

Abstract
Background
Adult animals with a neonatal ventral hippocampal lesion (NVHL) exhibit deficits in working memory and sensorimotor gating similar to those observed in schizophrenia. As cognitive deficits in this disorder are typically associated with changes in cortical metabolic levels, we investigated here whether an NVHL affects metabolic responses to ventral tegmental area (VTA) activation, a procedure that elicits abnormal cell firing in the prefrontal cortex (PFC) of NVHL animals.
Methods
Prefrontal cortex metabolic activity was determined by measuring cytochrome oxidase I (CO-I) staining. Cytochrome oxidase I levels were quantified by densitometry in pre- and postpubertal sham-operated and lesioned rats that received one or three series of fifteen 20-Hz trains of VTA stimuli every 20 seconds.
Results
Ventral tegmental area stimulation yielded higher levels of PFC CO-I in NVHL animals when compared with the sham-operated group, an effect that appeared only after puberty. Increasing the series of burst stimulations further elevated CO-I in sham-operated, but not in NVHL animals.
Conclusions
Increased PFC CO-I activity after VTA burst stimulation in NVHL rats highlights the enhanced energy demand that could be linked to the exaggerated response to stress observed in these animals. The inability to further increase the response with higher mesocortical activity, as observed in sham-operated animals, could be expression of a reduced PFC functional capacity in lesioned animals. Thus, a hyperexcitable PFC with a reduced ability to further increase activity could be a plausible pathophysiological scenario for schizophrenia. Human functional studies could be interpreted in the light of this conceptual framework.
Keywords: Cytochrome oxidase, hypofrontality, neonatal ventral hippocampal lesion, prefrontal cortex, schizophrenia, ventral tegmental area
The prefrontal cortex (PFC) is critically affected in schizo- formation phrenia,adisorder and dopamine thatalsosystems involves(for thehippocampal review, see O’Donnell and Grace 1998). A neonatal ventral hippocampal lesion (NVHL) has been proposed as a developmental model of schizophrenia (Lipska and Weinberger 1998, 2000), and it may reproduce an altered PFC function. This manipulation yields a variety of behavioral deficits, including hyperlocomotion and excessive reactivity to stress (measured with repeated intraperitoneal saline injection, inescapable foot shock, or a swimming test), as well as impairments in sensorimotor gating, social interactions, and working memory; all these manifestations are evident only after puberty (Becker et al 1999; Chambers et al 1996; Chrapusta et al 2003; Lipska et al 1993, 1995, 2002). Although the mechanisms underlying these changes remain unknown, treatment with antipsychotic drugs effectively reverses some of the abnormal behavioral responses that are associated with the NVHL (Le Pen and Moreau 2002; Lipska and Weinberger 1994), suggesting that the mesolimbic and the mesocortical dopaminergic systems are compromised in these animals. Indeed, we have shown that PFC pyramidal neurons in NVHL rats exhibit an abnormal firing increase (instead of the typical decrease) in response to mesocortical activation (O’Donnell et al 2002). This effect was observed in postpubertal NVHL animals but not with analogous lesions performed in adulthood, indicating that the altered PFC response may result from abnormal postnatal developmental changes within the cortico–mesocortical system (Lipska and Weinberger 2000; O’Donnell et al 2002). Prefrontal cortex lesions can reverse NVHL-induced behavioral deficits (Lipska et al 1998) as well as abnormal mesolimbic responses in the nucleus accumbens (Goto and O’Donnell 2004). Thus, an NVHL can cause PFC anomalies that appear after adolescence providing a parallel with the timing of symptom emergence in schizophrenia.
The electrophysiological findings reviewed above point to a hyperreactive PFC in animals with an NVHL. It is likely that the abnormal electrophysiological responses are reflected in local PFC metabolic activity. Thus, by assessing cytochrome oxidase I (CO-I) histochemistry, we investigated whether ventral tegmental area (VTA) stimulation affects PFC metabolic levels and how these changes become altered in NVHL animals. This method was preferred because CO-I levels are tightly coupled to neuronal firing (Hirsch et al 2000; Wong-Riley 1989) and provide a better anatomical resolution than that obtained from 2-deoxyglucose autoradiography (Hevner et al 1995).
Methods and Materials
Animals
Pregnant Sprague–Dawley rats were obtained at 18 days of gestation from Taconic Farms (Germantown, NY). At postnatal day 6 (PD 6), male pups (15–19 g) were randomly separated in two groups, either to be lesioned with ibotenic acid or to receive a sham injection of artificial cerebrospinal fluid (aCSF). All experimental protocols were performed according to the USPHS Guide for the Care and Use of Laboratory Animals and had been approved by the Albany Medical College Institutional Animal Care and Use Committee.
Neonatal Ventral Hippocampal Lesion
Male pups (PD 6–7) were anesthetized with hypothermia by being placed in wet ice for 10–15 minutes. They then were secured onto a platform on a stereotaxic frame (O’Donnell et al 2002). A cannula was lowered into the ventral hippocampus, and .3 μL o f ibotenic acid (10μg/μL i n aCSF) or of aCSF (sham) was delivered at a rate of .15 μL/min with a minipump. This procedure was repeated in the contralateral hippocampus. After surgery, animals were warmed up and returned to their cages until weaning. The entire rostrocaudal extent of damage (areas with cell loss and disorganization) was estimated in all animals by measuring the damage extent across different coronal sections that were Nissl-stained coronal sections.
Ventral Tegmental Area Stimulation and Tissue Preparation
Ventral tegmental area stimulation procedures were similar to what has been published elsewhere (Lewis and O’Donnell 2000). Briefly, rats (prepubertal: PD 33–35; adult: PD >61) were anesthetized with chloral hydrate (400 mg/kg intraperitoneally) and placed in a stereotaxic frame. Bupivacaine (.25%) was applied subcutaneously before any skin incision was made, and burr holes were drilled in the skull for stimulating electrode placement in the VTA (5.8 mm posterior to bregma, .5 mm lateral from the midline, and 8.3 mm below the brain surface; Paxinos and Watson 1997). Concentric bipolar electrodes with .5 mm between tips were used for VTA stimulation. Fifteen 20-Hz trains of five pulses mimicking dopamine (DA) cell burst firing were delivered every 20 seconds. Current pulses were driven by a Master 8 Stimulator (AMPI, Jerusalem, Israel) and were controlled by a computer. In control animals (both sham-operated and NVHL), the stimulating electrode was placed in the VTA, but no current pulses were delivered. Five minutes after the last train of stimuli, the electrode was slowly removed and the rat was decapitated. The brain was quickly removed, frozen in dry ice–acetone at −25°C, and stored at −80°C. Coronal 16-μm-thick tissue sections were cut in a cryostat and thaw-mounted onto double gelatin-coated glass slides. Tissue sections were dehydrated at room temperature and stored at −80°C until processing.
Cytochrome Oxidase Histochemistry and Data Analysis
Histochemistry of CO-I was performed according to the protocol of Wong-Riley (1979), as modified by Murer et al (2000). Slides were incubated for 90 minutes at 37°C in .1 M phosphate buffer (pH 7.4) containing .50 g/L of 3,3′-diaminobenzidine (Sigma-Aldrich, St. Louis, Missouri), .33 g/L of horse heart cytochrome c (Sigma-Aldrich), 44 g/L of sucrose (Sigma-Aldrich), and .2 g/L of catalase (hydrogen peroxidase oxidoreductase; Sigma-Aldrich). After incubation (90 min), slides were rinsed three times in phosphate buffer, dehydrated and coverslipped. Measurements of CO-I intensity were performed by digitizing the stained section using a slide scanner (Coolscan IV; Nikon, Japan), converting the image to grayscale, and using the scanner software (Nikon Scan v.3.1) to determine optical density. These measures were taken by an investigator blind to the experimental condition. The mean relative optical density (ROD) per pixel was determined by subtracting the optical density of the background from that of the structure of interest (i.e., the prelimbic and infralimbic regions of the medial PFC) in coronal sections within 3.2 to 2.5 mm rostral to bregma (Paxinos and Watson 1997). Background OD was measured at the level of the forceps minor or corpus callosum. For each animal, a single value per structure was obtained by averaging measurements from several (6–8) sections.
Statistical Analyses
All data are expressed as mean ± SD. Student’s t test was used for two-group comparisons involving a single continuous variable. To compare the effect of NVHL on CO-I activity along two or more variables, two-way ANOVA followed by Tukey’s post hoc test was preferred. Differences between experimental conditions were considered to be statistically significant when p< .05.
Results
The NVHL procedure caused neuronal loss and cellular disarray in the ventral hippocampus as described elsewhere (Lipska et al 1993; O’Donnell et al 2002; Figure 1). Only animals that showed a bilateral lesion that was restricted to the ventral hippocampus were included for analysis. All measures were taken from adult (PD >61) or prepubertal (PD 33–35) animals.
Figure 1.
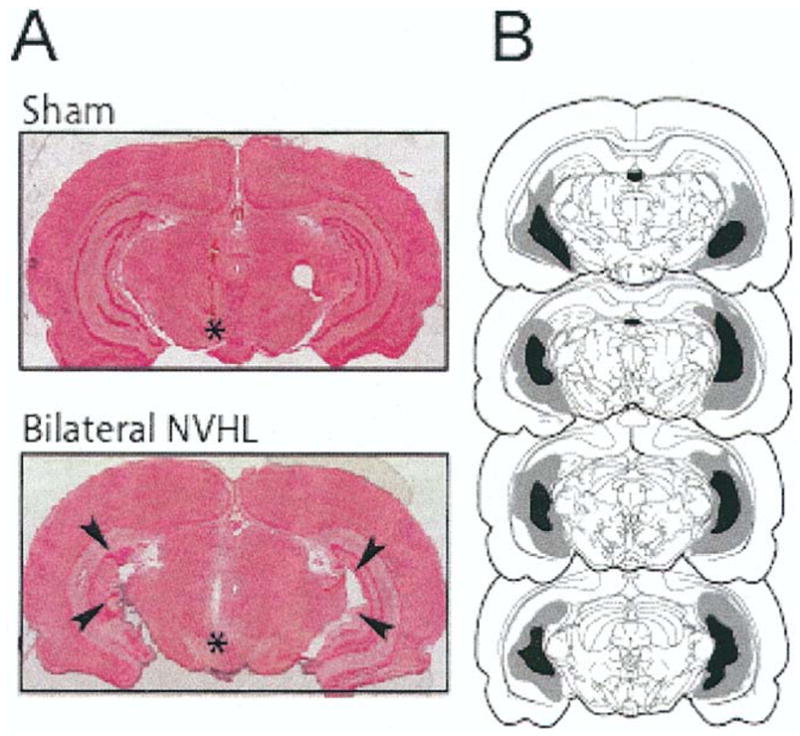
(A) Coronal section (Nissl staining) showing the ventral hippocampus after a sham operation (top) and a neonatal ventral hippocampal lesion (NVHL; bottom). The ventral hippocampus (arrowheads) of the lesioned animal is characterized by loss of neurons, cellular disarray, and enlarged ventricles. *Placement of ventral tegmental area–stimulating electrode track. (B) Drawings illustrating the extension of the lesion. Gray and dark areas indicate maximal and minimal extents of damage, respectively.
We first investigated the range of CO-I staining at different incubation times by using slides that contained normal control PFC sections. Prefrontal cortex CO-I staining increased with incubation time, reaching a saturation level after 120–150 minutes (Figure 2). Therefore, the most appropriate incubation times to confidently detect changes in CO-I signal were 60 and 90 minutes (Figure 2). All measurements in this study were conducted in brain sections incubated for 90 minutes.
Figure 2.
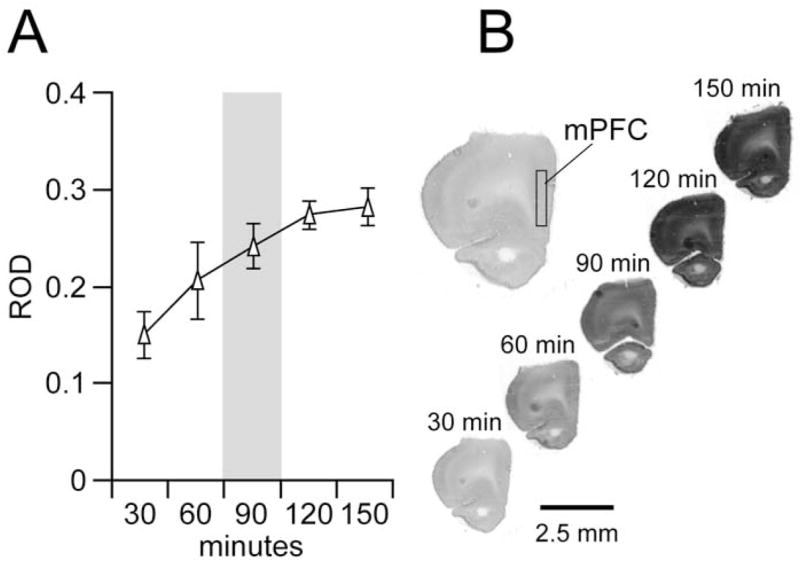
(A) Relationship between incubation time and prefrontal cortex (PFC) cytochrome oxidase I (CO-I) levels in slides containing normal nonstimulated brain sections (mean ± SD; n = 4; 5– 6 sections per animal). After incubating for 120 –150 minutes, PFC CO-I staining reached saturation level. As indicated in gray, 90 minutes of incubation was the most appropriate time point at which to assess the effect of neonatal ventral hippocampal lesion and ventral tegmental area stimulation on PFC CO-I activity. The variability of CO-I staining was lower than that from 60 minutes, and therefore it allows a better relative optical density range to detected CO-I changes in the PFC and other cortical and subcortical regions. (B) Representative examples of coronal forebrain sections at the level of the PFC showing CO-I staining after 30, 60, 90, 120, and 150 minutes of incubation.
Prefrontal cortex CO-I activity increased with VTA stimulation in NVHL animals. Adult NVHL and sham-operated animals exhibited similar basal PFC CO-I activity. Relative optical density in the PFC was .23 ± .02 in NVHL (n = 6) and .24 ± .02 in sham-operated rats (n = 5) when VTA stimulation was not delivered. After 15 repetitions of VTA burst stimulation (sets of 5 pulses at 20 Hz every 30 s), animals with an NVHL yielded significantly higher PFC CO-I levels when compared with the sham-operated group. Relative optical density in the PFC of stimulated NVHL animals was .26 ± .01 (n = 7) and .21 ± .02 in stimulated sham-operated animals (n = 8; Figure 3A). A two-way ANOVA analysis did not reveal any significant effect of stimulation or lesion as independent factors; however, a significant interaction between stimulation and lesion was observed [F(1,22) = 11.8, p< .003], indicating that VTA stimulation elicited different effects in sham-operated and lesioned animals. A similar analysis of other brain regions including the somatosensory cortex, dorsal striatum, and nucleus accumbens (core and shell) did not reveal differences between sham-operated and lesioned animals (Figure 3B). The higher VTA-evoked metabolic response in the PFC was not present in prepubertal (PD 28–36) NVHL rats. Prepubertal NVHL and sham-operated animals exhibited similar PFC CO-I levels: basal PFC ROD was .21 ± .01 in NVHL (n = 6) and .21 ± .02 in sham-operated (n = 6) animals. Electrical VTA stimulation also resulted in similar CO-I staining in younger animals, irrespective of lesion condition: PFC ROD was .21 ± .01 in NVHL (n = 5) and .21 ± .02 in sham-operated (n = 5) animals. These data suggest that VTA-driven abnormal PFC CO-I increase in the lesioned animals is restricted to the mesocortical system and emerges after puberty.
Figure 3.
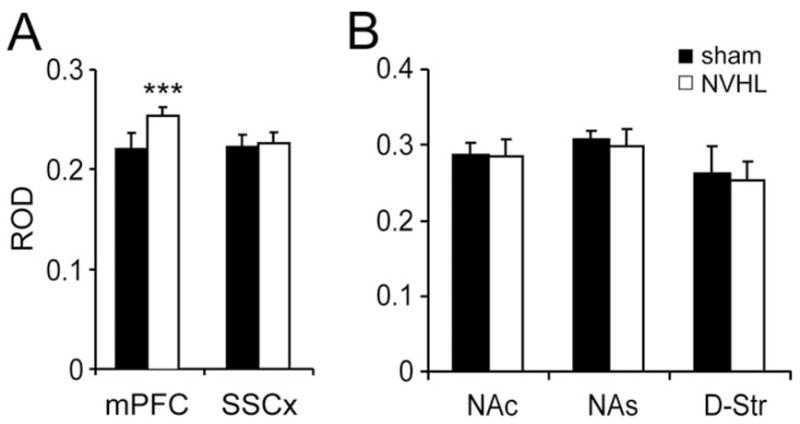
(A) Effects of ventral tegmental area (VTA) burst stimulation on medial prefrontal cortex (mPFC) and somatosensory cortex (SSCx) cytochrome oxidase I (CO-I) activity. A significantly higher CO-I level was found in the medial PFC of adult (postnatal day > 62) neonatal ventral hippocampal lesioned animals when VTA stimulation was delivered (*** p< .002, compared with sham-operated, Tukey post hoc test after significant analysis of variance assessment). In contrast, VTA stimulation failed to modify the CO-I activity in the SSCx. (B) Effect of VTA stimulation on CO-I levels in three different subcortical regions that receive important DA inputs: the nucleus accumbens core (NAc) and shell (NAs), and the dorsal striatum (D-Str). No statistical differences were observed between sham-operated and NVHL animals in any of these regions.
We also examined the metabolic responses to more protracted mesocortical stimulation in pre and postpubertal sham-operated and lesioned animals. The elevated PFC CO-I response observed in NVHL animals (compared with stimulated sham-operated animals) that received a single set (S1) of 15 trains was not observed after delivering three series (S3) of 15 trains. In contrast, PFC CO-I responses increased to a similar extent in both groups after S3 stimulations (PFC ROD in sham-operateds: .26 ± .01, n = 5; in NVHL: .25 ± .02, n = 7). When the VTA was not stimulated, PFC ROD was .23 ± .03 in sham-operated (n = 5) and .24 ± .03 in NVHL (n = 6) animals. Sham animals that received three series of trains in the VTA exhibited a significantly higher PFC ROD than was exhibited in S1 stimulated sham-operated rats (p< .01, Tukey post hoc test after significant ANOVA). However, an S3 train series did not yield higher PFC ROD than did S1 stimulation in NVHL animals. The higher metabolic response to S3 VTA train stimulation was not evident in the PFC of prepubertal animals (Figure 4). Prefrontal cortex ROD in S3-stimulated prepubertal sham-operated and lesioned rats were .22 ± .02 (n = 4) and .21 ± .01 (n = 6), respectively. These values were similar to those obtained with one set of train stimulation (see previous paragraph) as well as to the ROD levels when S3 VTA stimulation were not delivered (sham operated: .21 ± .02, n = 9; NVHL: .20 ± .02, n = 8). When all PFC data were normalized to the average CO-I ROD value in nonstimulated conditions (for both sham operated and NVHL rats separately; Figure 4), the different impact on PFC metabolic activity that was induced by S1 and S3 VTA stimulation in both treatment groups become evident. Ventral tegmental area stimulation had minimal effects in prepubertal animals from either group. However, in adult animals, a single series of VTA stimuli caused metabolic activity approximately 10% higher than baseline in NVHL (which was significant) and <10% lower in sham-operated animals (not significative). Increasing the series of VTA stimulation (S3) resulted in higher CO-I levels in sham-operated animals (Figure 4). The difference in metabolic activation by VTA stimulation observed with S1 in sham-operated vs. NVHL animals was not observed using an S3 protocol. Altogether, these data indicate that adult, not prepubertal, NVHL animals exhibited altered PFC metabolic response to increasing mesocortical activity.
Figure 4.
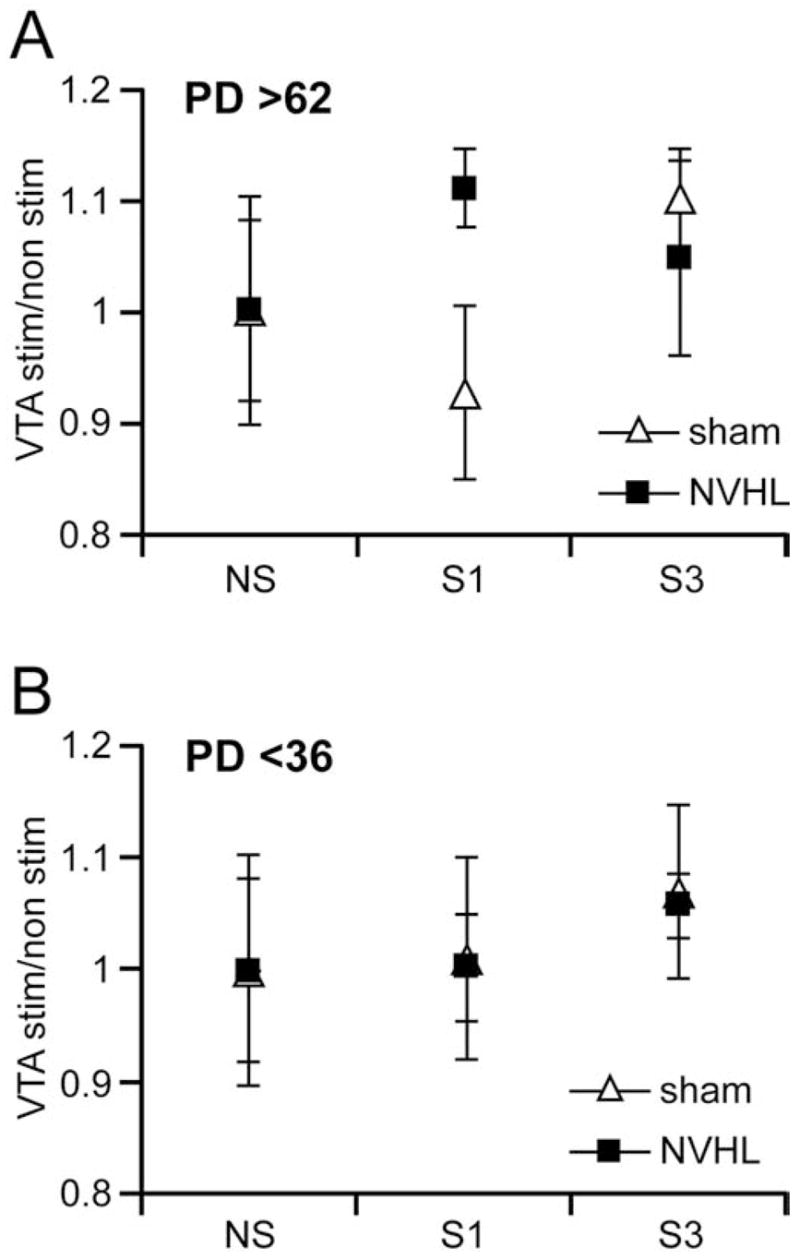
Medial prefrontal cortex (PFC) cytochrome oxidase I (CO-I) ratio induced by one (S1; 15 train of 5 pulses at 20 Hz every 30 s) and three (S3; 3 repetitions of S1 every 20 min) sets of repetitions of ventral tegmental area (VTA) stimulation in adult (A) and prepubertal (B) sham-operated and neonatal ventral hippocampal lesioned (NVHL) animals. An opposite PFC CO-I ratio response to S1 VTA stimulation was found in adult sham-operated and lesioned animals whereas no consistent changes were observed in the PFC of prepubertal rats. Increasing the repetitions of burst stimulation to S3 effectively enhanced the ratio of CO-I level in the adult PFC of sham-operated animals as compared with that induced with S1. In contrast to what was observed in the sham-operated group, S3 stimulation failed to further increase the CO-I ratio obtained in the PFC of adult NVHL animals with S1. PD, postnatal day; NS, no stimulation.
Discussion
We investigated the effect of VTA stimulation on PFC metabolism of NVHL animals by measuring the density of CO-I histochemistry staining. Sham and lesioned animals exhibited similar PFC CO-I levels when VTA stimulation was not delivered. After 15 trains of burst (5 pulses at 20 Hz) stimulation to the VTA, significantly higher CO-I levels were observed in the PFC of adult, not prepubertal, lesioned animals, and slightly lower PFC metabolic activity, in sham-operated animals. These changes were not observed in other cortical and subcortical regions. Increasing the number of bursts delivered to the VTA (3 sets of 15 trains) failed to potentiate the already elevated PFC metabolic level obtained with one set of 15 repetitions in the lesioned group but did increase metabolic levels in the PFC of sham-operated animals, suggesting a ceiling effect in lesioned animals. These results indicate that PFC metabolic responses to VTA activation are compromised in NVHL animals, probably reflecting an inefficient functional capacity of the mesocortical system that becomes evident after puberty.
Energy production in the brain is strongly dictated by local neuronal network activity and afferent inputs. Cytochrome oxidase I i s the terminal enzyme of the mitochondrial electron-transport chain that provides most of the adenosine triphosphate that is used in the brain (for review, see Wong-Riley 1989) and it is a useful marker of brain metabolic activity. Wong-Riley and co-workers have shown that a mono-ocular injection of tetrodotoxin, a procedure used to inhibit neuronal activity in the geniculate body, also induced a profound decrease in local CO-I activity (Wong-Riley 1979). Both CO-I protein levels and mRNA coding for the subunits of CO-I are responsive to changes in neuronal activity, allowing analyses of regional, cellular, and subcellular functional levels (Hevner and Wong-Riley 1990, 1991; Wong-Riley 1989). Expression of CO-I correlated with neuronal firing in the basal ganglia after chronic nigrostriatal lesions (Hirsch et al 2000; Vila et al 2000). In addition, the anatomical resolution of CO-I histochemistry is better than that of 2-deoxyglucose autoradiography (Hevner et al 1995; Wong-Riley 1989). Thus, our measures of CO-I in cortical and subcortical regions are likely to reflect cell firing and overall neuronal activity patterns that are evoked by the stimulation protocols used.
Electrical VTA stimulation with a burst pattern identical to the one used here elicits different firing responses in PFC pyramidal neurons in NVHL and sham-operated or naïve animals. Stimulation of VTA with trains of pulses mimicking DA neurons burst firing typically evokes prolonged plateau depolarizations with suppression of cell firing in PFC pyramidal neurons recorded in vivo from naïve animals (Lewis and O’Donnell 2000). This response was dramatically altered in animals with an NVHL, in which VTA stimulation caused exaggerated cell firing, but only in adult animals (O’Donnell et al 2002). The increased PFC metabolism driven by VTA stimulation that was observed in this study may be related to the VTA-evoked firing recorded in these animals. In sham-operated animals, as in naïve rats, VTA stimulation decreased pyramidal cell firing (O’Donnell et al 2002). This effect could be related to the slight decrease in CO-I activity we observed in sham-operated animals. Thus, CO-I levels in the PFC after VTA stimulation match quite closely the changes in firing rate that previously have been reported with similar stimulation patterns.
Increasing the number of stimuli in the VTA induced significantly higher CO-I levels in the PFC of sham-operated animals than those obtained with S1-stimulation protocol. This suggests that activation of the mesocortical pathway can lead to neuronal firing increase in the normal PFC, depending on the afferent demand. In contrast, a hyperreactive PFC (detected as increased spike firing and metabolic activity in response to VTA stimulation) was observed in NVHL animals with less intense stimulation. Increasing repetitions of VTA train stimulation failed to further increase PFC metabolic response in NVHL animals. This can be interpreted as evidence of a reduced functional capacity of the PFC in NVHL animals. Thus, a postpubertal acquisition of this abnormal PFC condition could be linked to the exaggerated response to stress (Lipska et al 1993) and the expression of cognitive deficits involving executive control (e.g., working memory) that typically emerge after puberty in NVHL animals (Lipska and Weinberger 1998).
At a cellular level, it recently has been shown that DA modulation of PFC glutamatergic transmission matures after puberty (Tseng and O’Donnell 2005) and involves multiple cellular mechanisms, including activation of fast-spiking interneurons (Tseng and O’Donnell 2004). It is possible that an early inactivation of the hippocampal formation alters the postnatal maturation of a subpopulation of inhibitory interneurons in the PFC, the parvalbumin-containing GABAergic interneurons. Consequently, the abnormal and exaggerated VTA-evoked PFC metabolic response that is observed in neonatally lesioned animals could reflect a developmental disruption of PFC GABAergic modulation by DA. These changes may result in reduction of a local inhibitory tone that normally matures during adolescence (Tseng et al 2005). Although this hypothesis remains to be examined, recent studies showed that NVHLs selectively down-regulate the expression of glutamate decarboxylase-67 mRNA in the PFC, the rate-limiting enzyme for GABA synthesis (Lipska et al 2003a, 2003b). Therefore, a functional attenuation of local GABAergic transmission could contribute to establishing the hyperexcitable PFC and higher metabolic activity observed in these animals.
The NVHL is proposed as a model of PFC deficits in schizophrenia, a disorder characterized by hypofrontality. Although the neural bases that underlie cortical dysfunction in schizophrenia remain unclear, most neuroimaging findings have been interpreted as indicating a hypofunctional PFC state during working-memory testing. Our findings of increased PFC cell firing (O’Donnell et al 2002) and of increased metabolism in response to VTA stimulation in NVHL animals do not at first appear consistent with the hypofrontality concept. Although hypofrontality refers to the lack of PFC activation during working-memory tasks (for review, see Manoach 2003), studies also showed absence of changes or even increased activation of the dorsolateral PFC (Callicott et al 2000, 2003a, 2003b; Honey et al 2002; Manoach et al 1999, 2000). Thus, PFC dysfunction and working memory deficits in schizophrenia are variable and complex. It is well known that PFC activity increases with working memory load until the demand exceeds the functional capacity of the system, during which the activation decreases (Braver et al 1997; Callicott et al 1999; Goldberg et al 1998; Manoach 2003). A similar inverted-U relationship between PFC activity and task demand exists in schizophrenia, but one shifted toward the left (Manoach 2003). Therefore, the loads required to perform the task may determine whether cortical metabolism increases or decreases.
The abnormal PFC activity observed in NVHL animals was driven by VTA burst stimulation. This manipulation may be reproducing the effect of salient stimuli and the sustained activity of some cells during working memory tasks, which is dependent on DA (Goldman-Rakic 1996). A critical element in the role of mesocortical system on working memory and other cognitive functions is the ability to enhance salient inputs and reduce unwanted activity (O’Donnell 2003). The hyperreactive PFC observed in NVHL animals could reflect a breakdown of this contrast-enhancement property of mesocortical inputs, resulting in inappropriate engagement of PFC units during a particular task. In summary, an abnormal PFC metabolic response to mesocortical activation in animals with an NVHL becomes evident after puberty and appears concurrently with many mesocortically dependent behavioral abnormalities that are observed in these animals. These changes could be related to the altered PFC function observed in schizophrenia, a cortical pathophysiological condition that the NVHL has been proposed to model (Lipska and Weinberger 1998).
Acknowledgments
This work was supported by National Institutes of Health Grant No. MH57683 (P.O’D) and by a National Alliance for Research on Schizophrenia and Depression (NARSAD) Independent Investigator Award (P.O’D). We thank Ms. Maureen O’Keeffe for her excellent technical assistance.
References
- Becker A, Grecksch G, Bernstein HG, Hollt V, Bogerts B. Social behaviour in rats lesioned with ibotenic acid in the hippocampus: Quantitative and qualitative analysis. Psychopharmacology (Berl) 1999;144:333–338. doi: 10.1007/s002130051015. [DOI] [PubMed] [Google Scholar]
- Braver TS, Cohen JD, Nystrom LE, Jonides J, Smith EE, Noll DC. A parametric study of prefrontal cortex involvement in human working memory. Neuroimage. 1997;5:49 – 62. doi: 10.1006/nimg.1996.0247. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Bertolino A, Mattay VS, Langheim FJ, Duyn J, Coppola R, et al. Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb Cortex. 2000;10:1078 –1092. doi: 10.1093/cercor/10.11.1078. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Egan MF, Mattay VS, Bertolino A, Bone AD, Verchinksi B, et al. Abnormal fMRI response of the dorsolateral prefrontal cortex in cognitively intact siblings of patients with schizophrenia. Am J Psychiatry. 2003a;160:709 –719. doi: 10.1176/appi.ajp.160.4.709. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Mattay VS, Bertolino A, Finn K, Jones K, Frank JA, et al. Physiological characteristics of capacity constraints in working memory as revealed by functional MRI. Cereb Cortex. 1999;9:20 –26. doi: 10.1093/cercor/9.1.20. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Mattay VS, Verchinski BA, Marenco S, Egan MF, Weinberger DR. Complexity of prefrontal cortical dysfunction in schizophrenia: More than up or down. Am J Psychiatry. 2003b;160:2209 –2215. doi: 10.1176/appi.ajp.160.12.2209. [DOI] [PubMed] [Google Scholar]
- Chambers RA, Moore J, McEvoy JP, Levin ED. Cognitive effects of neonatal hippocampal lesions in a rat model of schizophrenia. Neuropsychopharmacology. 1996;15:587–594. doi: 10.1016/S0893-133X(96)00132-7. [DOI] [PubMed] [Google Scholar]
- Chrapusta SJ, Egan MF, Wyatt RJ, Weinberger DR, Lipska BK. Neonatal ventral hippocampal damage modifies serum corticosterone and dopamine release responses to acute footshock in adult Sprague-Dawley rats. Synapse. 2003;47:270 –277. doi: 10.1002/syn.10179. [DOI] [PubMed] [Google Scholar]
- Goldberg TE, Berman KF, Fleming K, Ostrem J, Van Horn JD, Esposito G, et al. Uncoupling cognitive workload and prefrontal cortical physiology: A PET rCBF study. Neuroimage. 1998;7:296 –303. doi: 10.1006/nimg.1998.0338. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic P. Regional and cellular fractionation of working memory. Proc Natl Acad Sci USA. 1996;93:13473–13480. doi: 10.1073/pnas.93.24.13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, O’Donnell P. Prefrontal lesion reverses abnormal mesoaccumbens response in an animal model of schizophrenia. Biol Psychiatry. 2004;55:172–176. doi: 10.1016/s0006-3223(03)00783-2. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Liu S, Wong-Riley MT. A metabolic map of cytochrome oxidase in the rat brain: Histochemical, densitometric and biochemical studies. Neuroscience. 1995;65:313–342. doi: 10.1016/0306-4522(94)00514-6. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Wong-Riley MT. Regulation of cytochrome oxidase protein levels by functional activity in the macaque monkey visual system. J Neurosci. 1990;10:1331–1340. doi: 10.1523/JNEUROSCI.10-04-01331.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevner RF, Wong-Riley MT. Neuronal expression of nuclear and mitochondrial genes for cytochrome oxidase (CO) subunits analyzed by in situ hybridization: Comparison with CO activity and protein. J Neurosci. 1991;11:1942–1958. doi: 10.1523/JNEUROSCI.11-07-01942.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Perier C, Orieux G, Francois C, Feger J, Yelnik J, et al. Metabolic effects of nigrostriatal denervation in basal ganglia. Trends Neurosci. 2000;23(10 Suppl):S78 –S85. doi: 10.1016/s1471-1931(00)00021-5. [DOI] [PubMed] [Google Scholar]
- Honey GD, Bullmore ET, Sharma T. De-coupling of cognitive performance and cerebral functional response during working memory in schizophrenia. Schizophr Res. 2002;53:45–56. doi: 10.1016/s0920-9964(01)00154-2. [DOI] [PubMed] [Google Scholar]
- Le Pen G, Moreau JL. Disruption of prepulse inhibition of startle reflex in a neurodevelopmental model of schizophrenia: Reversal by clozapine, olanzapine and risperidone but not by haloperidol. Neuropsychopharmacology. 2002;27:1–11. doi: 10.1016/S0893-133X(01)00383-9. [DOI] [PubMed] [Google Scholar]
- Lewis BL, O’Donnell P. Ventral tegmental area afferents to the prefrontal cortex maintain membrane potential “up” states in pyramidal neurons via D1 dopamine receptors. Cereb Cortex. 2000;10:1168 –1175. doi: 10.1093/cercor/10.12.1168. [DOI] [PubMed] [Google Scholar]
- Lipska BK, al-Amin HA, Weinberger DR. Excitotoxic lesions of the rat medial prefrontal cortex. Effects on abnormal behaviors associated with neonatal hippocampal damage. Neuropsychopharmacology. 1998;19:451–464. doi: 10.1016/S0893-133X(98)00045-1. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Aultman JM, Verma A, Weinberger DR, Moghaddam B. Neonatal damage of the ventral hippocampus impairs working memory in the rat. Neuropsychopharmacology. 2002;27:47–54. doi: 10.1016/S0893-133X(02)00282-8. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Chrapusta SJ, Egan MF, Weinberger DR. Neonatal excitotoxic ventral hippocampal damage alters dopamine response to mild repeated stress and to chronic haloperidol. Synapse. 1995;20:125–130. doi: 10.1002/syn.890200205. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: A potential animal model of schizophrenia. Neuropsychopharmacology. 1993;9:67–75. doi: 10.1038/npp.1993.44. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Lerman DN, Khaing ZZ, Weickert CS, Weinberger DR. Gene expression in dopamine and GABA systems in an animal model of schizophrenia: Effects of antipsychotic drugs. Eur J Neurosci. 2003a;18:391– 402. doi: 10.1046/j.1460-9568.2003.02738.x. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Lerman DN, Khaing ZZ, Weinberger DR. The neonatal ventral hippocampal lesion model of schizophrenia: Effects on dopamine and GABA mRNA markers in the rat midbrain. Eur J Neurosci. 2003b;18:3097–3104. doi: 10.1111/j.1460-9568.2003.03047.x. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. Subchronic treatment with haloperidol and clozapine in rats with neonatal excitotoxic hippocampal damage. Neuropsychopharmacology. 1994;10:199 –205. doi: 10.1038/npp.1994.22. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. Prefrontal cortical and hippocampal modulation of dopamine-mediated effects. Adv Pharmacol. 1998;42:806 – 809. doi: 10.1016/s1054-3589(08)60869-8. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: Schizophrenia as a reality test. Neuropsychopharmacology. 2000;23:223–239. doi: 10.1016/S0893-133X(00)00137-8. [DOI] [PubMed] [Google Scholar]
- Manoach DS. Prefrontal cortex dysfunction during working memory performance in schizophrenia: Reconciling discrepant findings. Schizophr Res. 2003;60:285–298. doi: 10.1016/s0920-9964(02)00294-3. [DOI] [PubMed] [Google Scholar]
- Manoach DS, Gollub RL, Benson ES, Searl MM, Goff DC, Halpern E, et al. Schizophrenic subjects show aberrant fMRI activation of dorsolateral prefrontal cortex and basal ganglia during working memory performance. Biol Psychiatry. 2000;48:99 –109. doi: 10.1016/s0006-3223(00)00227-4. [DOI] [PubMed] [Google Scholar]
- Manoach DS, Press DZ, Thangaraj V, Searl MM, Goff DC, Halpern E, et al. Schizophrenic subjects activate dorsolateral prefrontal cortex during a working memory task as measured by fMRI. Biol Psychiatry. 1999;45:1128 –1137. doi: 10.1016/s0006-3223(98)00318-7. [DOI] [PubMed] [Google Scholar]
- Murer MG, Dziewczapolski G, Salin P, Vila M, Tseng KY, Ruberg M, et al. The indirect basal ganglia pathway in dopamine D(2) receptor-deficient mice. Neuroscience. 2000;99:643– 650. doi: 10.1016/s0306-4522(00)00223-2. [DOI] [PubMed] [Google Scholar]
- O’Donnell P. Dopamine gating of forebrain neural ensembles. Eur J Neurosci. 2003;17:429 – 435. doi: 10.1046/j.1460-9568.2003.02463.x. [DOI] [PubMed] [Google Scholar]
- O’Donnell P, Grace AA. Dysfunctions in multiple interrelated systems as the neurobiological bases of schizophrenic symptom clusters. Schizophr Bull. 1998;24:267–283. doi: 10.1093/oxfordjournals.schbul.a033325. [DOI] [PubMed] [Google Scholar]
- O’Donnell P, Lewis BL, Weinberger DR, Lipska BK. Neonatal hippocampal damage alters electrophysiological properties of prefrontal cortical neurons in adult rats. Cereb Cortex. 2002;12:975–982. doi: 10.1093/cercor/12.9.975. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 3. London: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Kloc M, Lewis BL, O’Donnell P. Changes in D2 dopaminergic modulation of prefrontal cortical GABAergic interneurons during adolescence and in a developmental animal model of schizophrenia. Society for Neuroscience Abstracts. 2005;31:444.1. [Google Scholar]
- Tseng KY, O’Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci. 2004;24:5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O’Donnell P. Postpubertal emergence of prefrontal cortical up states induced by D1-NMDA co-activation. Cereb Cortex. 2005;15:49 –57. doi: 10.1093/cercor/bhh107. [DOI] [PubMed] [Google Scholar]
- Vila M, Perier C, Feger J, Yelnik J, Faucheux B, Ruberg M, et al. Evolution of changes in neuronal activity in the subthalamic nucleus of rats with unilateral lesion of the substantia nigra assessed by metabolic and electrophysiological measurements. Eur J Neurosci. 2000;12:337–344. doi: 10.1046/j.1460-9568.2000.00901.x. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT. Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res. 1979;171:11–28. doi: 10.1016/0006-8993(79)90728-5. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MTT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94 –101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]