

Abstract
Hyperglycemia-induced oxidative stress is an inciting event in the development of diabetic complications including diabetic neuropathy. Our observations of significant oxidative stress and morphological abnormalities in mitochondria led us to examine manganese superoxide dismutase (SOD2), the enzyme responsible for mitochondrial detoxification of oxygen radicals. We demonstrate that over expression of SOD2 decreases superoxide (O2•−) in cultured primary dorsal root ganglion (DRG) neurons and subsequently blocks caspase-3 activation and cellular injury. Under expression of SOD2 in dissociated DRG cultures from adult SOD2+/− mice results in increased levels of O2•−, activation of caspase-3 cleavage and decreased neurite outgrowth under basal conditions that are exacerbated by hyperglycemia. These profound changes in sensory neurons led us to explore the effects of decreased SOD2 on the development of diabetic neuropathy (DN) in mice. DN was assessed in SOD2+/− C57BL/6J mice and their SOD2+/+ litter mates following streptozotocin (STZ) treatment. These animals, while hyperglycemic, do not display any signs of DN. DN was observed in the C57BL/6Jdb/db mouse, and decreased expression of SOD2 in these animals increased DN. Our data suggest that SOD2 activity is an important cellular modifier of neuronal oxidative defense against hyperglycemic injury.
Keywords: neuropathy, oxidative stress, SOD2
INTRODUCTION
The idea that oxidative stress contributes to diabetic complications is widely acknowledged (Cameron et al., 1998; Vincent et al., 2004c; Low et al., 1997; Cameron et al., 1993; Hounsom et al., 2001;Brownlee, 2001). Our work focuses on understanding how hyperglycemia-induced oxidative stress contributes to diabetic neuropathy (DN). A unifying mechanism of DN is the ability of both metabolic and vascular insults to increase cellular oxidative stress and impair the function of the peripheral nervous system (Brownlee et al., 2004; Leinninger et al., 2006). An important mediator of oxidative stress is reactive oxygen species (ROS). ROS include oxygen radicals such as superoxide (O2•−) and the hydroxyl radical (.OH), and non-radical derivatives of molecular oxygen such as hydrogen peroxide (H2O2).
ROS are a normal result of cellular respiration; however, excess glucose may cause over-production of ROS and subsequently deplete cellular antioxidant capacity (Brownlee, 2005). Work in clinical diabetes supports this concept. Measures of oxidative stress including TRAP (Total Radical Antioxidant Potential), thiobarbituric acid reactive substances (TBARS, a measure of oxidized lipids), and advanced glycation end products (AGEs) are all elevated in diabetic patients (Rosenson, 2004; Matteucci et al., 2004;Siemionow and Demir, 2004) and in experimental models of diabetes (Toth et al., 2004; Hounsom et al., 2001).
Several lines of evidence point toward glucose-mediated accumulation of ROS as an inciting event in the development of DN (Jeffcoate, 2005;Wada and Yagihashi, 2005; Obrosova et al., 1993). In neurons, high glucose impairs the mitochondrial electron transfer chain, leading to the excess production of O2•− (Russell et al., 2002;Vincent et al., 2004c; Bolanos et al., 2004). ROS are produced when electrons escape from complexes I and III of the electron transfer chain and react with molecular oxygen to form O2•−. O2•− attacks the iron sulfur centers of complexes I and III, further limiting their function (Vincent et al., 2004c). In addition to O2•−, .OH, peroxynitrite, alkoxyl, peroxyl, and hydroperoxyl radicals also form, further damaging proteins, lipids and nucleic acids (Vincent et al., 2004c; Vincent and Feldman, 2004; Cellek, 2004).
Superoxide dismutases are essential, ubiquitous enzymes that detoxify highly reactive O2•− .by catalysis into H2O2, which in turn is reduced to H2O in the mitochondria by glutathione (Wallace, D. C. 02). The mitochondrial form of the enzyme, Mn+SOD or SOD2 is essential for cellular development and survival, since homozygous knockout of this gene is perinatally lethal (Lebovitz et al., 1996). Heterozygous expression of SOD2 leads to depletion of mitochondrial glutathione and increased oxidative stress in mice (Williams et al., 1998). Furthermore, SOD2+/− mice also display decreased activity of aconitase and NADH oxidoreductase and increased levels of intra-mitochondrial protein carbonyls and damaged mitochondrial DNA (Williams et al., 1998). They also display evidence of decreased acetylcholine-induced arterial relaxation (Ohashi et al., 2006) and increased myocardial degeneration (Strassburger et al., 2005) that both lead to impaired blood flow.
Current rodent models of DN fail to develop changes that closely resemble the human disease (Whiteley and Tomlinson, 1985; Brussee et al., 2004). The reasons for this failure are likely multiple, and include the absence of genetic susceptibility genes and a short life-span (Breyer et al., 2005). Since SOD mimetics can both decrease rodent nerve conduction deficits (Coppey et al., 2001) and restore blood flow (Schnackenberg and Wilcox, 2001) in diabetes, we hypothesized that decreased expression of SOD would accelerate oxidative injury in hyperglycemia and magnify DN. To address this question, we first altered the level of SOD2 in an in vitro model of DN, primary dorsal root ganglion (DRG) neurons. Next, we examined dissociated DRG cultures from the adult SOD2+/− mouse and then used the SOD2+/− mouse to determine the effects of decreased SOD2 gene in two models of diabetes; streptozotocin (STZ)-induced type 1 diabetes and genetic type 2, dbdb mice. DN was assessed over 24 weeks of diabetes. These experiments were aimed at confirming our in vitro observations and generating a mouse model of DN that experiences enhanced oxidative injury in response to hyperglycemia.
MATERIALS AND METHODS
Adenoviral Transfection of Primary DRG Neurons
DRG were harvested from E15 Sprague-Dawley rats, dissociated in 1% trypsin, and cultured on rat-tail collagen-coated plates in growth media. All culture reagents are from Gibco (Grand Island, NY) unless stated otherwise. Growth media were prepared using Neurobasal media supplemented with 1X B-27 additives, 50 ng/ml nerve growth factor (NGF) (Harlan Bioscience, Indianapolis, IN), 40 μM 5-fluoro-2-deoxyuridine (FUDR) (Sigma, St. Louis, MO), and 1,000 U/ml penicillin/streptomycin/neomycin (ABX) solution. Initial plating media contained 2 μM glutamine. DRG neurons were re-fed after 24 h in fresh media containing all additives except glutamine. On day 2, cells were rinsed then re-fed using treatment media (Neurobasal media containing 4 ng/ml selenium, 4 ng/ml hydrocortisone, 0.01 mg/ml transferrin, 3 ng/ml β-estradiol, 50 ng/ml NGF, 40 μM FUDR, and ABX). Experiments were performed on DRG neurons on day 3 in culture in the absence of B-27 additives.
Adenovirus (Ad) constructs containing cDNA for SOD1, SOD2, oxomaleate carrier protein (OMC) or green fluorescent protein (GFP) were prepared as previously described (Hong et al., 2001; Du et al., 2000; Vincent et al., 2004a; Vincent et al., 2004b) and purified at the University of Iowa Gene Transfer Vector Core. At 24 h, 95–100% of DRG neurons were infected using 1,000 plaque forming units (pfu) per cell, determined by counting GFP positive neurons. DRG neurons were cultured as described above, infected on day 2 with Ad, and experiments performed on day 3. To produce a hyperglycemic insult, 20 mM additional glucose (total 45 mM glucose) was added to the media for the period specified in individual experiments. This model of hyperglycemic insult has been described previously (Russell et al., 1999; Russell et al., 2002; Vincent et al., 2005a). Media glucose concentrations did not significantly decrease during the treatment periods as determined by standard gas chromatography-mass spectrometry (data not shown).
Primary Dissociated DRG Cultures of SOD2+/− DRG from Adult Mice
C57BL/6J SOD2+/− and littermate control animals were purchased from Jackson Laboratories (Bar Harbor, Maine). DRG were collected from C57BL/6J control and C57BL/6J SOD2+/− mice when they were 6–8 weeks of age. Coverslips were coated with 0.01% poly-L-ornithine (0.1mg/ml) (Sigma) overnight, washed, dried and then further coated with rat tail collagen as previously described (Vincent et al., 2004b). Mouse DRG were extracted from adult mice and placed in L-15 media during dissection, dissociated in papain (2 mg/ml) and 2.5% collagenase for 30 min, and then quenched in calf serum. DRG were then extracted by centrifugation, placed in warm plating medium, and triturated 20–30 times with a calf serum-coated glass pipet. Adult mouse DRG neurons were cultured in Neurobasal medium (Sigma) containing 25 mM glucose (optimal basal glucose for neurons) (Vincent et al., 2004b; Russell et al., 1999; Russell and Feldman, 1999), with 1X B27 (Sigma), 0.14 mM L-glutamine, and 40 μM FUDR to remove contaminating Schwann cells. Hyperglycemia was induced as described above for rat primary DRG cultures.
Neurite Outgrowth
DRG neurons were cultured as described above and fixed in 4% paraformaldehyde (PFA, in phosphate buffered saline, 0.1 M, pH 7.4) 12 h following plating. Neurites were assessed by measuring the longest neurite on each of 20 randomly selected neurons per condition using a graduated microscope reticule per our published protocol (Russell et al., 1999).
In Vitro Measures of Oxidative Stress and Cell Death
We previously published the time-course of oxidative stress and markers of DRG neuron injury in our hyperglycemia model (Vincent et al., 2004b; Russell et al., 2002; Vincent et al., 2005a). The time-points used in the present studies are based on the previous experiments. We previously demonstrated that DCFDA oxidation peaked at 3–5 h, caspase-3 activation at 5–6 h, and differences in neurite growth peaked at 12 h. After 3 d in culture, H2O2 generation was assessed by loading DRG neurons with the H2O2 probe CM-H2-DCFDA (Russell et al., 2002; Vincent et al., 2005a). Reduced CM-H2-DCFDA is nonfluorescent. Following oxidation by H2O2, green fluorescent 2’,7’-dichlorofluorescein (DCF) is generated. Stock CM-H2-DCFDA is dissolved in DMSO at 5 mg/ml and diluted in HBSS (10 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2) to 17 μg/ml. The diluted CM-H2-DCFDA was applied to the culture medium at a 1:50 dilution 15 min prior to the end of the 5 h glucose treatment period. The cells were rinsed in HBSS and examined in a fluorescence plate reader. DCF fluorescence was assessed at 485 nm excitement and 520 emission with 1 sec integration (Vincent et al., 2005a;Vincent et al., 2005b). In experiments involving Ad.GFP infected DRG neurons, the GFP signal was assessed prior to glucose exposure and this value was subtracted from the final green fluorescence. Increases in green fluorescence in DRG neurons not loaded with CM-H2-DCFDA were not significant over 5 h. Thus, the increase in green fluorescence from initial to final readings was a measure of CM-H2-DCFDA oxidation.
After 5 h exposure to 20 mM excess glucose, 1 coverslip from each dose was treated with fluorescent CaspaTag reagent per the manufacturer’s protocol (Chemicon, San Francisco, CA) and our previous report (Vincent et al., 2005b). This kit contains a cell-permeable, fluorescence-tagged caspase-3 substrate that binds irreversibly to the active site of caspase-3. After incubation with the substrate for 1 h and fixation in 4% PFA, cells were prepared according to the manufacturer’s instructions. Neurons were counterstained with bisbenzimide to identify the nuclei. DRG neurons with active caspase-3 were identified using fluorescence microscopy; the green fluorescent signal is a direct measure of the amount of active caspase-3 present and must be clearly localized to the neuronal cytosol to score positive. Random fields were counted containing at least 10 neurons; 10 fields were counted per coverslip and 3 coverslips per condition.
Mitochondrial Membrane Potential
Tetramethylrhodamine ester (TMRM, Molecular Probes, Eugene, OR) was used to examine mitochondrial polarization. TMRM (50 nM) was applied for 30 min at 37oC followed by fluorimetry (Fluoroskan Ascent II plate reader, Helsinki, Finland) with 485 nm excitation and 590 nm emission. The results were corrected for DRG protein concentration. This assay has been validated using 10 μM valinomycin (a depolarizing agent) and 10 μM oligomycin (a hyperpolarizing agent) (Russell et al., 2002).
SOD Activity Assay
DRG neurons were scraped off the plate in sterile water, then immediately assayed using a kit (Bioxytech SOD-525, Catalog No. 21010 from Oxis Health Products, Foster City, CA). Lysates were divided in two and one set was extracted with ethanol-chloroform EtOH/CHCl3 (62.5/37.5 v/v) to remove SOD2 activity per manufacturer’s instructions.
Amplex Red Determination of O2•− Generation
Adult DRG neurons cannot be cultured in sufficient numbers or cell density for direct fluorescence probes to be used. Therefore, the generation of O2•− was detected in DRG neuron cultures from adult mice using an Amplex Red-linked assay according to the manufacturer’s information and kit (Molecular Probes, Eugene, OR). In brief, DRG neurons following experimental treatments were scraped from the coverslip in Reaction Buffer (0.25 M sodium phosphate, pH 7.4) and passed through a 26-gauge needle 2–3 times. Samples were prepared in the presence or absence of SOD (Sigma, St. Louis, MO) in a 96-well plate. Standards were prepared using a range of 0–20 μM H2O2 ± SOD. The reaction was initiated by application of a solution of Amplex Red and horse-radish peroxidase as outlined by the manufacturer. Fluorescence was measured using 544 excitation and 590 emission filters immediately and after 30 min. The amount of O2•− in the samples was calculated from the difference between the concentration of H2O2 detected in the presence or absence of SOD.
Transgenic SOD2+/− Mice
Mice [B6.Cg-m+/+Leprdb/J a.k.a. db/db, C57BL/6J, C57BL/6J SOD2+/−] were purchased from Jackson Laboratories (Bar Harbor, Maine). Breeding colonies were established at the University of Michigan to provide the animals used in this study. All animals were genotyped 4 weeks after birth. Animals were cared for following the University of Michigan Committee on the Care and Use of Animals guidelines.
Induction of Diabetes
Following an overnight fast, male C57BL/6J SOD2+/+ and C57BL/6J SOD2+/−mice were injected i.p. with 55 mg/kg of STZ (Sigma Aldrich, St. Louis, MO) dissolved in citrate buffer (pH 5.5) for 5 days. Diabetes was defined as blood glucose over 200 mg/dl. B6.Cg-m+/+Leprdb/J (db/db) mice but not db+ mice become diabetic at approximately 4 weeks of age. Onset of diabetes was confirmed at 8 weeks and both male and female db+ and db/db mice were included. These animals were crossed with C57BL/6J SOD2+/− mice to form the following groups: C57BL/6db+SOD2+/+, C57BL/6db/dbSOD2+/+, C57BL/6db+SOD2+/−, C57BL/6db/dbSOD2+/−.
Monthly blood glucose levels were measured in all animals following a 6 h fast. One drop of tail blood was analyzed using a standard glucometer (One Touch Profile, LIFESCAN, Inc. Milpitas, CA, number 6 strips). Glycated hemoglobin (GHb) was measured using the Helena Laboratories Test Kit, Glyco-Tek Affinity Column Method (Catalog #5351). This test measures any stable form of glycosylated hemoglobin. Interassay variations are 8.8% at 6.0% GHb, 3.8% at 19.5 % GHb. All analyses and procedures described were performed in compliance with protocols established by the Animal Models of Diabetic Complications Consortium (AMDCC), see http://www.amdcc.org. Each group contained 10 animals, unless otherwise indicated.
Tail Flick Analgesia
Mice were placed in an acrylic holder atop a tail flick analgesia meter (Model 336TG Life Sciences, Woodland Hills, CA). The time from activation of the beam to animal response was detected and recorded electronically. This method does not result in sensitization (Lee et al., 1990).
Nerve Conduction Studies (NCS)
Measures of nerve conduction velocity (NCV) were performed per our published protocols (Stevens et al., 2004) and in compliance with protocols established by the AMDCC (http://www.amdcc.org). Mice were anesthetized with 30/0.75 mg/kg ketamine/acepromazine by peritoneal injection. Tail and limb temperatures were maintained at 32–34°C using a heating pad. Hind limb injections were avoided to prevent nerve or muscle injury. The platinum needle electrodes designed for mice were cleaned with 70% alcohol between animals to maintain pathogen-free status. The recording/stimulating electrodes in the tail were placed 30 mm apart. For the sciatic nerve the recording electrodes were placed in the dorsum of the foot and the stimulating electrodes at the knee and sciatic notch. For stimulation, the cathode was distal, the anode was placed along the length of the nerve, 5 mm from the cathode. The frequency band was inclusive of two, 10 Hz for muscle potential recordings and ten, 2 Hz for sensory potential recordings. Tail sensory NCV (TSNCV) was an orthodromic measurement determined by stimulating the tail 30 mm proximal to recording electrodes on the tail. NCV was calculated by dividing the distance (30 mm) by the take-off latency (msec) of the sensory nerve action potential. Sciatic-tibial motor NCV (SMNCV) was determined by recording in the dorsum of the foot and stimulating with supramaximal stimulation first at the knee, then at the sciatic notch. Latencies were measured in each case from the initial onset of the compound muscle action potential. The SMNCV was calculated by dividing the distance between the cathode placements by the difference calculated by subtracting the motor distal latency at the knee from the sciatic notch.
Tissue Harvest
Tissues were harvested 24 weeks post-induction of diabetes. Fresh tissues were dissected following an overdose of sodium pentobarbital and flash frozen in liquid nitrogen. A blood sample (100–300 μl) was collected into K3 EDTA Vacutainer tubes (Becton Dickinson, Franklin Lakes, NJ), and centrifuged at 100 g for 15 min at 4°C. Plasma was collected and stored at −80°C.
Total Radical Antioxidant Potential (TRAP)
TRAP is determined by a luminometric method (Alho et al., 1998; Vincent et al., 2005a). DRG neurons are lysed in ultra-pure water, then sonicated to complete the breakdown of cellular structures. Lysates are centrifuged at 10,000 g for 5 min, then 5 μL of lysate is loaded on a 96-well plate; serial doubling dilutions of a further 5 μL aliquot are also loaded in wells of the 96-well plate. 200 μL of luminol (ECL, Amersham, Arlington Heights, IL) are added, followed by 50 μL of the oxidizing agent 100 mM 2,2’-azobis(amidinopropane) dihydrochloride (ABAP). The plate is immediately placed in a luminometric plate reader (Fluoroskan Ascent II, Labsystems, Helsinki, Finland) and the mean lag to the ABAP-induced oxidation of luminol that is attributed to the antioxidant potential of the cell lysate is measured by plotting luminescence against time (Alho et al., 1998).
Intraepidermal Nerve Fiber Density (IENF)
Foot pads were collected from the plantar surface of the hind paw, postfixed in Zamboni’s (2 % PFA, 1 % picric acid in 0.1 M phosphate buffered saline solution) overnight, rinsed in 5, 10 and 20% sucrose in 50 mM sodium phosphate buffer, cryoembedded, sectioned (35 μm) and processed for PGP 9.5 immunohistochemistry (1:2000 Chemicon, Temecula, CA), a pan-axonal marker (McCarthy et al., 1995). The entire papillae in three non-adjacent sections were imaged per sample using an Olympus FluoView 500 confocal microscope with a 60 X 1.2 water immersion objective at a resolution of 800 X 600 pixels. The optical section thickness was 0.5 μm. Forty images per stack were flattened using MetaMorph (version 6.14) arithmetic option. Integrated morphometry analysis was used to exclude extraneous signals. The data are presented as the percent area of PGP 9.5 positive fibers per area of epidermis across one entire papilla (Christianson et al., 2003).
Statistical Analysis
In cell culture studies, all experimental paradigms were performed in triplicate on three separate occasions with different cell cultures, giving a final n=9 for each data point. In mouse studies, all experimental groups began with 10 mice per group. In vivo testing of nerve function was performed on 10 mice giving n=10. Following euthanasia, 3 mice were perfusion fixed for histological analysis, giving n=3 for IENFD. The remaining 7 mice were prepared fresh, so TRAP assays were n=7. Data analyses were performed using Prism, version 3 (GraphPad Software, Inc.). The Mann Whitney U test was used to compare SOD2+/+ db/db and SOD2+/− db/db or STZ-treated C57BL/6J SOD2+/+ and SOD2+/− diabetic animals to test our hypothesis that SOD2 under expression would exacerbate diabetic complications (Snedecor and Cochran, 1989). The same test was used to compare SOD2+/+ and SOD2+/− control on both genetic backgrounds to determine the effect of SOD2 under expression in nondiabetic otherwise healthy mice.
Assumptions about the Gaussian distribution of data and rules for transformation of non-normative data were made as previously described (Russell et al., 1999; Russell et al., 2002). Comparison of dependent variables was performed using factorial analysis of variance (ANOVA) with 95% confidence intervals. All measurements were made by an observer blinded to the experimental condition. Bar graphs illustrate the mean ± standard error of the mean (SEM).
RESULTS
Adenoviral Transfection of Primary DRG Neurons
To assess the ability of increased SOD2 expression to alter the course of DRG neuron injury in hyperglycemia, DRG neurons were infected with 1,000 pfu/cell on day 2 in culture with Ad.GFP or Ad.SOD2 per our published protocol (Vincent et al., 2004a). After 24 h, greater than 90% of neurons were transfected, as determined by counting GFP co-expression. To confirm that the Ad.SOD2 construct performed in a similar manner as the other Ad. constructs, DRG neurons were infected with either Ad.GFP, Ad.OMC (a mitochondrial protein unrelated to glucose metabolism), Ad.SOD1 or Ad.SOD2 then processed for SOD activity assay (Fig. 1A) or Western blot (Fig. 1B). Total SOD activity (Fig 1A) was increased in DRG neurons infected with Ad.SOD1 or Ad.SOD2. Extraction of the sample with ethanol: chloroform removed SOD2, so the increased SOD activity was lost following extraction in the Ad.SOD2 but not the Ad.SOD1-infected cells. Equal amounts of protein loaded on the Western blotting (Fig. 1B) indicate that DRG neurons infected with Ad.SOD2 display a marked increase in the SOD2 band compared with the Ad.GFP and Ad.SOD1 infected cells. The blot suggests that SOD1 is moderately downregulated in the presence of high levels of SOD2. For comparison, in DRG neurons probed for SOD1, the band in the Ad.SOD1-infected DRG neurons was markedly increased compared to Ad.SOD2 or GFP-infected DRG neurons. These data provide strong evidence that the adenoviral constructs specifically increased expression of the enzymes they code for and did not produce a non-specific increase in antioxidant potential.
Figure 1. SOD2 Protects Sensory Neurons from O2•− Mediated Damage.
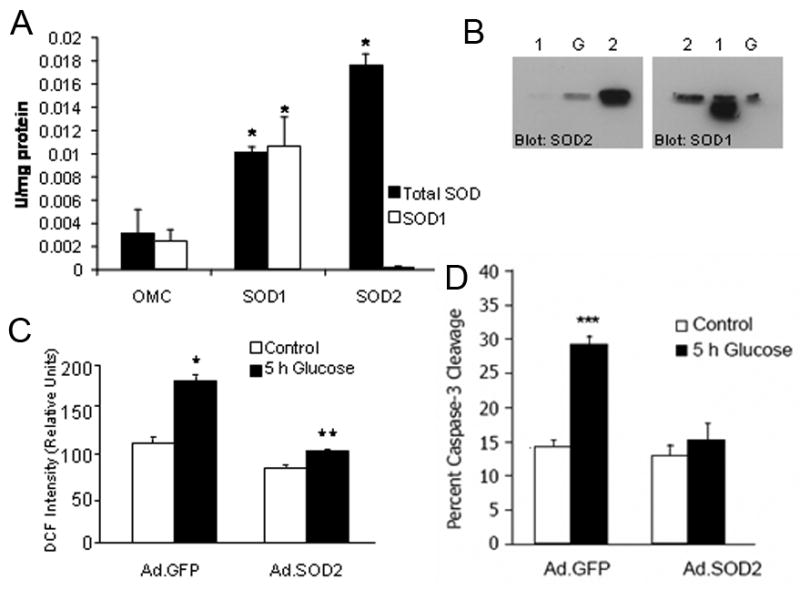
A) Adenoviral infection specifically increases SOD1 or SOD2 in DRG neurons. The graph shows SOD activity in lysates from Ad.OMC (a comparison mitochondrial protein), Ad.SOD1, or Ad.SOD2-infected DRG neurons. Ad.SOD2 specifically upregulated the activity of SOD2 but not SOD1. *p<0.01 compared to Ad.OMC-infected DRG neurons. B) Western blots of lysates of (1) Ad.SOD1-infected, (2) Ad.SOD2-infected, or (G) Ad.GFP-infected DRG neurons that are probed for SOD1 or SOD2. C) Oxidation of non-fluorescent CM-H2DCFDA to green fluorescent DCF indicates an increase in oxidative stress, particularly H2O2 generation following treatment with 20 mM added glucose that is blocked in SOD2 transfected cells. *p<0.01 compared with untreated control; **p<0.01 compared with Ad.GFP/glucose. D) SOD2, but not GFP overexpression, prevents 20 mM glucose-induced caspase-3 activation. ***p < 0.001 compared to untreated control.
In Ad.GFP infected DRG neurons, 5 h of 20 mM added glucose resulted in increased ROS generation (Fig. 1C), p < 0.001. Overexpression of SOD2 using Ad infection decreased basal levels of ROS and completely prevented the increase in ROS after 5 h exposure to 20 mM added glucose (Fig. 1C). Caspase-3 activation was observed in approximately 15% of Ad.GFP or Ad.SOD2 infected DRG neurons, similar to control neurons (Vincent et al., 2004b) (Fig. 1D). Following the addition of 20 mM glucose, activated caspase-3 increased almost 3-fold after 5 h in the Ad.GFP transfected neurons, p < 0.001. Glucose-induced caspase-3 activation was prevented in Ad.SOD2 infected neurons (Fig. 1D). These data suggest that mitochondrial O2•− is a critical mediator of DRG injury and that SOD2 overexpression prevents glucose-mediated damage in DRG neurons.
Primary Dissociated DRG Cultures from SOD2+/− Adult Mice
Since increased SOD2 prevented DRG neuron oxidative stress and injury in hyperglycemia, then decreased SOD2 should promote hyperglycemia-induced DRG neuron injury. To test this hypothesis, DRG neurons were dissociated and cultured from adult C57BL/6J SOD2+/− mice. Dissociated DRG cultures from SOD2+/+ littermates were used as controls. In preliminary experiments, an Amplex Red-linked assay was used to indicate O2•− generation in the DRG neuron cultures. SOD2+/− DRG neurons displayed approximately twice the level of O2•− as SOD2+/+ cultures under basal conditions, p < 0.01 (Fig. 2A). 20 mM added glucose for 5 h induced a parallel increase in levels of O2•−.production in both the SOD2+/+ and SOD2+/− cultures, p < 0.05.
Figure 2. Decreased Expression of SOD2 Increases Glucose-Mediated O2•− and Apoptosis.
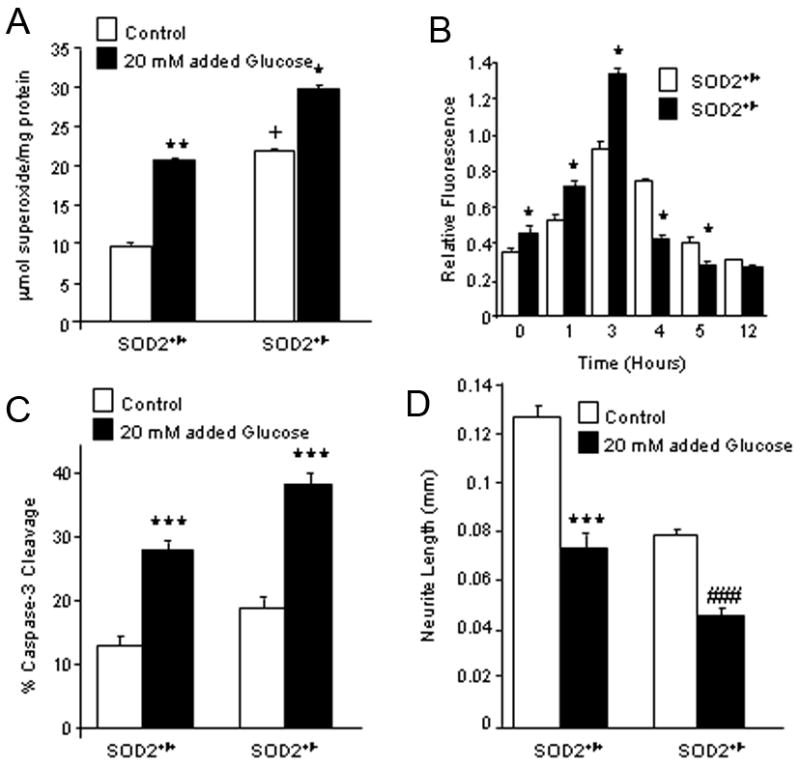
DRG neurons from adult SOD2+/+ and SOD2+/− mice were exposed to 20 mM added glucose. A) O2•− generation was measured using an in vitro Amplex Red oxidation reaction at 5 h (Vincent et al., 2005a). **p < 0.001 compared to SOD2+/+ in basal glucose; +p < 0.01 compared to SOD2+/+ in basal glucose; *p < 0.05 compared to SOD+/− in basal glucose. B) DRG neurons were loaded with TMRM (50 nM), then mitochondrial membrane potential assessed through the increase in red fluorescence. In the presence of 20 mM glucose, hyperpolarization was greater and depolarization occurred earlier in SOD2+/− compared to SOD2+/+ neurons. C) Caspase-3 activation was determined by counting the percent of DRG neurons labeled with a fluorescent caspase-3 substrate (CaspaTag). Glucose-induced caspase-3 activation was increased in both the SOD2+/−and SOD+/+ cultures, compared to basal glucose, ***p < 0.001. D) Neurites were measured in DRG neurons 12 h after plating in control or hyperglycemia (20 mM added glucose) media. Mean neurite length was shorter in 20 mM added glucose than basal glucose, ***p < 0.001 for SOD2+/+ and ###p < 0.05 for SOD2+/−.
Formation of ROS, especially O2•−, cause mitochondrial damage and depolarization. We previously demonstrated rapid mitochondrial hyperpolarization followed by depolarization in wild-type DRG neurons (Russell et al., 1999; Vincent et al., 2004b). TMRM was used to examine the effect of SOD2 under expression on mitochondrial depolarization in response to excess glucose. SOD2+/− DRG neurons display significantly increased hyperpolarization and depolarization compared to SOD2+/+ DRG neurons (Fig. 2B).
Subsequent to ROS formation and mitochondrial depolarization, DRG neurons displayed increased caspase-3 activation (Fig. 2C) and decreased neurite outgrowth (Fig. 2D). SOD2+/− cultures under basal conditions demonstrated nearly 3 times more caspase-3 cleavage than SOD2+/+ cultures. The level of activated caspase-3 was substantially increased in SOD2+/+ cultures following the addition of 20 mM glucose. The glucose-induced increase in the SOD2+/− neurons was less dramatic than SOD2+/+ cultures, but the overall caspase-3 activation in glucose-exposed SOD2+/− neurons was higher than in wild-type or basal glucose conditions. This indicates that SOD2+/− neurons are more susceptible to glucose-induced injury (Fig. 2C).
Neurite outgrowth was used as a measure of neuronal health. Under basal conditions, SOD2+/− neurons extended shorter neurites than control cultures. This effect was exacerbated when cells were cultured for 12 h in the presence of 20 mM added glucose (Fig. 2D), however, as with levels of H2O2 and caspase-3 activation, the difference between basal and 20 mM added glucose was more dramatic in the SOD2+/+ neurons.
SOD2+/+ and SOD2+/− STZ-Treated Mice
The profound differences in oxidative stress detected in the SOD2+/− adult DRG neurons led us to examine DN in the SOD2+/− mouse. These experiments were conducted within the framework of the AMDCC murine neuropathy guidelines. The animals used in the initial mouse experiments were all male C57BL/6J SOD2+/+ or C57BL/6J SOD2+/−. Previous studies revealed that C57BL/6J female mice were resistant to induction of diabetes by STZ injection (Leiter et al., 1987). The mice were genotyped and assigned to one of 4 groups, SOD2+/+ non-diabetic control (C), SOD2+/+ diabetic (D), SOD2+/− C, SOD2+/− D. Monthly 6 h fasting tail blood glucose measurements confirmed the onset and maintenance of diabetes. Diabetes was induced by STZ injections per the AMDCC protocol when the animals were 4 weeks old. Diabetic animals remained hyperglycemic throughout the 6 month time period as evidenced by glycated hemoglobin (GHb) measurements at 24 weeks (Table 1). In addition to elevated blood glucose, the diabetic animals lost weight compared to healthy controls (Table 1). Weight loss was significantly different between the control and diabetic animals but was not affected by SOD2 under expression (Table 1).
Table 1.
Animal Weights and Glycated Hemoglobin
| SOD2+/+ C | SOD2+/+ D | SOD2+/− C | SOD2+/− D | SOD2+/+ db+ | SOD2+/+ db/db | SOD2+/− db+ | SOD2+/− db/db | |
|---|---|---|---|---|---|---|---|---|
| Fasting Blood Glucose, 8 Weeks (mg/dL) | NA | NA | NA | NA | 125 ± 6
(10) |
186 ± 24
(7) |
117 ± 7
(9) |
155 ± 20
(6) |
| Weight, Initial (g) (8 weeks for db+, db/db group) | NA | 27.6 + 0.7
(10) |
NA | 26.5 + 0.6
(10) |
24.0 ± 1.1
(10) |
39.6 ± 1b
(7) |
22.9 ± 1
(9) |
34.8 ± 4.4d
(6) |
| Weight, 24 weeks (g) | 32.7 ± 1.2
(10) |
25.7 ± 1.3a
(8) |
35 ± 1.3
(9) |
27.1 ± 0.9a
(9) |
29.8 ± 1.4
(10) |
65.5 ± 2.4b
(7) |
29.7 ± 1.5
(8) |
60.9 ± 3.1e
(6) |
| Blood Glucose 24 weeks (non-fasting) (mg/dL) | 114.3 ± 3.6
(9) |
366.8 ± 73.4
(8) |
114.2 ± 6.7
(10) |
370.7 ± 36.4b
(8) |
77.2 ± 13.5
(10) |
110.9 ± 30
(7) |
54.6 ± 11.7
(8) |
97.5 ± 17.6
(6) |
| Glycated Hemoglobin 24 weeks (% standard) | 4.9 ± 0.3
(10) |
11.2 ± 0.3c
(8) |
4.7 ± 0.1
(10) |
9.6 ± 0.7b
(8) |
4.0 ± 0.1
(10) |
5.1 ± 0.2e
(7) |
4.1 ± 0.2
(8) |
5.6 ± 0.6
(6) |
Table 1 SOD2+/+ and SOD2+/− STZ treated mice lost weight (aP < 0.003 between SOD2+/+ C and D and SOD2+/− C and D) and had elevated blood glucose (bP< 0.0001 SOD2+/− C and D) and elevated glycosylated hemoglobin (cP< 0.0002, bP<0.0001). NA = Initial weights were taken to determine STZ dose, mice not receiving STZ were not weighed and their initial fasting blood glucoses were assumed to be normal. All of the C57Bl6 mice came from the same vendor at the same time and were the same age and sex. Db+ and db/db mice became hyperglycemic at approximately 8 weeks of age. SOD2 +/+ and +/− db/db were heavier at the beginning of the experiment (bP < 0.0001, dP< 0.036) and continued to gain weight up to 24 weeks (bP < 0.0001, eP< 0.0007). Initial and final blood glucose levels were not significantly different between the SOD2+/+ or +/− db+ or db/db mice; however, glycated hemoglobin was significantly different between the SOD2+/+ db+ and db/db mice.
Neuropathy in C57BL/6J SOD2 Mice Treated With STZ
To assess the presence of DN, tail flick nerve conduction studies were performed at 12 and 24 weeks. Significant differences in tail flick latencies were not observed with diabetes or with decreased SOD2 expression (Fig. 3A). Measures of nerve conduction velocities also demonstrated no statistical difference between diabetic and control, and the SOD2+/− mice were not significantly different from SOD2+/+ mice. There were no differences in sciatic motor nerve conduction velocity (SMNCV) between the experimental groups (Fig. 3B). Overall antioxidant potential (TRAP) was not significantly different in the DRG between any of the 4 groups of animals (Fig. 3C).
Figure 3. Measures of Nerve Function and Antioxidant Potential in SOD2+/+ and SOD2+/− Mice on a C57BL/6 Background.
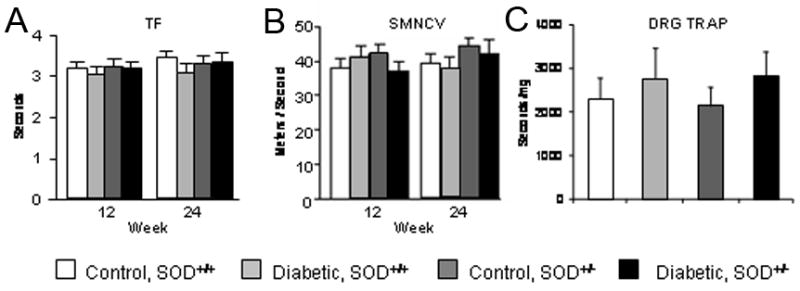
A) Tail flick (TF) latencies are measured at 12 and 24 weeks post induction of diabetes. B) Sciatic Motor Nerve Conduction Velocity (SMNCV) was assessed at 12 and 24 weeks. C) Total Radical Antioxidant Potential (TRAP) was measured in DRG after 24 weeks. No differences were found between the four experimental groups. n=10 for each experimental group.
SOD2 db+ and db/db Transgenic Mouse Model
Because we did not detect DN in the STZ-treated C57BL/6J mouse (regardless of SOD2 expression), our next approach was to examine the effect of reduced SOD2 expression in a spontaneously diabetic mouse, the db/db animal. These mice are obese, insulin resistant and hyperglycemic (Hummel et al., 1966; Sima and Shafrif, 2000) [Jackson Laboratories http://jaxmice.jax.org (stock #000697)]. The groups included SOD2+/+ db+, SOD2+/+ db/db, SOD2+/− db+and SOD2+/− db/db. The glycemic profiles of these mice are presented in Table 1. When fed a high fat diet, the db/db mice maintain elevated blood glucose and die at approximately 16 weeks of age. The mice in the current study were fed a standard diet in order to extend the experimental period to 24 weeks. After 16 weeks, tail blood glucoses in the db/db mice, regardless of SOD2 genotype, began to normalize and by 24 weeks had attained essentially normoglycemic levels. The 24 week GHb data parallel the loss of hyperglycemia (Table 1).
Neuropathy in SOD2+/− db/db Mice
In the SOD2+/+ and SOD2+/− db/db mice, tail flick latencies were significantly increased at both 8 and 12 weeks following the onset of diabetes when compared to the nondiabetic lean control db+ animals (Fig. 4A, p < 0.001). At 24 weeks, SMNCV were lower in the SOD2+/+ db/db than the SOD2+/+ db+ mice but this difference did not reach statistical difference. However the difference between the SOD2+/− db+ and SOD2+/−db/db mice was statistically significant (Fig. 4B, p < 0.05), indicating that diabetes in the presence of decreased SOD2 expression may affect nerve function. There were intriguing differences in DRG TRAP between the experimental groups at 24 weeks (Fig. 4C). The values in SOD2+/+ mice was around 10-fold higher than the SOD2+/− mice regardless of diabetic state. There was a non-significant tendency for TRAP to be lower in SOD2+/+ db/db compared with SOD2+/+ db+ mice. Diabetes produced a significant decrease in TRAP in the SOD2+/− mice compared to non-diabetic litter mates.
Figure 4. Measures of Nerve Function and Antioxidant Potential in SOD2+/+ and SOD2+/− Mice on a db+ or db/db Background.

Nerve function and antioxidant potential was assessed using tail flick latency (TF) at 8 and 12 weeks (A), Sciatic Motor Nerve Conduction Velocity (SMNCV) at 12 and 24 weeks (B), and DRG TRAP at 24 weeks (C). In A, TF latencies are significantly longer in db/db, *p < 0.001. In B, a significant difference in SMNCV is only detected at 24 weeks between the db+ and db/db SOD2+/−mice (*p < 0.001). In C, TRAP was significantly lower in db/db mice compared with db+ (+p<0.05). n=10 for each experimental group.
Intraepidermal Nerve Fiber Density in SOD2+/− Mice
Differences in intraepidermal fiber density (IENFD) were not detected in the C57BL/6J SOD2+/+ or SOD2+/− mice with or without STZ treatment (data not shown). This finding is unsurprising considering the lack of any other signs of neuropathy in these mice.
As seen in Fig. 4, diabetes alone resulted in a sensory deficit in both the SOD2+/+ db/db and SOD2+/− db/db mice. Decreased SMNCV was paralleled by a decrease in PGP 9.5 immunoreactive fibers within the epidermis of the hind paw (Fig. 5). In the control SOD2+/+ db+ animals, fine, varicose, PGP 9.5 positive fibers branch from large bundles in the dermis at almost 90o angles to innervate the epidermis (Fig. 5A). In diabetic mice (Fig. 5B) PGP 9.5 positive nerve bundles were observed at the dermis/epidermis junction, but few fibers extended into the epidermis. The difference in epidermal area occupied by PGP 9.5 positive fibers between SOD2+/+ db+ and SOD2+/+ db/db mice was significant (p < 0.01), indicating the expected loss of nerve fibers in db/db animals with DN (Fig. 5C). SOD2 under expression further increased this loss of IENFD. The SOD2+/− db/db mice had a significantly lower IENFD when compared to the SOD2+/+ db/db animals (p < 0.05).
Figure 5. Measures of IENFD in SOD2+/+ and SOD2+/− Mice on a C57BL/6 db+ or db/db Background.
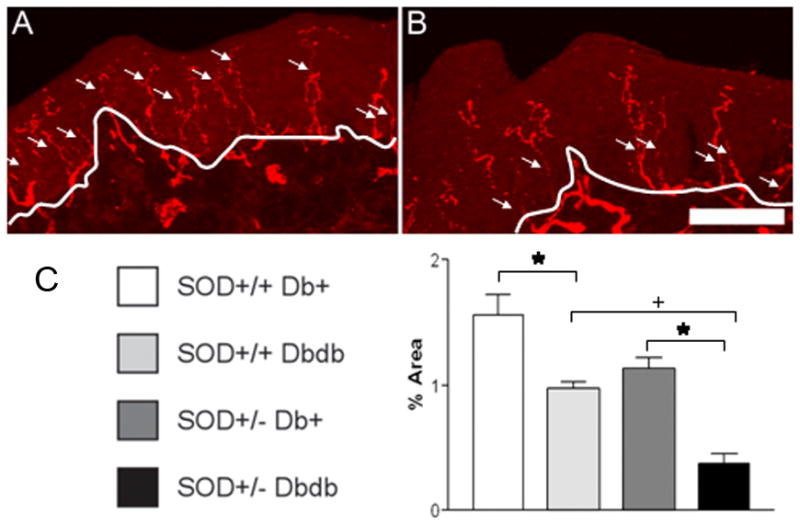
Representative SOD2+/+ db+ (A) and SOD2+/−db/db (B) images are shown to illustrate the flattened confocal mouse footpad sections processed for PGP 9.5 immunofluorescence. The white line in A identifies the division between dermis and epidermis and the arrows indicate IENFD that cross this division. Fewer fibers are evident in the diabetic footpad (B) compared with the non-diabetic control (A). Bar = 50 μm. In C, IENFD expressed as percent of total epidermal area. *p < 0.01 between SOD2+/+ db+ and SOD2+/+ db/db, and between SOD2+/−db+ compared to SOD2+/− db/db. +p < 0.05 between SOD2+/+ db/db compared to SOD2+/− db/db. n=3 for each experimental group.
DISCUSSION
We previously reported a fundamental link between hyperglycemia-induced ROS and DN (Vincent et al., 2005b; Russell et al., 2002; Leinninger et al., 2004;Russell et al., 1999). This link supports the findings of other studies in experimental and clinical diabetes (Cameron et al., 1993; Low et al., 1997; Cameron et al., 1998; Hounsom et al., 2001). The current study explores the hypothesis that accelerating the accumulation of ROS in the peripheral nervous system will in turn accelerate the development of DN. We reasoned that genetically decreasing the ability to detoxify ROS by altering the expression of a specific mitochondrial enzyme involved in oxidative defense, Mn+SOD or SOD2, would provide a logical way to accelerate ROS accumulation. Our data show that 1) over expression of SOD2 in primary DRG cultures protects against the formation of mitochondrial ROS and subsequent cleavage of caspase-3, 2) under expression of SOD2 in vitro increases oxidative stress under basal conditions and exacerbates hyperglycemia-induced oxidative stress, 3) under expression of SOD2 in vivo can increase morphological and physiological manifestations of DN.
We began our experiments by measuring the levels of ROS and cellular injury in primary DRG neuron cultures that overexpressed SOD2. Transfection of DRG neurons with Ad.SOD2 reduced the baseline level of ROS below that of control/untreated DRG with concomitant decrease in the activation of caspase-3. Increased ROS and caspase-3 activation normally induced by 20 mM excess glucose were not only blocked by overexpression of SOD2 but also reduced below control levels. This protective ability of SOD2 in DRG in vitro is consistent with other studies using SOD2 overexpression. SOD2 overexpression eliminates pro-oxidant-induced mitochondrial ROS in PC-12 cells and decreases cell injury (Pias et al., 2003). Similarly, in retinal pigment epithelial cells, the gene dosage of SOD2 directly relates to the degree of mitochondrial injury and apoptosis induced by H2O2 (Kasahara et al., 2005). In hemopoietic cells, SOD2 transgene decreases ionizing radiation-induced mitochondrial membrane permeability, cytochrome c release and caspase-3 activation (Guo et al., 2003). Collectively, these data provide strong evidence that increased mitochondrial ROS in the presence of pro-oxidant stressors, including hyperglycemia, can be prevented by enhancing mitochondrial SOD2 activity.
We noted that SOD1 activity was downregulated in the presence of increased SOD2 expression. This underscores the dynamic regulation of antioxidant enzymes in order to maintain a redox balance. Since ROS can act as second messengers, too much radical scavenging activity could be deleterious to cells under basal conditions (Tang et al., 2005). Overall in the SOD2 overexpressing cells, the total SOD activity was elevated compared to cells not overexpressing SOD2, thus they maintained an increased antioxidant potential that was protective in the face of a hyperglycemic insult.
Because our data demonstrated that increasing SOD2 expression in DRG blocked glucose-mediated injury in vitro, we reasoned that decreasing SOD2 expression would enhance glucose-mediated injury. For these experiments, we compared cultured DRG neurons from adult mice with full SOD2 expression (SOD2+/+) to DRG from mice heterozygous for SOD2 expression (SOD2+/−). Mice homozygous for SOD2 knockout (SOD2−/ −) could not be used as these mice die shortly after birth with severe mitochondrial abnormalities in skeletal and cardiac muscle and brain (Lebovitz et al., 1996; Li et al., 1995; Melov et al., 1999). As anticipated, under basal culture conditions, increased levels of O2− • and cleaved caspase-3 were present in DRG neurons from SOD2+/− mice when compared to neurons from SOD2+/+ mice. Upon treatment with 20 mM added glucose, O2− • and cleaved caspase-3 increased in both the SOD2+/+ and in the SOD2+/−DRG neurons. However, relative to SOD2+/+ DRG neurons, mitochondrial depolarization was significantly elevated and neurite outgrowth was significantly decreased in SOD2+/− DRG neurons. These data support our initial contention that decreasing SOD2 expression in DRG neurons may enhance glucose-mediated injury. Reddy and colleagues performed similar studies in cultured lens epithelium from SOD2+/+ and SOD2+/− mice. Epithelium from SOD2+/− mice are exquisitely more sensitive to oxidative stress than epithelium from SOD2+/+ mice and show profound mitochondrial damage, cytochrome c release, caspase-3 activation and apoptosis (Reddy et al., 2004). In further agreement with our findings, astrocytes cultured from SOD2+/−mice survive under basal conditions, but are exquisitely sensitive to paraquat when compared to astrocyte cultures from SOD2+/+ mice (Liu et al., 2006). Taken together, these findings continue to support the idea that altered SOD2 expression modulates cellular defense against oxidative stress.
We next turned to animal models of altered SOD2 expression. The first mouse study utilized the commercially available SOD2+/− mouse on the C57BL/6J strain. Over the course of 6 months of STZ-induced diabetes, male C57BL/6J SOD2+/+ and SOD2+/−mice did not develop DN. Measures of sensory function and neurophysiology remained normal. The foot pad innervation study in the male C57BL/6J SOD2+/+ and SOD2+/−mice confirmed that there was no hind paw neuropathy. This anatomical assessment of neuropathy is part of the standard neuropathy phenotyping recommended by the AMDCC (www.amdcc.org) and is sensitive and reproducible (Levy et al., 1989; Kennedy et al., 1996; Arezzo, 1999). The TRAP values suggest that these mice did not develop significant oxidative stress in the DRG, so we may conclude that loss of one copy of SOD2 was not sufficient to increase oxidative stress in vivo, despite our observations in vitro. It is possible that the mice adapted to the loss of one copy of the SOD2 gene by upregulating other antioxidant defense systems, although in non-stressed control SOD2+/−mice no changes were found in antioxidant enzymes in a survey of several mouse organs (Van Remmen et al., 1999). While these results were at first unexpected, these same animals had no evidence of diabetic nephropathy or retinopathy [data available on www.amdcc.org, and reviewed in (Breyer et al., 2005)].
Since mouse strain or the method of diabetes induction may be the parameters that determine the development of DN, we next pursued our studies in a genetic model of type 2 diabetes, the db/db mouse. In contrast to the STZ C57BL/6J animals, in the first 12 weeks of diabetes, SOD2+/+ db/db and SOD2+/− db/db mice displayed behavioral evidence of DN, with prolonged tail flick times of greater than 10 sec, highly statistically significant from nondiabetic SOD2+/+ db+ and SOD2+/− db+ mice. In addition, sciatic motor nerve conduction velocities declined in the SOD2+/+ db/db compared with non-diabetic litter mates and there was a robust further decline in the SOD2+/− db/db animals. The SOD2+/+ mice displayed 10-fold higher TRAP in the DRG than the SOD2+/− mice regardless of the diabetic state. This was a surprising result suggesting that these mice may be highly dependent upon the regulation of SOD2 activity to resist obesity-induced oxidative stress. In SOD2+/− mice, db/db animals displayed a significant decrease in DRG TRAP compared with db+ mice, suggesting that the antioxidant capacity is overwhelmed in diabetes in these mice and may contribute to the development of DN. Our TRAP data are in agreement with other studies that demonstrate a decrease in TRAP in diabetes that correlates with complications (Vincent et al., 2004c). The SOD2+/− db/db mice had fewer PGP 9.5 positive fibers i.e. a lower IENFD than the SOD2+/+ db/db mice (p < 0.05). The anticipated differences between epidermal nerve fiber densities in diabetic and control animals (regardless of SOD2 expression) were also in accordance with published reports in human patients (Yasuda et al., 1985; Levy et al., 1989; Kennedy et al., 1996; Arezzo, 1999) and experimental models of DN (Christianson et al., 2003).
The fact that SOD2+/− mice on the db/db genetic background have a more severe DN phenotype than SOD2+/+ on this same background supports our contention that oxidative stress is important in the pathogenesis of DN. Our results are broadly supported by over a decade of research in SOD2+/− and SOD2−/ − mice. While the homozygous SOD2−/ − mice die shortly after birth (Lebovitz et al., 1996; Li et al., 1995; Melov et al., 1999), the heterozygous SOD2+/− mice develop normally, but have increased susceptibility to toxic injury and pro-oxidant stress. For example, while the mitochondrial toxin 3-nitropropionic acid increases striatal excitotoxicity and oxidative stress in mouse models of Huntington’s disease, these effects are amplified in Huntington SOD2+/− transgenic mice (Kim and Chan, 2002). When compared to SOD2+/+ mice, SOD2+/− mice display greater hepatotoxicity after nimesulide treatment (Ong et al., 2006), poorer survival following ischemic stroke (Chan, P. H. 05) and increased susceptibility to toxin induced seizures (Liang and Patel, 2004). Increased cellular injury in these mouse models is prevented by SOD mimetics, confirming the direct role of SOD2 in enhancing disease phenotypes (Patel, 2003; Ali et al., 2004). Such evidence has led to heightened interest in the clinical application of antioxidants for neurological disorders, including DN (Paolisso et al., 1994; Natarajan et al., 2002; Henriksen, 2006; Coverley and Baxter, 1997). One caveat is that the SOD2+/− mice also display evidence of cardiovascular insufficiency (Ohashi et al., 2006; Strassburger et al., 2005). Reduced nerve blood flow also may contribute to DN through mechanisms that only partially involve oxidative stress (Ishii et al., 1998).
In summary, over production of SOD2 is protective in an in vitro model of DN while under production of SOD2 renders the peripheral nervous system more susceptible to hyperglycemia-induced oxidative stress. In the C57BL/6J mouse, diabetes alone via low dose STZ injection produces the anticipated hyperglycemia, but no parallel increases in DN. Superimposing enhanced oxidative stress with the SOD2+/− genotype did not make these animals more prone to DN, perhaps because of some antioxidant compensation since TRAP was unchanged. In contrast, in vivo reduction of SOD2 gene expression (SOD2+/−) did contribute to the development of neuropathy in the db/db mouse. These mice had markedly lower TRAP values in the DRG. By comparing the two groups of animals, we can conclude that mouse strain is an important determinant of the development of neuropathy. One notable difference between the two groups in the current study, though, is that the db/db mice were hyperlipidemic (data not shown, but compare body weights and also see www.amdcc.org). The contribution of plasma free fatty acids to neuronal or nerve injury may be an important additional area that requires investigation. We conclude that on a susceptible genetic background, additional oxidative stress through decreasing the intrinsic mitochondrial antioxidant enzyme SOD2 can promote the development of DN. Thus: (1) therapies aimed at decreasing nervous system oxidative stress have therapeutic potential in DN; (2) we can probe the mechanisms of DN in rodent models of the disease by genetically modifying their ability to resist oxidative stress.
Acknowledgments
This work was supported by the National Institutes of Health (NS36778, NS38849, NS42056, DK60994, DK20572), the Juvenile Diabetes Research Foundation (JDRF) Center for the Study of Complications in Diabetes, the Office of Research Development (Medical Research Service), the Department of Veterans Affairs, and the Program for Neurology Research and Discovery.
The authors wish to acknowledge Ms. Julie Erwin for expert manuscript preparation, Dr. Steven Lentz of the Michigan Diabetes Research and Training Center for his assistance with confocal imaging, and Ms. Alice Mentzer, Ms. Erin Fredrickson and Dr. Chao Gong for experimental assistance. Immunohistochemical studies were conducted within the Morphometry Core of the JDRF Center for the Study of Complications in Diabetes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alho H, Leinonen JS, Erhola M, Lonnrot K, Aejmelaeus R. Assay of Antioxidant Capacity of Human Plasma and CSF in Aging and Disease. Restor Neurol Neurosci. 1998;12:159–165. [PubMed] [Google Scholar]
- Ali SS, Hardt JI, Quick KL, Kim-Han JS, Erlanger BF, Huang TT, Epstein CJ, Dugan LL. A biologically effective fullerene (C60) derivative with superoxide dismutase mimetic properties. Free Radic Biol Med. 2004;37:1191–1202. doi: 10.1016/j.freeradbiomed.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Arezzo JC. New developments in the diagnosis of diabetic neuropathy. Am J Med. 1999;107:9S–16S. doi: 10.1016/s0002-9343(99)00008-x. [DOI] [PubMed] [Google Scholar]
- Bolanos JP, Cidad P, Garcia-Nogales P, gado-Esteban M, Fernandez E, Almeida A. Regulation of glucose metabolism by nitrosative stress in neural cells. Mol Aspects Med. 2004;25:61–73. doi: 10.1016/j.mam.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Breyer MD, Bottinger E, Brosius FC, III, Coffman TM, Harris RC, Heilig CW, Sharma K. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- Brussee V, Cunningham FA, Zochodne DW. Direct insulin signaling of neurons reverses diabetic neuropathy. Diabetes. 2004;53:1824–1830. doi: 10.2337/diabetes.53.7.1824. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA, Horrobin DH, Tritschler HJ. Effects of alphalipoic acid on neurovascular function in diabetic rats: interaction with essential fatty acids. Diabetologia. 1998;41:390–399. doi: 10.1007/s001250050921. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA, Maxfield EK. Anti-oxidant treatment prevents the development of peripheral nerve dysfunction in streptozotocin-diabetic rats. Diabetologia. 1993;36:299–304. doi: 10.1007/BF00400231. [DOI] [PubMed] [Google Scholar]
- Cellek S. Point of NO return for nitrergic nerves in diabetes: a new insight into diabetic complications. Curr Pharm Des. 2004;10:3683–3695. doi: 10.2174/1381612043382792. [DOI] [PubMed] [Google Scholar]
- Chan PH. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann NY Acad Sci. 2005;1042:203–209. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- Christianson JA, Riekhof JT, Wright DE. Restorative effects of neurotrophin treatment on diabetes-induced cutaneous axon loss in mice. Exp Neurol. 2003;179:188–199. doi: 10.1016/s0014-4886(02)00017-1. [DOI] [PubMed] [Google Scholar]
- Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Salvemini D, Yorek MA. Effect of M40403 treatment of diabetic rats on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. Br J Pharmacol. 2001;134:21–29. doi: 10.1038/sj.bjp.0704216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coverley JA, Baxter RC. Phosphorylation of insulin-like growth factor binding proteins. Mol Cell Endocrinol. 1997;128(1–2):1–5. doi: 10.1016/s0303-7207(97)04032-x. [DOI] [PubMed] [Google Scholar]
- Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci USA. 2000;97:12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Seixas-Silva JA, Jr, Epperly MW, Gretton JE, Shin DM, Bar-Sagi D, Archer H, Greenberger JS. Prevention of radiation-induced oral cavity mucositis by plasmid/liposome delivery of the human manganese superoxide dismutase (SOD2) transgene. Radiat Res. 2003;159:361–370. doi: 10.1667/0033-7587(2003)159[0361:porioc]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Henriksen EJ. Exercise training and the antioxidant alpha-lipoic acid in the treatment of insulin resistance and type 2 diabetes. Free Radic Biol Med. 2006;40:3–12. doi: 10.1016/j.freeradbiomed.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Hong Y, Fink BD, Dillon JS, Sivitz WI. Effects of adenoviral overexpression of uncoupling protein-2 and -3 on mitochondrial respiration in insulinoma cells. Endocrinology. 2001;142:249–256. doi: 10.1210/endo.142.1.7889. [DOI] [PubMed] [Google Scholar]
- Hounsom L, Corder R, Patel J, Tomlinson DR. Oxidative stress participates in the breakdown of neuronal phenotype in experimental diabetic neuropathy. Diabetologia. 2001;44:424–428. doi: 10.1007/s001250051638. [DOI] [PubMed] [Google Scholar]
- Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153:1127–1128. doi: 10.1126/science.153.3740.1127. [DOI] [PubMed] [Google Scholar]
- Ishii H, Koya D, King GL. Protein kinase C activation and its role in the development of vascular complication in diabetes mellitus. J Mol Med. 1998;76:21–31. doi: 10.1007/s001090050187. [DOI] [PubMed] [Google Scholar]
- Jeffcoate WJ. Abnormalities of vasomotor regulation in the pathogenesis of the acute charcot foot of diabetes mellitus. Int J Low Extrem Wounds. 2005;4:133–137. doi: 10.1177/1534734605280447. [DOI] [PubMed] [Google Scholar]
- Kasahara E, Lin LR, Ho YS, Reddy VN. SOD2 protects against oxidation-induced apoptosis in mouse retinal pigment epithelium: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2005;46:3426–3434. doi: 10.1167/iovs.05-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy WR, Wendelschafer-Crabb G, Johnson T. Quantitation of epidermal nerves in diabetic neuropathy. Neurology. 1996;47:1042–1048. doi: 10.1212/wnl.47.4.1042. [DOI] [PubMed] [Google Scholar]
- Kim GW, Chan PH. Involvement of superoxide in excitotoxicity and DNA fragmentation in striatal vulnerability in mice after treatment with the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab. 2002;22:798–809. doi: 10.1097/00004647-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Layton BE, Sastry AM, Wang H, Sullivan KA, Feldman EL, Komorowski TE, Philbert MA. Differences between collagen morphologies, properties and distribution in diabetic and normal BioBreeding and Sprague-Dawley rat sciatic nerves. Journal of Biomechanics. 2004;37:879–888. doi: 10.1016/j.jbiomech.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Lebovitz RM, Zhang H, Vogel H, Cartwright J, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Cox DJ, Mook DG, McCarty RC. Effect of hyperglycemia on pain threshold in alloxan-diabetic rats. Pain. 1990;40:105–107. doi: 10.1016/0304-3959(90)91057-P. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Backus C, Sastry AM, Yi Y-B, Wang C-W, Feldman EL. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiol Dis. 2006;23:11–22. doi: 10.1016/j.nbd.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Russell JW, van Golen CM, Berent A, Feldman EL. Insulin-like growth factor-I (IGF-I) regulates glucose-induced mitochondrial depolarization and apoptosis in human neuroblastoma. Cell Death Differ. 2004;11:885–896. doi: 10.1038/sj.cdd.4401429. [DOI] [PubMed] [Google Scholar]
- Leiter EH, Le PH, Coleman DL. Susceptibility to db gene and streptozotocin-induced diabetes in C57BL mice: control by gender-associated, MHC-unlinked traits. Immunogenetics. 1987;26:6–13. doi: 10.1007/BF00345448. [DOI] [PubMed] [Google Scholar]
- Levy DM, Karanth SS, Springall DR, Polak JM. Depletion of cutaneous nerves and neuropeptides in diabetes mellitus: an immunocytochemical study. Diabetologia. 1989;32:427–433. doi: 10.1007/BF00271262. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2(−/+) mice. Free Radic Biol Med. 2004;36:542–554. doi: 10.1016/j.freeradbiomed.2003.11.029. [DOI] [PubMed] [Google Scholar]
- Liu J, Narasimhan P, Song YS, Nishi T, Yu F, Lee YS, Chan PH. Epo protects SOD2-deficient mouse astrocytes from damage by oxidative stress. Glia. 2006;53:360–365. doi: 10.1002/glia.20289. [DOI] [PubMed] [Google Scholar]
- Low PA, Nickander KK, Tritschler HJ. The roles of oxidative stress and antioxidant treatment in experimental diabetic neuropathy. Diabetes. 1997;46(Suppl 2):S38–S42. doi: 10.2337/diab.46.2.s38. [DOI] [PubMed] [Google Scholar]
- Matteucci E, Malvaldi G, Fagnani F, Evangelista I, Giampietro O. Redox status and immune function in type I diabetes families. Clin Exp Immunol. 2004;136:549–554. doi: 10.1111/j.1365-2249.2004.02470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy BG, Hsieh ST, Stocks A, Hauer P, Macko C, Cornblath DR, Griffin JW, McArthur JC. Cutaneous innervation in sensory neuropathies: evaluation by skin biopsy. Neurology. 1995;45:1848–1855. doi: 10.1212/wnl.45.10.1848. [DOI] [PubMed] [Google Scholar]
- Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan SK, Lakshmi S, Punitham R, Arokiasamy T, Sukumar B, Ramakrishnan S. Effect of oral supplementation of free amino acids in type 2 diabetic patients-- a pilot clinical trial. Med Sci Monit. 2002;8:CR131–CR137. [PubMed] [Google Scholar]
- Obrosova IG, Julius UA. Role for poly(ADP-ribose) polymerase activation in diabetic nephropathy, neuropathy and retinopathy. Curr Vasc Pharmacol. 2005;3:267–283. doi: 10.2174/1570161054368634. [DOI] [PubMed] [Google Scholar]
- Ohashi M, Runge MS, Faraci FM, Heistad DD. MnSOD deficiency increases endothelial dysfunction in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2331–2336. doi: 10.1161/01.ATV.0000238347.77590.c9. [DOI] [PubMed] [Google Scholar]
- Ong MM, Wang AS, Leow KY, Khoo YM, Boelsterli UA. Nimesulide-induced hepatic mitochondrial injury in heterozygous Sod2(+/−) mice. Free Radic Biol Med. 2006;40:420–429. doi: 10.1016/j.freeradbiomed.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Paolisso G, D'Amore A, Balbi V, Volpe C, Galzerano D, Giugliano D, Sgambato S, Varricchio M, D'Onofrio F. Plasma vitamin C affects glucose homeostasis in healthy subjects and in non-insulin-dependent diabetics. Am J Physiol. 1994;266:E261–E268. doi: 10.1152/ajpendo.1994.266.2.E261. [DOI] [PubMed] [Google Scholar]
- Patel MN. Metalloporphyrins improve the survival of Sod2-deficient neurons. Aging Cell. 2003;2:219–222. doi: 10.1046/j.1474-9728.2003.00055.x. [DOI] [PubMed] [Google Scholar]
- Pias EK, Ekshyyan OY, Rhoads CA, Fuseler J, Harrison L, Aw TY. Differential effects of superoxide dismutase isoform expression on hydroperoxide-induced apoptosis in PC-12 cells. J Biol Chem. 2003;278:13294–13301. doi: 10.1074/jbc.M208670200. [DOI] [PubMed] [Google Scholar]
- Reddy VN, Kasahara E, Hiraoka M, Lin LR, Ho YS. Effects of variation in superoxide dismutases (SOD) on oxidative stress and apoptosis in lens epithelium. Exp Eye Res. 2004;79:859–868. doi: 10.1016/j.exer.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Rosenson RS. Statins in atherosclerosis: lipid-lowering agents with antioxidant capabilities. Atherosclerosis. 2004;173:1–12. doi: 10.1016/S0021-9150(03)00239-9. [DOI] [PubMed] [Google Scholar]
- Russell JW, Feldman EL. Insulin-like growth factor-I prevents apoptosis in sympathetic neurons exposed to high glucose. Horm Metab Res. 1999;31:90–96. doi: 10.1055/s-2007-978704. [DOI] [PubMed] [Google Scholar]
- Russell JW, Golovoy D, Vincent AM, Mahendru P, Olzmann JA, Mentzer A, Feldman EL. High glucose induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002;16:1738–1748. doi: 10.1096/fj.01-1027com. [DOI] [PubMed] [Google Scholar]
- Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis. 1999;6:347–363. doi: 10.1006/nbdi.1999.0254. [DOI] [PubMed] [Google Scholar]
- Schnackenberg CG, Wilcox CS. The SOD mimetic tempol restores vasodilation in afferent arterioles of experimental diabetes. Kidney Int. 2001;59:1859–1864. doi: 10.1046/j.1523-1755.2001.0590051859.x. [DOI] [PubMed] [Google Scholar]
- Siemionow M, Demir Y. Diabetic neuropathy: pathogenesis and treatment. J Reconstr Microsurg. 2004;20:241–252. doi: 10.1055/s-2004-823112. [DOI] [PubMed] [Google Scholar]
- Sima AAF, Shafrif E. Animal Models in Diabetes: A Primer Taylor and Francis. Amsterdam: 2000. [Google Scholar]
- Snedecor GW, Cochran WG. Statistical methods. Iowa State University Press; Ames, Iowa: 1989. [Google Scholar]
- Srinivasan S, Hatley ME, Bolick DT, Palmer LA, Edelstein D, Brownlee M, Hedrick CC. Hyperglycaemia-induced superoxide production decreases eNOS expression via AP-1 activation in aortic endothelial cells. Diabetologia. 2004;47:1727–1734. doi: 10.1007/s00125-004-1525-1. [DOI] [PubMed] [Google Scholar]
- Stevens MJ, Dananberg J, Feldman EL, Lattimer SA, Kamijo M, Thomas TP, Shindo H, Sima AAF, Greene DA. The linked roles of nitric oxide, aldose reductase and (Na+,K+)-ATPase in the slowing of nerve conduction in the streptozotocin diabetic rat. J Clin Invest. 1994;94:853–859. doi: 10.1172/JCI117406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens MJ, Lattimer SA, Feldman EL, Helton ED, Millington DS, Sima AAF, Greene DA. Acetyl-L-carnitine deficiency as a cause of altered nerve myo-Inositol content, Na, K-ATPase activity and motor conduction velocity in the streptozotocin-diabetic rat. Metabolism. 1996;45:865–872. doi: 10.1016/s0026-0495(96)90161-4. [DOI] [PubMed] [Google Scholar]
- Strassburger M, Bloch W, Sulyok S, Schuller J, Keist AF, Schmidt A, Wenk J, Peters T, Wlaschek M, Lenart J, Krieg T, Hafner M, Kumin A, Werner S, Muller W, Scharffetter-Kochanek K. Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic Biol Med. 2005;38:1458–1470. doi: 10.1016/j.freeradbiomed.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Tang XQ, Feng JQ, Chen J, Chen PX, Zhi JL, Cui Y, Guo RX, Yu HM. Protection of oxidative preconditioning against apoptosis induced by H2O2 in PC12 cells: mechanisms via MMP, ROS, and Bcl-2. Brain Res. 2005;1057:57–64. doi: 10.1016/j.brainres.2005.07.072. [DOI] [PubMed] [Google Scholar]
- Toth C, Brussee V, Cheng C, Zochodne DW. Diabetes mellitus and the sensory neuron. J Neuropathol Exp Neurol. 2004;63:561–573. doi: 10.1093/jnen/63.6.561. [DOI] [PubMed] [Google Scholar]
- Van Remmen H, Salvador C, Yang H, Huang TT, Epstein CJ, Richardson A. Characterization of the antioxidant status of the heterozygous manganese superoxide dismutase knockout mouse. Arch Biochem Biophys. 1999;363:91–97. doi: 10.1006/abbi.1998.1060. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Feldman EL. New insights into the mechanisms of diabetic neuropathy. Rev Endo Metabol Dis. 2004;5:227–236. doi: 10.1023/B:REMD.0000032411.11422.e0. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Feldman EL, Song DK, Jung V, Schild A, Zhang W, Imperiale MJ, Boulis NM. Adeno-associated viral-mediated insulin-like growth factor delivery protects motor neurons in vitro. Neuromolecular Medicine. 2004a;6:79–86. doi: 10.1385/NMM:6:2-3:079. [DOI] [PubMed] [Google Scholar]
- Vincent AM, McLean LL, Backus C, Feldman EL. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. FASEB J. 2005a;19:638–640. doi: 10.1096/fj.04-2513fje. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Olzmann JA, Brownlee M, Sivitz WI, Russell JW. Uncoupling proteins prevent glucose-induced neuronal oxidative stress and programmed cell death. Diabetes. 2004b;53:726–734. doi: 10.2337/diabetes.53.3.726. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr Rev. 2004c;25:612–628. doi: 10.1210/er.2003-0019. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Stevens MJ, Backus C, McLean LL, Feldman EL. Cell culture modeling to test therapies against hyperglycemia-mediated oxidative stress and injury. Antioxidant and Redox Signaling. 2005b;7:1494–1506. doi: 10.1089/ars.2005.7.1494. [DOI] [PubMed] [Google Scholar]
- Wada R, Yagihashi S. Role of advanced glycation end products and their receptors in development of diabetic neuropathy. Ann NY Acad Sci. 2005;1043:598–604. doi: 10.1196/annals.1338.067. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA. Methods in Molecular Biology. 2002;197:3–54. doi: 10.1385/1-59259-284-8:003. [DOI] [PubMed] [Google Scholar]
- Whiteley SJ, Tomlinson DR. Motor nerve conduction velocity and nerve polyols in mice with short-term genetic or streptozotocin-induced diabetes. Exp Neurol. 1985;89:314–321. doi: 10.1016/0014-4886(85)90092-5. [DOI] [PubMed] [Google Scholar]
- Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem. 1998;273:28510–28515. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Kikkawa R, Hatanaka I, Kobayashi N, Taniguchi Y, Shigeta Y. Skin biopsy as a beneficial procedure for morphological evaluation of diabetic neuropathy. Acta Pathol Jpn. 1985;35:1–8. doi: 10.1111/j.1440-1827.1985.tb02201.x. [DOI] [PubMed] [Google Scholar]