


Abstract
The repair of DNA by nucleotide excision repair (NER) and non-homologous end joining (NHEJ) is essential for maintenance of genomic integrity and cell viability. Examination of NHEJ and NER in vitro using cell-free extracts has led to a deeper understanding of the biochemical mechanisms that underlie these processes. Current methods for production of whole-cell extracts (WCEs) to investigate NER and NHEJ start with one or more liters of culture containing 1–5 × 109 cells. Here, we describe a small-scale method for production of WCE that can be used to study NER. We also describe a rapid, small-scale method for the preparation of WCE that can be used in the study of NHEJ. These methods require less time, 20- to 1000-fold fewer cells than large-scale extracts, facilitate examination of numerous samples and are ideal for such applications as the study of host–virus interactions and analysis of mutant cell lines.
INTRODUCTION
The chemistry of DNA makes it highly resistant, but not impervious, to damage. Environmental and intracellular agents can cause lesions that can lead to misinformation or break the flow of genetic information. In addition, DNA replication and somatic cell recombination introduce double-strand breaks (DSBs) in DNA that challenge genomic continuity. To maintain the proper flow of correct genetic information DNA repair mechanisms have evolved to identify and repair base lesions and strand breaks.
The nucleotide excision repair (NER) pathway repairs bulky DNA adducts or intrastrand cross-links caused by exposure to UV light or alkylating agents. This process encompasses two sub-pathways that survey the entire genome (global genome repair, GGR) and the transcriptionally active part of the genome (transcription coupled repair, TCR) for base lesions that block the elongating RNA polymerase (1,2).
The importance of DSB repair is highlighted by the observation that failure to repair even a single DSB can result in the loss of genetic information, chromosomal translocation and even cell death (3). Non-homologous end joining (NHEJ) is an important pathway employed by mammalian cells in the repair of adventitious DSBs and also for the repair of programmed DSBs made during somatic cell recombination (2–4). Defects in NHEJ can result in gross chromosomal aberrations such as translocations and studies in mouse models have shown that such defects can lead to events that initiate or propagate tumorigenesis (3,4).
Investigation of the molecular mechanisms that underlie NER and NHEJ was stimulated by the development of cell-free extracts that supported these processes in vitro (5–8). Unfortunately, the requirement of 1–5 × 109 cells for these methods limits the utility of standard protocols. Here, we report the development of small-scale, cell-free extract protocols to study NER and NHEJ in vitro.
MATERIALS AND METHODS
Cell culture
HeLa cells (Biovest International Inc., National Cell Culture Center) were grown in suspension to 106 cells/ml in Joklik's MEM, 5% newborn calf serum, 100 U/ml penicillin and 100 µg/ml streptomycin (PenStrep). 293 cells were cultured as monolayers in EMEM, 10% fetal bovine serum (FBS) and PenStrep in 25 cm2 flasks. CHO AA8 (wild type), CHO UV5 (XPD) (gifts from Dr Michael Seidman) and CHO UV41 (XPF) (ATCC) cells were grown in minimal essential medium alpha modification, 10% FBS and PenStrep in 175 cm2 flasks. Adherent CHO AA8 cells were grown to large scale using CellSTACK culture chambers (Corning); a 10-layer chamber yields ∼109 cells.
Viral infections
293 monolayers were grown in 25 cm2 flasks; the medium was removed and was replaced with 1 ml fresh media with virus (MOI = 10 p.f.u./cell). Virus was allowed to adsorb (2 h, 37°C, with gentle agitation) after which the inoculum was aspirated away and replaced with fresh medium. Infected cells were cultured for times indicated.
Large-scale extracts for NER
Large-scale preparations of NER-competent whole-cell extracts (WCE) were carried out essentially as previously described (8–10). Briefly, cells were harvested and rinsed with phosphate-buffered saline (PBS). Subsequent steps were carried out at 4°C. Cells were resuspended in four packed cell volumes (PCV) of hypotonic lysis buffer (HLB: 10 mM Tris–HCl pH 8.0, 1 mM EDTA) with 5 mM DTT and incubated on ice for 20 min. Protease inhibitors were added to 1 mM PMSF, 10 µg/ml aprotinin, 5 µg/ml leupeptin and 100 µg/ml soybean trypsin inhibitor and cells were homogenized [20 strokes, Potter–Elvehjem 10 ml glass tube with a Teflon pestle (Kontes)]. The homogenate was transferred to a beaker in an ice bath. Using a magnetic stirrer, 4 PCV of sucrose-glycerol buffer [50 mM Tris–HCl pH 8, 10 mM MgCl2, 2 mM DTT, 25% sucrose (w/v), 50% glycerol (v/v)] and 1 PCV of saturated (NH4)2SO4 (pH 7.0) were added and the solution was mixed slowly for 30 min. Ultracentrifugation (SW 41, 3 h, 125 000g) was used to remove solid material. A total of 0.33 g per ml finely ground solid (NH4)2SO4 was added to the supernatant with 10 µl of 1 M NaOH/g of (NH4)2SO4 to maintain neutral pH and the extract was allowed to stir for 30 min. Precipitated proteins were collected (centrifugation, 45 min, 12 000g, 4°C) and the pellet was resuspended in a minimal volume of NER-dialysis buffer (25 mM HEPES pH 7.9, 100 mM KCl, 12 mM MgCl2, 0.5 mM EDTA, 2 mM DTT, 12% glycerol), dialyzed overnight against NER-dialysis buffer and insoluble material was removed [microcentrifugation, 10 min, 11 337g (13 000 r.p.m.), 4°C]. The resulting WCE was aliquoted, frozen on liquid nitrogen and stored at −80°C. Protein concentration: 15–20 mg/ml by BCA (Pierce Chemical Co.) or Bradford (Bio-Rad) protein assay systems.
Large-scale extracts for NHEJ
HeLa WCE was prepared essentially as previously described (5). Briefly, 5 × 109 cells were harvested, washed twice with PBS, resuspended in two PCV of HLB with 1 mM DTT and held on ice for 20 min. Cells were opened by dounce homogenization in the presence of protease inhibitors (1 mM PMSF, 2.2 ng/ml aprotinin, 1 ng/ml leupeptin, 1 ng/ml pepstatin A and 1 ng/ml chymostatin). High salt buffer (HSB: 83.5 mM Tris pH 7.5, 1.65 M KCl, 3.3 mM EDTA, 1 mM DTT) was added to adjust the final salt concentration to 0.33 M KCl and the sample was held on ice for 20 min. The sample was subject to ultracentrifugation (170 000g, 3 h, 4° C, Beckman SW41 Ti rotor), the supernatant was collected and dialyzed against 20 mM Tris pH 8.0, 0.1 M KOAc, 20% glycerol, 0.5 mM EDTA and 1 mM DTT for 2 h. The resulting WCE was snap frozen on liquid nitrogen, and stored at −80°C. Protein concentration: 15–20 mg/ml by Bradford assay.
Mini-NER–WCE (mNER–WCE)
Extracts were prepared from 5 × 107 CHO AA8, XPD or XPG cells (175 cm2 culture area) using a modified Manley procedure (8). Cells were harvested at 90–95% confluency by addition of trypsin to cover the plate surface and removal of the excess trypsin. The cells were resuspended in 40 ml of medium and pelleted (1000g, 10 min). The pellet was washed with 1 ml cold PBS, transferred to a silanized Eppendorf tube, pelleted [microcentrifugation, 200g (1600 r.p.m.), 5 min, Baxter Heraeus Biofuge 13] and lyzed by freezing on liquid nitrogen and thawing rapidly at 37°C. Cell pellets can also be stored at −80°C until extract preparation is convenient. Subsequent steps were carried out at 4°C and DTT was added fresh to each buffer on the day of use. The cell pellet (100 µl) was re-suspended with 10–20 strokes of a P-1000 pipette in 4 PCV (400 µl) of HLB with 5 mM DTT, 2 µg/ml aprotinin, 10 µg/ml leupeptin, 1 mM PMSF and 100 µg/ml soybean trypsin inhibitor and lysis was measured by trypan blue dye exclusion. Four PCV of sucrose–glycerol buffer were added and mixed slowly using a wide-bore pipette tip. One PCV (100 µl) of saturated (NH4)2SO4, pH 7.0 was added slowly and mixed by rotation (30 min, 4°C). Insoluble material was removed [microcentrifugation, 20 min, 11 500g (13 000 r.p.m.), 4°C] and the supernatant (∼800 µl) was transferred to a fresh silanized Eppendorf tube. Solid (NH4)2SO4 (finely ground) was added at 0.33 g of ammonium sulfate per milliliter of solution (0.264 g) with 1 µl of 0.1 M NaOH per 10 mg of (NH4)2SO4 to maintain pH 7.0 and the solution was mixed by rotation (20 min, 4°C). Precipitated proteins were collected [microcentrifugation, 20 min, 11 500g (13 000 r.p.m.), 4°C] and resuspended in 10 µl of NER dialysis buffer and dialyzed against 100 ml of NER dialysis buffer (6 h to overnight, 4°C) using Slide-A-Lyzer mini dialysis units 10 000 MWCO (Pierce, presoaked 20 min in deionized water to remove glycerol) to yield 80–100 µl of mNER-WCE per 100 µl PCV. Extracts may be concentrated by placing the dialysis unit on a bed of dry CM-Sephadex (Pharmacia) until the volume is ∼2/3–1/2 (∼15–30 min). We consistently observed increased excision activity using this concentration method, however the protein concentration does not increase as much as would be expected for a given volume reduction, presumably due to loss of protein on the dialysis membrane. mNER-WCE was aliquoted and frozen on liquid nitrogen and stored at −80°C. Protein concentration: 13 –14 mg/ml by Bradford assay.
One hundred and twenty-minute mini-NHEJ–WCE (mNHEJ–WCE)
Adherent cells were harvested at 70–80% confluency using a cell scraper, collected (centrifugation, 5 min, 800g, 4°C), washed twice in PBS, frozen on dry ice and stored at −80°C. Frozen cell pellets were resuspended in 2 PCV of HLB, incubated on ice for 20 min and lyzed by vortexing for 30 s. Nuclei were collected (microcentrifugation, 800g, 2 min, RT) and the supernatant was retained. Nuclei were resuspended in 1 PCV of nuclear extract buffer (25 mM Tris pH 8.0, 0.33 M KCl, 1.5 mM EDTA) and incubated on ice for 20 min. The retained supernatant was added back to the nuclei and cell debris was removed (microcentrifugation, 10 min, 16 500g, 4°C). The resulting supernatant was collected as mNHEJ–WCE and used without dialysis. Protein concentration: 5–10 mg/ml by Bradford assay. MNHEJ–WCE was aliquoted and frozen on liquid nitrogen and stored at −80°C up to 3 months without significant loss of activity.
Nucleotide excision assay
Repair substrate: oligonucleotides were synthesized using standard phosphoramidite chemistry on an Applied Biosystems Model 3400 DNA/RNA synthesizer, purified by SAX HPLC and desalted on a C-18 SEP PAK cartridge (Waters, Inc.). To prepare the platinated oligonucleotide: 26.5 nmol of oligonucleotide was incubated with 54 nmol cis-diamminedichloroplatinum (II) in 1.5 ml of 10 mM Tris pH 8.0, 1 mM EDTA (37°C, 16 h) then desalted on a C-18 SEP PAK cartridge, purified by SAX HPLC (linear gradient of 0.0–0.5 M NaCl in 50 mM Tris, pH 7.8 with 20% acetonitrile) and confirmed by MALDI-TOF mass spectrometry (m/z calculated.: 4078.64, observed: 4079.36). The platinated oligonucleotide was 32P labeled with T4-polynucleotide kinase (T4 PNK, New England Biolabs) and annealed with its complementary strand to create platinated duplex X (Table 1). The labeled substrate used to monitor NER was prepared by phosphorylation and ligation of component duplexes A−F + X (Table 1). Ligation products were gel purified (6% denaturing PAGE) and full-length individual strands were isolated then re-annealed by heating (90°C, 5 min), slow cooling to <30°C and stored (4°C). Nucleotide excision reactions were carried out essentially as previously described (10). Briefly, reactions (25 µl) were assembled in silanized tubes in 25 mM HEPES, pH 7.9, 4.4 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 70 mM KCl, 4 mM ATP, 200 µg/ml bovine serum albumin (BSA), 2.4–4.8% glycerol, 45 ng of pGEM3Z DNA and 5 µl of extract (90–100 µg of large-scale extract or 65–70 µg of mNER–WCE), incubated on ice for 10 min, started by addition of 10 fmol of 32P-labeled substrate and incubated at 30°C for 1 h. Reactions were stopped by addition of 10 µg of proteinase K in 0.3% SDS and deproteinized for 15 min at 37°C. Reactions were extracted once with phenol/chloroform/isoamyl alcohol (25:24:1 v/v) and once with chloroform/isoamyl alcohol (24:1). DNA was ethanol precipitated and the reaction products were separated by electrophoresis (10% sequencing gel). The gel was fixed (5% acetic acid, 5% methanol, 90% water) for 5 min and dried. Excision products were quantified using Image Quant 5.2 software. Quantification of excision products was done on samples in which saturation of the signal had not been reached.
Table 1.
Oligonucleotide duplexes used in this study
| Duplex | Sequence |
|---|---|
| A | 5′-TGCATCCTAACGTTAGGTCTC |
| 3′-CGTAGGATTGCAATCCAGAGGACTG | |
| B | 5′-CTGACTCAGGTGAAGCTTCAAGCCT |
| 3′-AGTCCACTTCGAAGTTCGGAGAAG | |
| C | 5′-CTTCCTAATCTGCCATGAAAA |
| X | 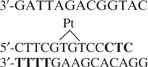 |
| D | 5′-ACGACCTGCAGTCTATA |
| 3′-GAGTGCTGGACGTCAGATATGTCCT | |
| E | 5′-CAGGATGTCAAGTCTAGACACTTC |
| 3′-ACAGTTCAGATCTGTGAAGTGACGG | |
| F | 5′-ACTGCCAAGTCTGAGTACTTGACTC |
| 3′-TTCAGACTCATGAACTGAGCC |
Ligation of these duplexes mediated by complementary overhangs (bold) resulted in ordering of the fragments as A-B-C-X-D-E-F.
NHEJ assay
Assays for NHEJ were carried out as described in Refs (5,11). Briefly, reactions (10 µl) were carried out in 50 mM HEPES pH 8.0, 40 mM KOAc, 0.5 mM Mg(OAc)2, 1 mM ATP, 1 mM DTT, 0.1 mg/ml BSA and HindIII-linearized 5′-32P-labeled pBluescript KS+ DNA (10 ng) with 20–40 µg of WCE or NHEJ–mWCE. Reactions were incubated for 2 h at 37°C, stopped by the addition of deproteinization solution (2 µl, 10 mg/ml Proteinase K, 50 mM EDTA, 0.1 M Tris pH 7.5, 2.5% SDS) and deproteinized for 30 min at 37°C. Products were separated by agarose gel electrophoresis (0.6%, TAE) and visualized by autoradiography. Rabbit anti-human-XRCC4 antiserum (Serotec, AHP387) was used to neutralize NHEJ in vitro at the specified dilutions. Wortmannin (SIGMA) was used at the specified concentrations.
RESULTS
Mini-WCEs for the study of NER in vitro: mini-NER–WCE
We have developed a mini-extract protocol that can be used in the study of NER, which reduces the number of cells required for analysis by 20-fold. As shown in Figure 1, when a DNA substrate that carries a site-specific 1,3 platinum lesion is treated with large-scale extracts prepared in the traditional manner from 109 cells we observe the expected cleavage events 5′ and 3′ of the platinum lesion to produce 25–30 nt long excision products. When the same substrate is treated with mini-NER–WCE (mNER–WCE) prepared from 5 × 107 cells we observed the same cleavage near the platinum modification, which indicates that the mNER–WCE contains all of the factors necessary for recognition and excision of a platinum lesion.
Figure 1.

Mini-NER Whole-Cell extracts (mNER–WCE) are active for excision repair. Top: Schematic diagram of the substrate containing a 32P label near a single 1,3 d-(GTG) platinum lesion. Filled circle indicates 32P label. Pt denoted location of platinum lesion. Bottom: The substrate was incubated with wild-type (AA8, lanes 2–4), NER mutant extracts (XPD and XPG, lanes 5 and 6), or complemented XPD and XPG extracts (D+G, lane 7), analyzed on a 10% sequencing gel and quantified as described. L, large-scale extract; M, mNER–WCE; [M], mNER–WCE concentrated as described; (−) indicates no mWCE.
Because the mNER–WCE is less concentrated than large-scale extracts the observed excision activity was lower than that observed with the large-scale extracts. Figure 1 shows that concentration of the mNER–WCE increased the excision activity and made it comparable to the large-scale extract.
To test the dependence of the mNER–WCE-catalyzed excision events on known excision repair factors, we prepared mNER–WCE from cells deficient in XPD or XPG. Mini-NER–WCE prepared from XPD or XPG deficient cells is unable to excise the platinum lesion. Combining the XPD and XPG mini-extracts restores excision activity. These data show that extracts prepared using our small-scale mNER–WCE method are catalytically active for NER and that this method is applicable to a variety of cell lines.
Mini-WCE for the study of NHEJ in vitro: 120-min mini-NHEJ–WCE
Establishment of an in vitro DNA ligase IV-dependent NHEJ assay enabled examination of the molecular mechanism of the ligation step of NHEJ. As shown in Figure 2A, NHEJ–WCE prepared using the conventional large-scale method catalyze end joining, which is sensitive to the PI3-kinase inhibitors LY294002 and wortmannin. End joining catalyzed by the large-scale NHEJ–WCE is also sensitive to treatment with neutralizing anti-XRCC4 antibodies (lane 3) but not to treatment with control anti-GST antibodies (lane 4), which demonstrates a specific requirement for the XRCC4 protein. The data presented in Figure 2 are consistent with the original description of this method (5).
Figure 2.

Mini-NHEJ extracts (mNHEJ–WCE) are active for NHEJ in vitro. (A) Large-scale whole cell extracts (Lg scale NHEJ–WCE, 40 µg) prepared from HeLa cells was assayed in vitro for end joining in the presence or absence of neutralizing anti-XRCC4 antibodies (lane 3 αX 1:300), control anti-GST antibodies (lane 4 αG 1:50), LY294002 (lanes 6 and 7, 2 and 10 µM) or wortmannin (Wort. lanes 9 and 10, 0.3 and 1 µM). (B) Mini-NHEJ–WCE (mNHEJ–WCE, 40 µg) prepared from 293 cells was assayed in vitro for end joining in the presence or absence of neutralizing anti-XRCC4 antibodies (αX lanes 3 and 4, 1:1500 and 1:300), LY294002 (lanes 6 and 7, 2 and 10 µM) or wortmannin (Wort. lanes 9 and 10, 0.3 and 1 µM).
To facilitate our investigation of the molecular mechanism of NHEJ we developed a mini-extract protocol that reduces the number of cells required for analysis from 5 × 109 (5) to 5 × 106 cells. Mini–NHEJ–WCEs were prepared from 293 cells and end joining was assayed for DNA end joining. As shown in Figure 2B, treatment with either anti-XRCC4 antibodies, LY294002 or wortmannin resulted in a significant decrease in ligation efficiency (Figure 2B), which indicated a requirement for both XRCC4 and DNA-PKcs and identified the DNA end joining catalyzed by mNHEJ-WCE as bona fide mammalian NHEJ. Treatment with control anti-GST antibodies had no effect and similar results were obtained with mini-extracts prepared from HeLa cells (data not shown).
Inhibition of NHEJ by human adenovirus (Ad5)
Studies have shown that infection with human Ad5 results in inhibition of NHEJ in vivo (12–16). The utility of the mini-NHEJ–WCE protocol is highlighted in Figure 3 where we carried out small-scale infections and observed the inhibition of NHEJ in vitro in extracts prepared from Ad5-infected cells. We also used the mNHEJ–WCE protocol to facilitate mutational analysis to assess the role of early viral proteins in inhibition of NHEJ.
Figure 3.

Utility of mini-NHEJ WCE: time course and mutational analysis of NHEJ inhibition during human adenovirus (Ad5) infection. (A) Time course. 293 cells were cultured in T25 flasks, Ad5- or Mock-infected and grown for the indicated time post-infection. At each time point, cells from a single T25 flask were harvested and stored at −80°C. At the end of the time course, mWCEs were prepared and 40 µg of mWCE was assayed for in vitro NHEJ activity. (B) Ad5 E4-deletion mutants used in this study. White spaces denote deletions. Only complete ORFs are expressed. All other adenoviral genes are intact. (C) Mutational analysis. NHEJ catalyzed by mWCEs prepared from 293 cells infected with E4-deletion mutants. MNHEJ–WCE (40 g) prepared from Mock- or Ad5-infected 293 cells or from 293 cells infected with the indicated E4-deletion mutants was assayed for in vitro NHEJ activity. (−) indicates no mWCE.
Ad5 infections of 293 cells were carried out in 5 ml flasks and mNHEJ–WCE was prepared to observe inhibition of NHEJ over 48 h. As shown in Figure 3A, we observed no change in the NHEJ catalyzed by mNHEJ–WCE prepared from mock-infected cells isolated over 48 h, while in Ad5-infected cells, NHEJ activity declined within 24 h post-infection.
The adenoviral E1b 55k–E4 34k protein complex and the E4 11k protein inhibit viral genome concatenation mediated by NHEJ in vivo and the E1b 55k–E4 34k protein complex additionally inhibits NHEJ on other substrates (12–16). The mechanism of NHEJ inhibition by the E1b 55k–E4 34k complex has been found to be degradation of DNA ligase IV (16) and complementation of extracts prepared from Ad5-infected cells partially restores NHEJ in vitro (data not shown). In contrast, the mechanism of E4 11k inhibition of NHEJ in vivo remains an open question. To assess the potential of the adenovirus early protein E4 11k to inhibit the ligation step of NHEJ we used the mini-NHEJ–WCE protocol to examine the effects of infection with various early region 4 (E4)-deletion mutants of Ad5 (Figure 3B). As shown in Figure 3C, infection with E4-deletion mutant dl1004, which lacks most of the E4 region, demonstrates that inhibition of NHEJ requires expression of E4 proteins. E4-deletion mutants dl1014 and dl1015 do not express E4 34k and provide a clear look at effect of E4 11k on NHEJ. Comparison of NHEJ catalyzed by mWCE prepared from 293 cells infected with either E4 11k-deficient (dl1014) or E4 11k-expressing (dl1015) virus (Figure 3C) indicated that expression of E4 11k had only a slight effect, if any, on DNA ligase IV-dependent joining in vitro. These data show that while E4 11k is capable of inhibiting NHEJ on the viral genome in vivo, the factors required for DNA ligase IV-dependent end joining are not impaired.
DISCUSSION
Here we report the development of a small-scale cell extract protocol that can be used to prepare extracts for the study of NER in vitro. This method uses 20-fold fewer cells than previously published methods (8–10). We show that mNER–WCEs produce plantinated lesion-directed cleavages that are characteristic of the NER pathway and require participation of known NER factors XPD and XPG. Importantly, this method can be used for complementation analysis of NER-deficient cell lines or to screen the participation of NER factors in the repair of different DNA lesions.
We also describe a rapid, small-scale method for the production of NHEJ-competent WCEs. This method reduces the number of cells used to prepare extracts for NHEJ analysis from 5 × 109 cells used in the large-scale method of Baumann and West (5) to as few as 5 × 106 cells. While this is not the first report of a small-scale NHEJ assay, the method described by Diggle et al. (17) requires 0.5 − 1 × 108 cells and requires ultracentrifugation and dialysis. The method described here requires 10- to 20-fold fewer cells than the Diggle method, takes ∼120 min and does not require ultracentrifugation or dialysis.
The usefulness of both of these methods comes primarily from a reduction in the number of cells required to produce the mini-extracts. Elimination of ultracentrifugation steps reduces the time required for extract preparation and relaxes the need for specialized equipment. In the 120-min mNHEJ-WCE protocol, elimination of the dialysis step saves time and reduces sample manipulation. All of these improvements make the mini-extract protocols more amenable to multiple sample preparation and examination of numerous variables. As we show in Figure 3, the mNHEJ–WCE protocol is particularly useful in the examination of host–virus interactions where large-scale infections would be cumbersome.
ACKNOWLEDGEMENTS
We thank Joyce Cheung, Timra Gilson, Brenda Salerno, Sumithra Jayaram, B.T. Rantipole and Bill Russ for many thoughtful discussions. We thank Drs Joyce Reardon and Aziz Sancar for helpful advice on preparing NER extracts; Ms Maggie Wear for assistance in oligonucleotide synthesis; and Dr Michael Seidman for generously providing CHO AA8 and CHO UV5 cell lines. UV41 cells were obtained from American type culture collection under grant number CA082785. HeLa cells were obtained from the National Cell Culture Center (Minneapolis, MN, USA), supported under grant number U42 RR05991 from the National Center for Research Resources, NIH. This work was supported by the National Institutes of Health (NIH) grants GM070639-1 (to L.A.H.), 5R01CA082127 (to G.K.) and CA082785 (to P.S.M.), and by the Johns Hopkins University Bloomberg School of Public Health Faculty Research Initiatives Fund (to L.A.H. and G.K.). Funding to pay the Open Access publication charges for this article was provided by National Institutes of Health (NIH) grants GM070639-1.
Conflict of interest statement. None declared.
REFERENCES
- 1.Yang W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair. 2006;5:654–666. doi: 10.1016/j.dnarep.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 3.Ferguson DO, Alt FW. DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene. 2001;20:5572–5579. doi: 10.1038/sj.onc.1204767. [DOI] [PubMed] [Google Scholar]
- 4.Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair. 2006;5:1042–1048. doi: 10.1016/j.dnarep.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 5.Baumann P, West SC. DNA end-joining catalyzed by human cell-free extracts. Proc. Natl Acad. Sci. USA. 1998;95:14066–14070. doi: 10.1073/pnas.95.24.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wood RD. Repair of pyrimidine dimer ultraviolet light photoproducts by human cell extracts. Biochemistry. 1989;28:8287–8292. doi: 10.1021/bi00447a005. [DOI] [PubMed] [Google Scholar]
- 7.Wood RD, Robins P, Lindahl T. Complementation of the xeroderma pigmentosum DNA repair defect in cell-free extracts. Cell. 1988;53:97–106. doi: 10.1016/0092-8674(88)90491-6. [DOI] [PubMed] [Google Scholar]
- 8.Manley JL, Fire A, Cano A, Sharp PA, Gefter ML. DNA-dependent transcription of adenovirus genes in a soluble whole-cell extract. Proc. Natl Acad. Sci. USA. 1980;77:3855–3859. doi: 10.1073/pnas.77.7.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wood R, Biggerstaff M, Shivji MKK. Detection and measurement of nucleotide excision repair synthesis by mammalian cell extracts in vitro. Campanion Methods Enzymol. 1995;7:163–175. [Google Scholar]
- 10.Reardon JT, Sancar A. Purification and characterization of Escherichia coli and human nucleotide excision repair enzyme systems. Methods Enzymol. 2006;408:189–213. doi: 10.1016/S0076-6879(06)08012-8. [DOI] [PubMed] [Google Scholar]
- 11.Hanakahi LA, Bartlet-Jones M, Chappell C, Pappin D, West SC. Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair. Cell. 2000;102:721–729. doi: 10.1016/s0092-8674(00)00061-1. [DOI] [PubMed] [Google Scholar]
- 12.Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;22:6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 14.Boyer J, Rohleder K, Ketner G. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology. 1999;263:307–312. doi: 10.1006/viro.1999.9866. [DOI] [PubMed] [Google Scholar]
- 15.Nicolas AL, Munz PL, Falck-Pedersen E, Young CS. Creation and repair of specific DNA double-strand breaks in vivo following infection with adenovirus vectors expressing Saccharomyces cerevisiae HO endonuclease. Virology. 2000;266:211–224. doi: 10.1006/viro.1999.0062. [DOI] [PubMed] [Google Scholar]
- 16.Baker A, Rohleder KJ, Hanakahi LA, Ketner G. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 2007;81:7034–7040. doi: 10.1128/JVI.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diggle CP, Bentley J, Knowles MA, Kiltie AE. Inhibition of double-strand break non-homologous end-joining by cisplatin adducts in human cell extracts. Nucleic Acids Res. 2005;33:2531–2539. doi: 10.1093/nar/gki528. [DOI] [PMC free article] [PubMed] [Google Scholar]