R.S., Brunet A., Greenberg M.E. Cellular survivala play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Deng C., Zhang P., Harper J.W., Elledge S.J., Leder P. Mice lacking p21/CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- El-Deiry W.S. p21/p53, cellular growth control and genomic integrity. Curr. Top. Microbiol. Immunol. 1998;227:121–137. doi: 10.1007/978-3-642-71941-7_6. [DOI] [PubMed] [Google Scholar]
- Elledge S.J. Cell cycle checkpointspreventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Florini J.R., Magri K.A., Ewton D.Z., James P.L., Grindstaff K., Rotwein P.S. “Spontaneous” differentiation of skeletal myoblasts is dependent upon autocrine secretion of insulin-like growth factor-II. J. Biol. Chem. 1991;266:15917–15923. [PubMed] [Google Scholar]
- Ghattas I.R., Sanes J.R., Majors J.E. The encephalomyocarditis virus internal ribosome entry site allows efficient coexpression of two genes from a recombinant provirus in cultured cells and in embryos. Mol. Cell. Biol. 1991;11:5848–5859. doi: 10.1128/mcb.11.12.5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gredinger E., Gerber A.N., Tamir Y., Tapscott S.J., Bengal E. Mitogen-activated protein kinase pathway is involved in the differentiation of muscle cells. J. Biol. Chem. 1998;273:10436–10444. doi: 10.1074/jbc.273.17.10436. [DOI] [PubMed] [Google Scholar]
- Guo K., Wang J., Andres V., Smith R.C., Walsh K. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol. Cell. Biol. 1995;15:3823–3829. doi: 10.1128/mcb.15.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy O., Novitch B.G., Spicer D.B., Skapek S.X., Rhee J., Hannon G.J., Beach D., Lassar A.B. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- Jacks T., Weinberg R.A. The expanding role of cell cycle regulators. Science. 1998;280:1035–1036. doi: 10.1126/science.280.5366.1035. [DOI] [PubMed] [Google Scholar]
- Jiang B.H., Aoki M., Zheng J.Z., Li J., Vogt P.K. Myogenic signaling of phosphatidylinositol 3-kinase requires the serine-threonine kinase Akt/protein kinase B. Proc. Natl. Acad. Sci. USA. 1999;96:2077–2081. doi: 10.1073/pnas.96.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B.H., Zheng J.Z., Vogt P.K. An essential role of phosphatidylinositol 3-kinase in myogenic differentiation. Proc. Natl. Acad. Sci. USA. 1998;95:14179–14183. doi: 10.1073/pnas.95.24.14179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J.I., Clemmons D.R. Insulin-like growth factors and their binding proteinsbiological actions. Endocr. Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- Kaliman P., Canicio J., Shepherd P.R., Beeton C.A., Testar X., Palacin M., Zorzano A. Insulin-like growth factors require phosphatidylinositol 3-kinase to signal myogenesisdominant negative p85 expression blocks differentiation of L6E9 muscle cells. Mol. Endocrinol. 1998;12:66–77. doi: 10.1210/mend.12.1.0047. [DOI] [PubMed] [Google Scholar]
- Kaliman P., Canicio J., Testar X., Palacin M., Zorzano A. Insulin-like growth factor-II, phosphatidylinositol 3-kinase, nuclear factor-kappaB and inducible nitric-oxide synthase define a common myogenic signaling pathway. J. Biol. Chem. 1999;274:17437–17444. doi: 10.1074/jbc.274.25.17437. [DOI] [PubMed] [Google Scholar]
- Kaliman P., Vinals F., Testar X., Palacin M., Zorzano A. Phosphatidylinositol 3-kinase inhibitors block differentiation of skeletal muscle cells. J. Biol. Chem. 1996;271:19146–19151. doi: 10.1074/jbc.271.32.19146. [DOI] [PubMed] [Google Scholar]
- Kane L.P., Shapiro V.S., Stokoe D., Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Curr. Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- Lassar A.B., Skapek S.X., Novitch B. Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr. Opin. Cell Biol. 1994;6:788–794. doi: 10.1016/0955-0674(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Lawlor M.A., Feng X., Everding D.R., Sieger K., Stewart C.E.H., Rotwein P. Dual control of muscle cell survival by distinct growth factor regulated signaling pathways. Mol. Cell. Biol. 2000;20:3256–3265. doi: 10.1128/mcb.20.9.3256-3265.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor M.A., Rotwein P. Insulin-like growth factor-mediated muscle cell survivalcentral roles for Akt and the cyclin-dependent kinase inhibitor, p21. Mol. Cell. Biol. 2000;20:8983–8995. doi: 10.1128/mcb.20.23.8983-8995.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Good J.A., Ziegler W.H., Parekh D.B., Alessi D.R., Cohen P., Parker P.J. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- Lee J.E., Pintar J., Efstratiadis A. Pattern of the insulin-like growth factor II gene expressin during early mouse embryogenesis. Development (Camb.). 1990;110:151–159. doi: 10.1242/dev.110.1.151. [DOI] [PubMed] [Google Scholar]
- LeRoith D., Werner H., Beitner-Johnson D., Roberts C.T., Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- Levkau B., Koyama H., Raines E.W., Clurman B.E., Herren B., Orth K., Roberts J.M., Ross R. Cleavage of p21Cip1/Waf1 and p27Kip1 mediates apoptosis in endothelial cells through activation of Cdk2role of a caspase cascade. Mol. Cell. 1998;1:553–563. doi: 10.1016/s1097-2765(00)80055-6. [DOI] [PubMed] [Google Scholar]
- Liu J.P., Baker J., Perkins A.S., Robertson E.J., Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- Mal A., Chattopadhyay D., Ghosh M.K., Poon R.Y.C., Hunter T., Harter M.L. p21 and retinoblastoma protein control the absence of DNA replication in terminally differentiated muscle cells. J. Cell Biol. 2000;149:281–292. doi: 10.1083/jcb.149.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarras D., Aurade F., Johnson T., Ilan J., Gros F., Pinset C. Autonomous differentiation in the mouse myogenic cell line, C2, involves a mutual positive control between insulin-like growth factor II and MyoD, operating as early as at the myoblast stage. J. Cell Sci. 1996;109:551–560. doi: 10.1242/jcs.109.3.551. [DOI] [PubMed] [Google Scholar]
- Musaro A., Rosenthal N. Maturation of the myogenic program is induced by postmitotic expression of insulin-like growth factor I. Mol. Cell. Biol. 1999;19:3115–3124. doi: 10.1128/mcb.19.4.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes C., Hall A. Regulation and function of the Rho subfamily of small GTPases. Curr. Opin. Genet. Dev. 1994;4:77–81. doi: 10.1016/0959-437x(94)90094-9. [DOI] [PubMed] [Google Scholar]
- Olson E.N., Klein W.H. bHLH factors in muscle developmentdead lines and commitments, what to leave in and what to leave out. Genes Dev. 1994;8:1–8. doi: 10.1101/gad.8.1.1. [DOI] [PubMed] [Google Scholar]
- Otten A.D., Firpo E.J., Gerber A.N., Brody L.L., Roberts J.L., Tapscott S.J. Inactivation of MyoD-mediated expression of p21 in tumor cell lines. Cell Growth Diff. 1997;8:1151–1160. [PubMed] [Google Scholar]
- Parker S.B., Eichele G., Zhang P., Rawls A., Sands A.T., Bradley A., Olson E.N., Harper J.W., Elledge S.J. p53-independent expression of p21/Cip1 in muscle and other terminally differentiating cells. Science. 1995;267:1024–1027. doi: 10.1126/science.7863329. [DOI] [PubMed] [Google Scholar]
- Poluha W., Poluha D.K., Chang B., Crosbie N.E., Schonhoff C.M., Kilpatrick D.L., Ross A.H. The cyclin-dependent kinase inhibitor p21/WAF1 is required for survival of differentiating neuroblastoma cells. Mol. Cell. Biol. 1996;16:1335–1341. doi: 10.1128/mcb.16.4.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell-Braxton L., Hollingshead P., Warburton C., Dowd M., Pitts-Meek S., Dalton D., Gillet N., Stewart T.A. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993;7:2609–2617. doi: 10.1101/gad.7.12b.2609. [DOI] [PubMed] [Google Scholar]
- Pullen N., Dennis P.B., Andjelkovic M., Dufner A., Kozma S.C., Hemmings B.A., Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279:707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- Ramocki M.B., White M.A., Konieczny S.F., Taparowsky E.J. A role for RalGDS and a novel Ras effector in the Ras-mediated inhibition of skeletal myogenesis. J. Biol. Chem. 1998;273:17696–17701. doi: 10.1074/jbc.273.28.17696. [DOI] [PubMed] [Google Scholar]
- Romashkova J.A., Makarov S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- Rommel C., Clarke B.A., Zimmermann S., Nunez L., Rossman R., Reid K., Moelling K., Yancopoulos G.D., Glass D.J. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738–1741. doi: 10.1126/science.286.5445.1738. [DOI] [PubMed] [Google Scholar]
- Sarbassov D.D., Jones L.G., Peterson C.A. Extracellular signal-regulated kinase-1 and -2 respond differently to mitogenic and differentiative signaling pathways in myoblasts. Mol. Endocrinol. 1997;11:2038–2047. doi: 10.1210/mend.11.13.0036. [DOI] [PubMed] [Google Scholar]
- Sarbassov D.D., Peterson C.A. Insulin receptor substrate-1 and phosphatidylinositol 3-kinase regulate extracellular signal-regulated kinase-dependent and -independent signaling pathways during myogenic differentiation. Mol. Endocrinol. 1998;12:1870–1878. doi: 10.1210/mend.12.12.0205. [DOI] [PubMed] [Google Scholar]
- Sizemore N., Leung S., Stark G.R. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol. Cell. Biol. 1999;19:4798–4805. doi: 10.1128/mcb.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart C.E.H., James P.L., Fant M.E., Rotwein P. Overexpression of insulin-like growth factor-II induces accelerated myoblast differentiation. J. Cell Physiol. 1996;169:23–32. doi: 10.1002/(SICI)1097-4652(199610)169:1<23::AID-JCP3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Stewart C.E.H., Rotwein P. Growth, differentiation and survivalmultiple physiological functions for insulin-like growth factors Physiol. Rev. 76 1996. 1005 1026a [DOI] [PubMed] [Google Scholar]
- Stewart C.E.H., Rotwein P. Insulin-like growth factor-II is an autocrine survival factor for differentiating myoblasts J. Biol. Chem. 271 1996. 1130 11338b [DOI] [PubMed] [Google Scholar]
- Takano H., Komuro I., Oka T., Shiojima I., Hiroi Y., Mizuno T., Yazaki Y. The Rho family of G proteins play a critical role in muscle differentiation. Mol. Cell. Biol. 1998;18:1580–1589. doi: 10.1128/mcb.18.3.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamir Y., Bengal E. Phosphoinositide 3-kinase induces the transcriptional activity of MEF2 proteins during muscle differentiation. J. Biol. Chem. 2000;275:34424–34432. doi: 10.1074/jbc.M005815200. [DOI] [PubMed] [Google Scholar]
- Tollefsen S.E., Lajara R., McCusker R.H., Clemmons D.R., Rotwein P. Insulin like growth factors (IGF) in muscle development J. Biol. Chem. 264 1989. 13810 13817a [PubMed] [Google Scholar]
- Tollefsen S.E., Sadow J.L., Rotwein P. Coordinate expression of insulin-like growth factor II and its receptor during muscle differentiation Proc. Natl. Acad. Sci. USA. 86 1989. 1543 1547b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virkamaki A., Ueki K., Kahn C.R. Protein–protein interactions in insulin signaling and the molecular mechanisms of insulin resistance. J. Clin. Invest. 1999;103:931–943. doi: 10.1172/JCI6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Walsh K. Resistance to apoptosis conferred by Cdk inhibitors during myocyte differentiation. Science. 1996;273:359–361. doi: 10.1126/science.273.5273.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L., Zhou W., Croissant J.D., Johansen F.-E., Prywes R., Balasubramanyam A., Schwartz R.J. RhoA signaling via serum response factor plays an obligatory role in myogenic differentiation. J. Biol. Chem. 1998;273:30287–30294. doi: 10.1074/jbc.273.46.30287. [DOI] [PubMed] [Google Scholar]
- Weinberg R.A. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Weintraub H. The MyoD family and myogenesisredundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- Wu Z., Woodring P.A., Bhakta K.S., Tamura K., Wen F., Feramisco J.R., Karin M., Wang J.J., Puri P.L. p38 and extracellular signal-regulated kinase regulate the myogenic program at multiple steps. Mol. Cell. Biol. 2000;20:3951–3964. doi: 10.1128/mcb.20.11.3951-3964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D., Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- Yang S.-H., Galanis A., Sharrocks A.D. Targeting the p38 mitogen-activated protein kinases to MEF2 transcription factors. Mol. Cell. Biol. 1999;19:4028–4038. doi: 10.1128/mcb.19.6.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zackenhaus E., Jiang Z., Chung D., Marth J.D., Phillips R.A., Gallie B.L. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 1996;10:3051–3064. doi: 10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]
- Zetser A., Gredinger E., Bengal E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J. Biol. Chem. 1999;274:5193–5200. doi: 10.1074/jbc.274.8.5193. [DOI] [PubMed] [Google Scholar]
- Zhang P., Wong C., Liu D., Finegold M., Harper J.W., Elledge S.J. p21(CIP1) and p57(KIP2) control muscle differentiation at the myogenin step. Genes Dev. 1999;13:213–224. doi: 10.1101/gad.13.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., New L., Kravchenko V.L., Kato Y., Gram H., Di Padova F., Olson E.N., Ulevitch R.J., Han J. Regulation of the MEF2 family of transcription factors by p38. Mol. Cell. Biol. 1999;19:21–30. doi: 10.1128/mcb.19.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]