


Abstract
The annotation of the Escherichia coli K-12 genome in the EcoCyc database is one of the most accurate, complete and multidimensional genome annotations. Of the 4460 E. coli genes, EcoCyc assigns biochemical functions to 76%, and 66% of all genes had their functions determined experimentally. EcoCyc assigns E. coli genes to Gene Ontology and to MultiFun. Seventy-five percent of gene products contain reviews authored by the EcoCyc project that summarize the experimental literature about the gene product. EcoCyc information was derived from 15 000 publications. The database contains extensive descriptions of E. coli cellular networks, describing its metabolic, transport and transcriptional regulatory processes. A comparison to genome annotations for other model organisms shows that the E. coli genome contains the most experimentally determined gene functions in both relative and absolute terms: 2941 (66%) for E. coli, 2319 (37%) for Saccharomyces cerevisiae, 1816 (5%) for Arabidopsis thaliana, 1456 (4%) for Mus musculus and 614 (4%) for Drosophila melanogaster. Database queries to EcoCyc survey the global properties of E. coli cellular networks and illuminate the extent of information gaps for E. coli, such as dead-end metabolites. EcoCyc provides a genome browser with novel properties, and a novel interactive display of transcriptional regulatory networks.
INTRODUCTION
This article presents the state of the annotation of the Escherichia coli K-12 genome as defined by the EcoCyc (1–3) database (DB). We use database queries to EcoCyc to find the gaps in our knowledge of E. coli, thus defining opportunities for future research, such as dead-end metabolites within the E. coli metabolic network. Without computable descriptions of the genome and cellular networks of E. coli, it would be difficult if not impossible to perform the analyses described here.
Reed et al. (4) developed the notion that genome annotations have the potential to be multidimensional. They define a one-dimensional genome annotation as consisting of a list of cellular components; a two-dimensional annotation adds descriptions of cellular networks. A three-dimensional annotation describes spatial orientations of the network components, and a four-dimensional annotation describes genome changes that occur during evolution (Note that EcoCyc does not provide the third or fourth dimensions defined by Reed et al.). Adding to this concept, several additional aspects of genome annotations should be considered. To what degree does the genome annotation incorporate experimental versus computational information, and does it clearly differentiate them? What fraction of the annotations has been validated experimentally? Does the genome annotation include comprehensive written reviews of the gene products and cellular interactions? Does the annotation include citations that support written statements and other information in the annotation? Does the annotation include assertions made with MultiFun, Gene Ontology and other standard controlled vocabularies?
We put forth EcoCyc as a model for the annotation of other genomes. Its first dimension of genome annotation includes the highest fraction of experimentally validated annotations of any free-living organism, and experimental annotations are identified as such. EcoCyc provides textual summaries for every gene for which published literature has been identified by our curation team, along with extensive literature citations. Controlled vocabularies are used extensively within EcoCyc, and the database schema behind EcoCyc provides an exceptional level of computability. EcoCyc describes the metabolic, transport and transcriptional regulatory networks of E. coli.
We argue that all genome annotations for important experimental organisms should be performed in the context of a larger model-organism DB project that couples ongoing computational reannotation of the genome with a literature-based curation effort for the genome, and for the cellular networks of the organism, so as to integrate the best computational and experimental information available within a single resource. We further argue that another key to producing a rich and comprehensive genome annotation is the underlying software and database schema: without a suitable vessel to hold it (i.e. a database schema that can accurately capture each dimension of the annotation), a genome annotation can attain only limited dimensionality. The Pathway Tools software (5) that is used to construct EcoCyc is being used by groups outside SRI to create EcoCyc-like DBs for more than 100 other organisms. Furthermore, SRI has created 370 EcoCyc-like DBs within our BioCyc DB collection (see BioCyc.org). We make these DBs available for adoption (see http://biocyc.org/intro.shtml#adopt) by scientists who will refine them through ongoing curation.
MATERIALS AND METHODS
The results in this article pertain to version 11.0 of the EcoCyc DB released in March 2007. Analyses were performed using DB queries written in the Common Lisp language.
Dead-end metabolites were identified by a Common Lisp program that considers a compound C to be a dead end if and only if the following conditions are true. (i) C or a parent compound class of C is a substrate in only one reaction of the set of small-molecule reactions occurring in EcoCyc. (ii) C is not transported into E. coli by transporters defined in EcoCyc, nor are parent classes of C. (iii) C is a small molecule. A parent class of C is a database object describing a class of chemical compounds. For example, the function of a protein that transports all known amino acids is modeled in EcoCyc by a transport reaction among whose substrates is the class Amino Acids. That class is a parent of every individual amino acid in EcoCyc. This approach allows EcoCyc to compactly describe enzymes and transporters of broad substrate specificity. An alternative way to construct condition (i) would be for C to be a substrate of reactions that only produce C, or that only consume C. However, because the reaction direction information in EcoCyc is incomplete, this approach does not yield acceptable results. The definition that we used, based on condition (i), will be incomplete in the sense that it will fail to detect those true dead-end metabolites that are only produced, or only consumed, by multiple reactions. Nonetheless, the definition that we used produced useful results.
RESULTS
Genes and gene products
EcoCyc contains 4465 gene objects, of which 4460 are assigned locations within the E. coli MG1655 complete genome sequence. Five genes are not mapped to the sequence because they are known only on the basis of phenotypes reported in the literature, not their location in the genome. The set of genes with assigned chromosomal locations is very close to being the same as the recently updated gene set defined by the E. coli genome annotation consortium (6), although EcoCyc has introduced several new genes that have since been reported in the literature. The nucleotide sequence, and the nucleotide start and stop positions (i.e. their coding region) for each gene shared with (6) are the same as those in (6) for the vast majority of genes, although in a few cases we have adjusted gene start and stop positions based on published experimental information.
Each gene is associated in EcoCyc with at least one gene product. In the case of gene products that are chemically modified, such as by phosphorylation, the DB contains a distinct object for each different chemical state of the protein. EcoCyc also contains a distinct object for each isoform coded for by a gene (for example, the two proteins generated from dnaX are separate DB objects). In contrast, post-translational processing of a protein, such as removal of signal sequences or starting methionines, is represented as features on the protein sequence using the EcoCyc protein feature ontology (see http://biocyc.org/ECOLI/NEW-IMAGE?object=Protein-Features). Each gene product within EcoCyc is of a given type, which is defined by its parent DB class. Table 1 shows the distribution of E. coli gene products among these types. EcoCyc does not include objects for mRNA species, although it does include objects for other RNAs, as reflected in Table 1.
Table 1.
Statistics on EcoCyc gene products
| Proteins | 4316 |
| tRNAs | 89 |
| rRNAs | 22 |
| Misc RNAs | 64 |
Because of programmed frame shifting and other causes discussed in the text, there are more total gene products than genes.
EcoCyc contains queryable descriptions of the multimeric structure of E. coli gene products as described in the experimental literature. A total of 774 multimeric protein complexes are defined in EcoCyc, ranging in complexity from simple homodimers, to the NADH dehydrogenase I complex of 13 gene products, to the ribosome. Because these complexes were derived from the experimental literature, they could be a valuable training set for programs that predict protein–protein interactions. When chemically modified forms of a protein exist, they are represented within EcoCyc as separate DB objects to allow their activities to be accurately encoded.
EcoCyc collects extensive sets of synonyms for E. coli genes and gene products. The DB contains 16 799 gene names and synonyms (an average of 3.8 names per gene), and it contains 21 330 names and synonyms for gene products (an average of 4.8 names per gene product).
Gene product summaries
EcoCyc contains an extensive collection of mini-reviews that describe the functions of individual gene products, and of the multimeric complexes formed by E. coli gene products. These summaries were written by the EcoCyc curators, and are found in the display pages for gene products (proteins and RNAs). They are not found in gene pages, although they are only one click away from gene pages (click on the name of the gene product). Summaries are written after reviewing the literature for a gene, protein or protein complex, and cover information such as gene product function, interactions with other gene products, mutant phenotypes, multimeric structure, crystal structure and regulation. Summaries are of particular importance in the genomics era, when researchers frequently work in areas outside their area of specialization, such as during the annotation of newly sequenced microbial genomes.
In the summer of 2006, EcoCyc reached an important milestone: as of that time, EcoCyc curators had performed literature searches for every known E. coli K-12 gene and had authored summaries for every gene for which experimental literature was found. A total of 3332 (75%) E. coli genes are ‘covered by a summary’, meaning that a summary is found in the gene-product DB object, or in the DB object for a multimeric complex in which the gene product is a subunit. The remaining 1133 EcoCyc genes contain a brief, standardized summary stating that no information about the gene has been found by the EcoCyc curators in the biomedical literature, and the date on which a literature search was last performed.
We recently introduced a DB field that records the date on which an EcoCyc gene was last curated, indicating when a curator last performed a systematic literature search for information about that gene. See the field ‘Last Curated’ within the gene product pages for that date of last curation.
Summaries cite the literature from which their information was derived. Because the number of articles per E. coli gene varies greatly, from zero to hundreds, summary lengths also vary, from one line of text to thousands of words. Table 2 presents the length in words of the summaries found in EcoCyc gene product and protein-complex objects.
Table 2.
Number of EcoCyc gene products for which the written gene product summary is of a given length in words
| Words in gene-product summaries | Number of gene products |
|---|---|
| 0–20 | 306 |
| 21–50 | 671 |
| 51–100 | 694 |
| 101–200 | 776 |
| 201–2000 | 812 |
The information in EcoCyc has been derived from the 14 950 publications that EcoCyc cites. This number is a small fraction of the 239 344 publications returned by a PubMed search on ‘Escherichia coli’, because whereas EcoCyc is principally concerned with the molecular biology and biochemistry of E. coli K-12, the latter set of articles covers medical aspects of E. coli, other E. coli strains and uses of E. coli as a cloning vector.
Over time, more references will be added as EcoCyc curators review and update older EcoCyc entries, and as we curate additional types of regulation information into EcoCyc. It would be particularly helpful to our efforts in adding older omitted references to EcoCyc if E. coli scientists would review EcoCyc gene product pages for genes within their expertise and alert us to missing references and to incomplete or incorrect information using the button ‘Report Errors or Provide Feedback’ that is found at the bottom of most EcoCyc pages.
Gene functions
How close is the scientific community to knowing the function of every E. coli gene? Biochemical functions have been assigned to 3384 (76%) of them; the remaining 1077 (24%) E. coli genes have no functional assignment, or only a partial assignment. By ‘partial functions’, we mean approximate descriptions of function, usually resulting from sequence similarity, such as ‘oxidoreductase’ or ‘ABC transporter of unknown substrate’.
Although some of the 3384 E. coli genes with known functions had their functions assigned by sequence analysis of the E. coli genome, most genes in EcoCyc had their functions elucidated experimentally, and reported in the literature. EcoCyc makes use of a controlled set of evidence codes (7) that allows the DB to capture what type of evidence supports the functions of E. coli genes, and to capture what types of evidence support the presence of operons and pathways. Of the EcoCyc genes with assigned functions, 2941 had their functions elucidated experimentally (87% of genes with assigned functions, and 66% of all E. coli genes). This value of 66% with experimentally defined functions is extremely high given that it is common in microbial genome projects to be unable to assign any gene function, even computationally, to half the genes in the genome.
The first gene ontology ever developed—MultiFun—was developed for E. coli (8,9), and the EcoCyc project has been assigning E. coli genes to MultiFun categories since 1993. In 2006, we also began curating GO term assignments for E. coli. We obtained an initial set of GO term assignments by running all MultiFun assignments within EcoCyc through a MultiFun-to-GO mapping (see http://geneontology.org/GO.indices.shtml). This mapping could not assign evidence codes to the GO terms in EcoCyc. Thereafter, our standard procedure for curation of genes in EcoCyc includes updating of both MultiFun and GO term assignments, which are displayed near the top of EcoCyc gene and protein pages. Rather than storing GO terms with gene objects in EcoCyc, they are stored in gene products and in complexes, allowing EcoCyc to annotate both monomeric gene products, and multimers, with GO terms in a uniform fashion.
EcoCyc contains a total of 8155 MultiFun term assignments for 3361 of its genes (2.4 terms assigned per gene on average). It contains 5939 GO term assignments for 2866 of its genes (2.1 terms assigned per gene).
Computational update to the E. coli genome annotation
As new sequence-analysis methods are developed, new genome sequences are published, and the functions of genes in organisms other than E. coli are elucidated, it is probable that it will become possible to predict the functions of some E. coli genes of unknown function.
To keep the annotation of E. coli genes in EcoCyc as up-to-date as possible, we will perform a regular annual computational re-annotation of the E. coli genome using the robust genome auto-annotation pipeline (10) at the J. Craig Venter Institute (JCVI). This pipeline runs a gene prediction algorithm, Glimmer 3, followed by various homology searches, including a BLAST-based search against a nonredundant protein database, HMM searches against TIGRFAM and PFAM protein family models, PROSITE motif searches, and a BLAST search against the NCBI COG database; and analyses of protein properties, including prediction of signal peptides, other sorting signals and transmembrane helices.
We ran an automated re-annotation of E. coli K-12 in December 2006. A comparison of the automated annotation with the manually curated E. coli annotation in EcoCyc showed that 3124 genes were identical or essentially identical in both annotations. Two hundred and seventeen genes in the EcoCyc annotation were missed by the auto annotation, as they were not called as open reading frames by the Glimmer 3 gene finder. Manual examination of this set of genes indicated that these were mostly very small genes and included 62 pseudogenes, 11 leader peptides, 43 phage-related genes or IS elements and 67 small hypothetical genes.
Seven hundred and ninety-six genes differed between the two annotations, with the auto annotation giving more general or generic gene calls than the existing E. coli K-12 annotation. For example, kdsD, encoding d-arabinose 5-phosphate isomerase, was called as a putative isomerase by the automated annotation. Manual inspection of these differences indicated that in all these instances, the original annotation was correct and frequently supported by experimental evidence.
One hundred and ninety-one genes differed, with the auto annotation either providing a functional call for a gene annotated as ‘hypothetical’ or ‘conserved hypothetical’ in the original annotation, or by giving a more precise function to a gene annotated with a generic function. The underlying evidence for the auto annotation of each of these 191 genes was manually examined by a curator. For 94 out of the 191, there was sufficient evidence to modify the existing annotation in EcoCyc with information from the automated annotation. In most cases, this involved updating annotation as ‘conserved hypothetical’ or ‘hypothetical’ to a generic prediction such as ‘putative lipoprotein’, ‘dehydrogenase’, ‘kinase’, ‘lipase’, ‘carboxylase’, ‘protease’, and so on. In some cases, it involved providing a more detailed prediction such as updating ‘putative transporter’ to ‘putative amino acid transporter’. While these updates do not provide exact functions for these genes, we felt they did contain better information that might be helpful for experimentalists trying to find functions of novel E. coli genes. We used our controlled evidence codes to label these functional assignments as speculative and based on predictions, to distinguish them from experimentally determined functions. For the remaining 97 out of 191 genes where no changes were made, 24 had experimental data that supported the original EcoCyc annotation, and for the other 73 there was insufficient evidence to support the automated annotation over the original assignments.
Comparison to experimentally elucidated gene fractions of other model organisms
How does the figure of 66% of E. coli genes with experimentally determined functions compare to other model organisms? The answer is found in Table 3 by comparing lines two (‘EcoCyc definition’) and four (‘Def 2’): E. coli has the highest absolute number of genes with experimental functions of all the model organisms. The E. coli genome also has a much higher fraction of experimentally elucidated genes than do the genomes of other model organisms.
Table 3.
Genes of experimentally defined function in the major model organisms
| Escherichia coli | Saccharomyces cerevisiae | Drosophila melanogaster | Mus musculus | Arabidopsis thaliana | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EcoCyc | SGD | FlyBase | MGI | TAIR | ||||||
| Total genes | 4460 | 6203 | 14 816 | 35 887 | 37 186 | |||||
| EcoCyc definition | 2941 | 66% | ||||||||
| Def 1. Strict evidence, leaf terms only | 953 | 15% | 369 | 2% | 947 | 3% | 630 | 2% | ||
| Def 2. Loose evidence, leaf terms only | 2319 | 37% | 614 | 4% | 1456 | 4% | 1816 | 5% | ||
| Def 3. Strict evidence, all terms | 1666 | 27% | 737 | 5% | 2053 | 6% | 1058 | 3% | ||
| Def 4. Loose evidence, all terms | 3627 | 58% | 1302 | 9% | 4080 | 11% | 3021 | 8% | ||
For comprehensiveness, we compare the EcoCyc definition of experimentally defined function with four alternative definitions based on the Gene Ontology, although we consider Definition 2 to be the closest to the EcoCyc definition.
The assessment of the number of genes whose functions were determined by each mechanism in a given database is determined by querying ‘evidence codes’, which are mechanisms for recording within a database the type of evidence that was used to determine a gene function. That is, was the function determined through a given type of laboratory experiment, or through a computational method such as a sequence similarity search? Different databases provide somewhat different types of evidence code mechanisms.
The EcoCyc E. coli data in this table were assembled from the evidence codes stored in EcoCyc protein and RNA objects. EcoCyc contains GO terms; however, most EcoCyc GO terms currently lack evidence codes. EcoCyc evidence codes (7) are encoded using a different and more detailed approach than used in GO. That approach is not exactly comparable to GO evidence codes; therefore, some effort was required to obtain a fair comparison to the GO evidence codes.
The non-E. coli data in Table 3 were assembled from the Gene Ontology (GO) (11) term assignments available for these model organisms from the GO website (12), using GO data downloaded on 6 December 2006. Because the evidence codes in EcoCyc are not precisely comparable to those for the other model organisms, Table 3 presents four alternative definitions of what it means for a gene in an organism other than E. coli to have an experimentally determined function. We believe that the EcoCyc notion of experimentally defined function is most similar to Definition 2 used in Table 3 because the vast majority of EcoCyc genes with experimental evidence codes have specific functions. The results in Table 3 are of course dependent on the completeness of the GO annotations for each organism with respect to the biomedical literature.
All four of the non-E. coli definitions that we present share the notions that the gene must not have a GO code assignment of GO:0003674 (‘unknown function’), and that the gene must have a GO code assignment supported by a GO experimental evidence code. All four definitions allow GO code assignments from any of the three GO hierarchies (molecular function, biological process and cellular location). The four definitions differ along two dimensions. The first dimension is that two definitions (Definitions 3 and 4) allow genes annotated with GO terms for general classes of function such as ‘DNA helicase activity’ or ‘protein binding’ activity, and for specific functions such as ‘pyruvate kinase activity’. General GO terms, called ‘non-leaf terms’, have child (more specific) terms in the ontology hierarchy (called ‘leaf terms’). In contrast, Definitions 1 and 2 allow only specific functions such as ‘pyruvate kinase activity’, that do not have child terms in the ontology hierarchy. The second dimension along which the definitions differ is as to whether they allow all GO experimental evidence codes (Definitions 2 and 4), or whether they allow only the IDA (‘inferred from direct assay’) evidence code (Definitions 1 and 3). The four definitions are:
Strict evidence, leaf terms only: Require that the gene has a leaf GO code assignment with the IDA (Inferred from direct assay) evidence code.
Loose evidence, leaf terms only: Require that the gene has a leaf GO code assignment with any of the following experimental evidence codes: IDA, IGI (‘inferred from genetic interaction’), IMP (‘inferred from mutant phenotype’), IPI (‘inferred from physical interaction’) or TAS (‘traceable author statement’).
Strict evidence, all terms: Require that the gene has a leaf or non-leaf GO code assignment with the IDA (Inferred from direct assay) evidence code.
Loose evidence, all terms: Require that the gene has a leaf or non-leaf GO code assignment with any of the following experimental evidence codes: IDA, IGI, IMP, IPI or TAS.
The transcriptional regulatory network of E. coli
Since 2001, RegulonDB (13,14) and EcoCyc have been integrated in the sense that all data entry and updating for RegulonDB occurs within EcoCyc. Regulatory data are exported from EcoCyc at each EcoCyc release, and combined with certain additional information to create a new release of RegulonDB. The information in RegulonDB that is not present in EcoCyc includes predictions of promoters, transcription factor binding sites, and operons; ribosome-binding sites; and weight matrices for some transcription factors.
EcoCyc contains extensive descriptions of the transcriptional regulatory network of E. coli, obtained from the experimental literature. The 4460 E. coli genes are assigned to 3063 transcription units (TUs), that is, sets of genes that are transcribed in a single transcript. A total of 729 of those TUs are supported by experimental evidence; the remaining TUs were predicted computationally by the Pathway Tools operon predictor (15). The average TU contains 1.7 genes because although some TUs contain as many as 15 genes, the majority of TUs (2057) contain only a single gene. Figure 1 shows the length frequency of all E. coli TUs. One hundred EcoCyc genes have not been assigned to any TU, for reasons such as large overlaps between genes that may have confused our operon predictor. We are now curating those genes in EcoCyc to provide TU assignments.
Figure 1.

Distribution of number of genes contained in EcoCyc transcription units (TUs). For example, 110 TUs are four genes in length.
The TUs contain 1230 promoters, of which 816 have experimental evidence codes (we believe the number 816 is an undercount, because a number of older entries lack evidence codes). The number of promoters with experimental evidence can exceed the number of transcription units with such evidence because a promoter may be known without the end of the TU being established, or a promoter may have been identified upstream of a predicted TU, or because curators neglected to assign experimental evidence codes to some TUs.
One thousand seven hundred and ninety transcription factor (TF) binding sites are defined for the preceding promoters, of which 1625 have experimental evidence. A total of 706 TUs contain at least one defined TF-binding site. Most EcoCyc TUs lack binding sites because most TUs were predicted computationally, and their TF-binding sites were not predicted. More than one-third of those TUs that contain identified binding sites contain only one binding site; however, 19 TUs contain more than 10 TF-binding sites each.
EcoCyc contains 171 TFs with literature-derived transcriptional regulatory relationships involving one or more TF-binding sites. TFs are highly variable in the number of TUs that they regulate; 44 factors regulate one TU each, whereas two factors each regulate more than 100 TUs. Figure 2 provides the distribution of TF regulon size and identifies the global regulators of E. coli. Figure 3 shows the converse relationship, namely, the distribution of the number of TFs that regulate each TU. A total of 156 TUs are regulated by three or more TFs.
Figure 2.

Histogram of the number of transcription factors (TFs) that regulate a given number of transcription units (TUs). For example, there are 13 TFs where each of those TFs individually regulates three TUs. Put another way, this figure shows the distribution of E. coli regulon sizes. This figure differs from the next figure in various ways; for example, this figure shows how many occurrences there are where three different TUs each contain promoter-proximal binding sites for the same TF (13), whereas the next figure shows how many occurrences there are of a given TU containing promoter-proximal binding sites for three different TFs. The latter cases are also called ‘in-coming’ interactions, whose distribution is known to follow the power law.
Figure 3.

Histogram of the number of TUs that are jointly regulated by a given number of TFs. For example, 31 TUs are regulated by four TFs, meaning that each of those 31 TUs contains promoter-proximal binding sites for four different TFs.
EcoCyc contains DB objects that define interactions between TFs and 48 small-molecule ligands that regulate TF activity. Table 4 shows the TF ligands that are defined in EcoCyc. A total of 109 EcoCyc TFs lack defined ligands. In many cases, this is because the TFs interact with sensor kinases that themselves interact with the ligands, but we expect that ligands remain to be curated or elucidated for many of these TFs.
Table 4.
Small-molecule ligands that regulate the activity of E. coli transcription factors
| 2-Methylcitrate | Cyanate | Glyoxylate | Melibiose |
|---|---|---|---|
| 3-(3-Hydroxyphenyl)propionate | Cyclic-AMP | Hypoxanthine | 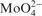 |
| 3-Phenylpropionate | Cytidine | l-arginine | N-acetyl-d-glucosamine |
| Adenosine 5′-phosphosulfate | d-ribose | l-ascorbate | N-acetylneuraminate |
| Allantoin | d-sorbitol | l-homocysteine | Na+ |
| Allolactose | d-xylose | l-leucine | Ni2+ |
| Alpha-l-arabinose | Formate | l-phenylalanine | O-acetyl-l-serine |
| An acyl-CoA | Fructose-1,6-bisphosphate | l-rhamnose | Phosphoenolpyruvate |
| ATP | Fructose-1-phosphate | l-tryptophan | Pyruvate |
| Beta-d-galactose | Fructuronate | l-tyrosine | S-adenosyl-l-methionine |
| Bio-5′-AMP | Gluconate | Maltotriose | Salicylate |
| Choline | Glycolate | Mannitol | Sn-glycerol-3-phosphate |
The interaction between each TF with each binding site is described by a separate interaction object within EcoCyc. These interaction objects also describe which sigma factor recognizes each promoter. Figure 4 shows the number of promoters controlled by each E. coli sigma factor, as reported in the literature.
Figure 4.

Number of promoters at which each sigma factor initiates transcription.
What fraction of the E. coli transcriptional network has been experimentally elucidated to date? Of the roughly 330 E. coli genes that encode TFs, at least one binding site has been experimentally determined for approximately half of them. It is likely that more binding sites for these TFs remain to be determined. A total of 23.0% of TUs have at least one experimentally determined binding site, and 23.8% of TUs have experimental evidence for the extent of the TU. Although our knowledge of the regulatory network is clearly incomplete, experimentalists have probably determined in the order of 25–30% of the regulatory interactions within the transcriptional regulatory network.
The Pathway Tools software (5) provides EcoCyc with several mechanisms for visualizing regulatory information. A new visualization of the full transcriptional regulatory network in EcoCyc, called the Regulatory Overview, is shown in Figure 5. The user can interactively explore regulatory relationships on this diagram through several different queries, such as displaying arrows linking gene G to all the genes that G regulates, or to all the genes that regulate G. In addition, the user can selectively highlight within a diagram of the full metabolic map of E. coli (see http://biocyc.org/ECOLI/NEW-IMAGE?type=OVERVIEW) those enzymes that are transcriptionally regulated by a specified transcription factor. These operations are currently available only through the desktop (locally installed) version of EcoCyc plus Pathway Tools; we plan to incorporate them in the Web version in fall 2007.
Figure 5.

EcoCyc Regulatory Overview. Each dot represents one gene. Genes are arranged in three rings. Genes in the innermost ring have no known regulators, but they regulate at least one other gene. Genes in the middle ring are known to be regulated by other genes, and also regulate other genes. Genes in the outermost ring have known regulators, but are not known to regulate any other genes. An arrow from gene A to gene B means that A regulates the transcription of B. The diagram shows all regulatory arrows for genes involved in the MultiFun categories of iron acquisition (green) and drug resistance/sensitivity (red).
Additional regulatory visualizations are available through both the Web and the desktop versions of EcoCyc. At the bottom of most EcoCyc gene pages is a diagram of the TU containing that gene. Clicking on the promoter displays a page of detailed information about the regulation of the TU. Clicking on a TF-binding site in the TU displays the EcoCyc protein page for the TF. This page shows every TU that the TF controls. Protein pages for sigma factors show all TUs that the sigma factor controls.
The EcoCyc project is beginning a new effort to encode many additional types of regulation, including attenuation, translational regulation and regulation by small RNAs. Our goal is for EcoCyc to capture and compute with all known types of regulation in E. coli.
The metabolic network of E. coli
A recent collaboration between the Karp and Palsson groups compared the metabolic model within EcoCyc with the model developed by the Palsson group. The comparison resulted in updates to both models (16,17). The resulting metabolic network within EcoCyc consists of 1008 reactions of small-molecule metabolism. A total of 722 of those reactions are assigned to one of the 194 metabolic pathways defined within EcoCyc; the remaining 286 reactions are not assigned to a pathway. Approximately 2−3 new metabolic pathways have been reported in the experimental literature per year for the past 5 years, indicating that our knowledge of the E. coli metabolic network is probably still incomplete.
The 1008 reactions of E. coli's small-molecule metabolism are catalyzed by 918 enzymes, of which 482 are monomers and 436 are multimers. A total of 354 of the multimers are homomultimers. The enzymes are formed from the products of 991 distinct genes (22% of the genome). One hundred and eighty five of the enzymes are multifunctional. Other properties of older versions of the metabolic network were discussed in (18).
Our ability to connect metabolism to genes in E. coli is incomplete. EcoCyc lists 41 E. coli metabolic enzymes whose activities have been demonstrated biochemically and appear in the literature, but whose genes have yet to be definitively identified—although computational hypotheses regarding the identities of some have been published (19,20). These enzymes are listed at http://ecocyc.org/enzymes.shtml, which is updated for every EcoCyc release.
Finally, we face the problem of dead-end metabolites within the E. coli metabolic network. A dead-end metabolite is a metabolite that is produced by the metabolic network and has no reactions consuming it, or that is only consumed by the metabolic network and has no reactions generating it, and in both cases has no identified transporter. We authored a program that searches EcoCyc according to a simplified version of the preceding definition (see Methods section) and found 169 dead-end metabolites. We focused our analysis on those 33 dead ends that are substrates in metabolic pathways, with the following results. See also analyses of E. coli dead ends by Reed et al. (20) (many of the results from that analysis were previously incorporated into EcoCyc) and by Kumar et al. (21) (which considered a broader set of dead ends, some of which are not found in pathways).
Analysis of dead-end compounds within a Pathway/Genome Database (PGDB) is a useful exercise to identify missing information in the database or in the experimental literature. In the case of EcoCyc, the classification of carbamate and H2CO3 as dead-end compounds led to the realization that the EcoCyc cyanate degradation pathway was incorrect. Rather than being depicted as an intermediate, carbamate was shown as a side product of the two-step pathway. The corrected pathway shows carbamate as an unstable intermediate that decomposes in a spontaneous reaction to ammonia and CO2. The final step, hydration of CO2 to bicarbonate by carbonic anhydrase, was shown to be an essential component of the pathway to prevent depletion of the intracellular bicarbonate pool (22).
A small number of compounds were classified as dead-end metabolites for trivial reasons. Several compounds are able to diffuse freely across the cytoplasmic membrane or are utilized in the periplasm, and thus there is no requirement for pathways or reactions to produce or import them (acetoacetate, dimethyl sulfoxide and dimethylsulfide are examples). Other compounds are known to participate in spontaneous reactions; this information allowed us to remove certain compounds (OH-, H2CO3, carbamate) from the list of dead ends. One compound, tetrahydropteroyltri-l-glutamate, lacked the proper chemical classification that would have allowed it to be included as a substrate in folate polyglutamylation.
The classification of a small number of compounds as dead-end metabolites occurred because information that is available in the experimental literature was missing within EcoCyc. Adding appropriate reactions and pathways that produce or utilize these compounds (5′-methylthioadenosine, undecaprenyl N-acetyl-glucosaminyl-N-acetyl-mannosaminuronate-4-acetamido-4,6-dideoxy-d-galactose pyrophosphate, 1,6-anhydro-N-acetylmuramate and GDP-l-fucose) to EcoCyc will be a priority in our further curation efforts.
Other cases of dead-end metabolites point to the need for further research. A number of metabolites require transport reactions to make them available to the cytoplasmic enzymes that utilize or degrade them (1,6-anhydro-N-acetylmuramate, 4-methyl-5-(beta-hydroxyethyl)thiazole, cobinamide, ethylene glycol, hydroxymethylpyrimidine, phenylethylamine, psicoselysine). For example, it is known that E. coli can grow on phenylethylamine, for which no transporter has been identified. Other compounds, such as cobinamide, are known to be required for biosynthesis of a metabolite (vitamin B12, in this case), but again no transporter has yet been identified. 1-Amino-propan-2-ol was thought to be its precursor in vitamin B12 biosynthesis, but this may be questionable. The entire biosynthesis route for vitamin B12 requires additional experimental work.
In some cases, it is known that a compound is a precursor for the synthesis of an essential metabolite, but no metabolic route has yet been identified. One such example is pimeloyl-CoA, which is required for biotin biosynthesis in E. coli. Because E. coli does not require pimeloyl-CoA for growth, it must be able to synthesize the compound, but the synthesis route and any enzymes that may be used are currently unknown. S-adenosyl-4-methylthio-2-oxobutanoate is a side product in biotin biosynthesis, but despite extensive searches of the available literature, we were unable to find information on the ultimate fate of this compound. The identities of the sulfur donors in the biotin synthase and lipoate synthase reactions are thought to be iron–sulfur clusters, but some uncertainty remains, and thus the donors were represented as stand-alone compounds in EcoCyc.
For several compounds (aminoacetaldehyde, dimethylbenzimidazole, oxalurate and urate), there is some suggestion or indirect evidence for synthesis or further metabolism, but no experimental confirmation in E. coli is available.
The transporters of E. coli
Two hundred and thirty-eight distinct transport reactions (transporter activities) are defined in EcoCyc. Those transport activities are assigned to 214 different transporters, which are encoded by 355 E. coli genes. A total of 180 additional E. coli genes are estimated to code for transporters of unknown function. The 180 substrates and classes of substrates for which E. coli transporters are known to exist are shown in Table 5.
Table 5.
Substrates known to be transported by E. coli
| (2-Aminoethyl)phosphonate | Curlin, major subunit | H+ | Mono-, di-, or trisaccharide <600 Da |
| 1,6-AnhMurNAc | Cyanate | H2O | |
| 2-Dehydro-3-deoxy-d-gluconate | Cytidine | Homoserine | Multidrug |
| 2-O-Alpha-mannosyl-d-glycerate | Cytosine | Homoserine lactone | N-acetyl-d-glucosamine |
| 3-(3-Hydroxyphenyl)propionate | d-alanine | Hydrophilic solute or ion <600 Da | N-acetylmuramate |
| 3-Aminopropylphosphonate | d-allose | Indole | N-acetylneuraminate |
| 3-Phospho-serine | d-cycloserine | Inosine | Na+ |
| 4-Aminobutyrate | d-galactarate | Iron (III) hydroxamate complex | NH4+ |
| A C4-dicarboxylate | d-galactonate | Isethionate | Ni2+ |
| A carbohydrate | d-glucarate | K+ | Nicotinamide Mononucleotide |
| A dipeptide | d-methionine | l-alanine | Nitrite |
| A drug | d-ribose | l-arginine | Non-specific ion/solute |
| A fatty acid | d-serine | l-ascorbate | O-acetyl-l-serine |
| A glucoronide | d-sorbitol | l-asparagine | O-antigen |
| A lipopolysaccharide | d-xylose | l-aspartate | Orotate |
| A macrolide antibiotic | Deoxyadenosine | l-carnitine | Pantothenate |
| A nucleoside | Deoxycytidine | l-cysteine | Phenylphosphonate |
| A peptide | Deoxyinosine | l-fucose | Phosphate |
| A peptide antibiotic | Deoxyuridine | l-glutamate | Phosphite |
| A quaternary amine | Enterobactin | l-glutamine | Propylphosphonate |
| Acetate | Ethylphosphonate | l-histidine | Protoheme IX |
| Adenosine | Fe2+ | l-idonate | Putrescine |
| Agmatine | Ferric coprogen | l-isoleucine | Pyridoxal |
| Alpha-ketoglutarate | Ferric dicitrate | l-lactate | Pyridoxamine |
| Alpha-l-arabinose | Ferric dihydroxybenzoylserine | l-leucine | Pyridoxine |
| Aminomethylphosphonate | Ferric enterobactin | l-lysine | Salicin |
| Ammonia | Folded proteins | l-methionine | Shikimate |
| An alkylphosphonate | Formate | l-ornithine | Sn-glycerol-3-phosphate |
| Arbutin | Fosmidomycin | l-phenylalanine | Spermidine |
| Arsenate | Fructose | l-proline | Succinate |
| Beta-alanine | Fructuronate | l-rhamnose | Sulfate |
| Beta-d-galactose | Fumarate | l-serine | Tartrate |
| Beta-d-glucose | Galactitol | l-threonine | Taurine |
| Beta-d-glucose-6-phosphate | Galacturonate | l-tryptophan | Thiamin |
| Ca2+ | Gamma-butyrobetaine | l-tyrosine | Thiosulfate |
| Cadaverine | GlcNAc-1,6-anhydro-MurNAc- l-Ala-gamma-d- Glu-meso-diaminopimelic acid-d-Ala | l-valine | Thymidine |
| Cd2+ | Gluconate | Lactose | Trehalose |
| Cellobiose | Glucosamine | Li+ | Uracil |
| Chitobiose | Glucuronate | Malate | Uridine |
| Chloride | Glutathione | Maltose | Xanthine |
| Choline | Glycerol | Mannitol | Xanthosine |
| Citrate | Glycine | Mannose | Zn2+ |
| Co2+ | Glycine betaine | Melibiose | |
| Cob(I)alamin | Glycolate | Methylphosphonate | |
| Cu+ | Glyoxylate | Mg2+ | |
| Cu2+ | Guanosine | Mn2+ |
Visualizing EcoCyc genome information
The Pathway Tools genome browser is one of several tools for visualizing EcoCyc genome information. It can be entered from any EcoCyc gene page (click on the Map Position line) to produce a genome view centered on that gene. It can also be entered from the BioCyc query page (see http://BioCyc.org/server.html) by selecting a chromosome in the Genome Browser line and clicking Submit. Figure 6 shows a view generated from the EcoCyc genome browser. The view shows a region of the genome that is wrapped across several lines; the right side of one line is continued at the left side of the line below it. The navigation panel in the upper left region allows the user to change the magnification of the diagram (vertical arrows) or to move the viewed region left or right (horizontal arrows), by smaller or larger degrees (single arrows versus double arrows). New views can also be selected by entering a gene name in the entry field near the top and clicking Go, or by entering start and stop base pairs and clicking Go. This genome browser has several novel aspects including the multiline presentation, and the clear visual designation of different gene types, of operons, and of promoters and terminators.
Figure 6.

Representative region of the E. coli genome generated by the EcoCyc genome browser.
Genes within the same operon are displayed in the same colour; operon boundaries are also indicated by the gray or green backgrounds behind genes. The green background denotes operons with experimental evidence; gray denotes computationally predicted operons. Transcription start sites and terminators are displayed at high magnifications. Protein-coding genes are displayed with isosceles arrowheads (such as ydiL in the second row), and RNA-coding genes are displayed with slanting heads (such as rprA in the second row). Pseudogenes contain a large X (see next to last row). (EcoCyc defines pseudogenes both as genes that are cryptic (not expressed), and as genes that are not able to encode a functional protein product.) Moving the mouse pointer over a given gene will display information about it in a pop-up window or at the bottom of the screen. For clarity, genes with significant overlaps are positioned above one another.
The Show Tracks button in the upper right allows display tracks to be created to display regions along the genome that are defined in a GFF file, as supported by other genome browsers such as Gbrowse (23) and the UC Santa Cruz Genome Browser (24). Figure 7 shows the use of tracks to display predicted promoters in a region of the E. coli genome.
Figure 7.

EcoCyc genome browser showing display tracks. The promoter locations highlighted below each gene were predicted computationally (25) and can be download from the Web (see http://regulondb.ccg.unam.mx/data/PromoterPredictionSet.txt).
Comparative views of several genomes for organisms within the BioCyc collection of 370 organism-specific PGDBs can easily be generated from within a BioCyc gene page from any organism by clicking on the button ‘Align in Multi-Genome Browser’ mid-way down the gene page, and then selecting the organisms for which a comparison is desired. The resulting visualization, such as that shown in Figure 8, aligns the genomes at the orthologs to the starting gene in the selected organisms, and displays the adjacent chromosomal regions to scale. The same navigation panel used in the regular genome browser can be used to scale and position the comparative genome browser. In the comparative genome browser, colour no longer encodes operon membership: genes drawn in the same colur are orthologs of one another (best bi-directional BLAST hits). Tens of other E. coli strains have been sequenced to date, and additional E. coli strains are being added to the BioCyc collection on an ongoing basis. We note that the K-12 strain described by EcoCyc is smaller, by more than 1000 genes, than other sequenced E. coli strains.
Figure 8.

The EcoCyc comparative genome browser centered at E. coli K-12 gene copA and its orthologs in Shigella flexneri and E. coli EDL933. This comparative view shows that K-12 copA is an ortholog of genes with no assigned functions in the two other strains, despite their orthology and similar length to copA. The comparative view also shows how the orthologous region continues for many genes upstream in K-12 and Shigella, but ends at ybaT for K-12 and EDL933.
DISCUSSION AND SUMMARY
EcoCyc provides a rich and multidimensional annotation of the E. coli genome that is coupled with a large complement of software tools for querying and visualization of the EcoCyc data.
EcoCyc contains a ‘living’ version of the E. coli annotation, subject to regular updates to gene sequences, functions and locations in response to advancing knowledge about this organism. We have recently performed the first in a regular series of computational reannotations of the E. coli genome using the JCVI automated annotation pipeline, resulting in modifications to the annotations of 94 genes in EcoCyc. These and all other updates to the genome can be visualized using the EcoCyc genome browser, which can also be used to compare related genome regions across multiple organisms, and to overlay other information on the genome via the tracks feature.
The dimensions of annotation within the current version (11.1) of EcoCyc are as follows.
The genome sequence is annotated with the positions of genes, promoters, terminators and transcription factor binding sites.
Functions are assigned to gene products, both as textual descriptions and as descriptions within the MultiFun Ontology and Gene Ontology.
Gene products are assigned to macromolecular complexes in which they function. Chemically modified states of gene products are defined when appropriate.
A long-term literature curation effort has populated EcoCyc with information from 15 000 publications. Every gene product for which experimental literature is likely available (3332 ORFs) contains a mini-review summary of what is known about the gene product, forming another dimension of annotation. Similar summaries are found in pathway objects and transcription unit objects within EcoCyc.
The functions of 3384 (76%) E. coli genes have been determined through wet-lab experimentation, which is significantly higher in both relative and absolute terms than for any other model organism. EcoCyc encodes the type of evidence supporting the functions of gene products, and of other entities including operons, promoters, transcription factor binding sites and pathways.
EcoCyc represents the transcriptional regulatory network of E. coli. The network describes transcriptional control of 718 TUs by 171 transcription factors. EcoCyc specifically represents the binding of these 171 factors to 1790 transcription factor binding sites, 1625 of which have experimental (wet lab) evidence. In addition, EcoCyc records the interactions by which 49 small-molecule ligands control the activities of the 171 transcription factors. A novel interactive visualization of the transcriptional regulatory network is available.
The metabolic network of E. coli as represented in EcoCyc consists of 194 metabolic pathways containing 722 reactions, plus an additional 286 reactions not assigned to a pathway. Database queries making use of this structured representation of E. coli metabolism identified gaps in the network, including reactions whose catalyzing enzymes have not been identified, and dead-end metabolites. These gaps guided additional curation efforts that either corrected representational concerns, found additional evidence in the literature, or identified matching gaps in our knowledge of E. coli biology.
Representation of transporters adds additional depth to the metabolic network in EcoCyc. The transport complement of E. coli consists of 214 transport proteins of known function that facilitate the transport of 180 substrates. As described above, an understanding of transporters is critical in evaluating the role of any metabolite in the overall metabolic network.
A number of other E. coli databases exist that provide annotated gene/protein content. Some provide complementary information while others share overlap. However, none provide the range of dimensional content and accessibility to the E. coli genome within EcoCyc. For example, EcoGene (26) focuses on corrections to the E. coli MG1655 sequence, and to gene start and stop positions. It also contains many literature citations, and a limited set of short gene summaries, most of which do not contain internal citations that provide sources for specific statements. The Coli Genetic Stock Center (see http://cgsc.biology.yale.edu/) provides valuable information on E. coli strains and mutations. It contains some short summaries that do not contain internal citations that provide sources for specific statements. KEGG (27) describes the E. coli genome annotation and its metabolic pathways. It contains no summaries or citations, and the user cannot determine what information in KEGG is literature curated versus predicted computationally. None of these databases provide evidence codes. EchoBASE (28) describes the E. coli genome annotation, providing some summaries that lack internal citations; and it provides high-throughput datasets for E. coli. UniProt (29) contains information about the E. coli proteome, including summaries and literature citations, although many functional assignments lack specific literature support. RegulonDB (14) describes the transcriptional regulatory network of E. coli. The RefSeq E. coli entry contains genome annotation information combined from several of the preceding databases. A number of other E. coli databases exist (see http://www.uni-giessen.de/~gx1052/IECA/ieca.html), although some have not been updated for many years.
The goal of EcoCyc is to accelerate scientific discovery and education. We recently surveyed more than 500 publications that cite EcoCyc, and found that their uses of EcoCyc fell into five major classes. (i) EcoCyc is a knowledge resource on the E. coli genome and cellular networks for experimentalists who work with E. coli, with other microbes, and with higher organisms. Scientists use it as an encyclopedic reference, particularly for analyzing large-scale datasets, such as for analyzing E. coli gene-expression data. (ii) Computational biologists use EcoCyc to answer biological questions using computational methods, such as assessing which metabolic enzymes are conserved between yeast and E. coli (30), studying the topological organization of the E. coli metabolic network (31), and studying the properties of the E. coli transcriptional regulatory network (32). (iii) Bioinformaticists use EcoCyc as a gold-standard data source for developing and validating new prediction algorithms, such as genome context algorithms for predicting functional relationships among proteins (33,34), operon predictors (34) and algorithms for predicting regulatory networks from gene expression data (35). (iv) Metabolic engineers consult EcoCyc when altering the metabolic network of E. coli (36). (v) University courses make use of EcoCyc in subjects ranging from microbial physiology through molecular genetics to microbial biotechnology.
The guiding principle of the EcoCyc project has been to store information about E. coli gene function and cellular networks within a structured ontology that makes those data readily accessible to symbolic systems biology computations (37). We have illustrated several such analyses in this article that use computational methods to summarize the content of EcoCyc, to assess its completeness, and to define areas for further research. Examples include the list of regulatory signals in Table 4, and the list of unsequenced E. coli enzymes at http://ecocyc.org/enzymes.shtml. Other programs assess the biological coherence of the data in EcoCyc, such as by propagating nutrients from known growth media through the EcoCyc metabolic network to assess whether the network can produce compounds essential for growth from a known minimal medium, and by searching for dead-end metabolites.
ACKNOWLEDGEMENTS
This work was supported by grants GM077678, GM075742 and GM071962 from the National Institutes of Health. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. We are grateful to Dr Robert Gunsalus for comments on this manuscript, and for his many suggestions regarding EcoCyc. Funding to pay the Open Access publication charges for this article was provided by the National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1.Keseler IM, Collado-Vides J, Gama-Castro S, Ingraham J, Paley S, Paulsen IT, Peralta-Gil M, Karp PD. EcoCyc: a comprehensive database resource for Escherichia coli. Nucleic Acids Res. 2005;33:D334–D337. doi: 10.1093/nar/gki108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karp PD, Riley M, Saier M, Paulsen IT, Collado-Vides J, Paley SM, Pellegrini-Toole A, Bonavides C, Gama-Castro S. The EcoCyc Database. Nucleic Acids Res. 2002;30:56–58. doi: 10.1093/nar/30.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karp PD, Riley M, Saier M, Paulsen IT, Paley SM, Pellegrini-Toole A. The EcoCyc and MetaCyc databases. Nucleic Acids Res. 2000;28:56–59. doi: 10.1093/nar/28.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reed JL, Famili I, Thiele I, Palsson BO. Towards multidimensional genome annotation. Nat. Rev. Genet. 2006;7:130–141. doi: 10.1038/nrg1769. [DOI] [PubMed] [Google Scholar]
- 5.Karp PD, Paley S, Romero P. The Pathway Tools software. Bioinformatics. 2002;18(Suppl. 1):S225–S232. doi: 10.1093/bioinformatics/18.suppl_1.s225. [DOI] [PubMed] [Google Scholar]
- 6.Riley M, Abe T, Arnaud MB, Berlyn MK, Blattner FR, Chaudhuri RR, Glasner JD, Horiuchi T, Keseler IM, et al. Escherichia coli K-12: a cooperatively developed annotation snapshot – 2005. Nucleic Acids Res. 2006;34:1–9. doi: 10.1093/nar/gkj405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karp PD, Paley S, Krieger CJ, Zhang P. An evidence ontology for use in pathway/genome databases. Pac. Symp. Biocomput. 2004:190–201. doi: 10.1142/9789812704856_0019. [DOI] [PubMed] [Google Scholar]
- 8.Riley M. Functions of the gene products of Escherichia coli. Microbiol. Rev. 1993;57:862–952. doi: 10.1128/mr.57.4.862-952.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serres MH, Riley M. MultiFun, a multifunctional classification scheme for Escherichia coli K-12 gene products. Genome Biol. 2000;5:205–222. doi: 10.1089/omi.1.2000.5.205. [DOI] [PubMed] [Google Scholar]
- 10.White O. Bacterial Genome Annotation at TIGR. In: Fraser CM, Read TD, Nelson KE, editors. Microbial Genomes. New Jersey: Humana Press: Totowa; 2004. [Google Scholar]
- 11.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.2007. Gene Ontology Current Annotations Page. [Google Scholar]
- 13.Salgado H, Santos-Zavaleta A, Gama-Castro S, Peralta-Gil M, Penaloza-Spinola MI, Martinez-Antonio A, Karp PD, Collado-Vides J. The comprehensive updated regulatory network of Escherichia coli K-12. BMC Bioinformatics. 2006;7:5. doi: 10.1186/1471-2105-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salgado H, Gama-Castro S, Peralta-Gil M, Diaz-Peredo E, Sanchez-Solano F, Santos-Zavaleta A, Martinez-Flores I, Jimenez-Jacinto V, Bonavides-Martinez C, et al. RegulonDB (version 5.0): Escherichia coli K-12 transcriptional regulatory network, operon organization, and growth conditions. Nucleic Acids Res. 2006;34:D394–D397. doi: 10.1093/nar/gkj156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romero PR, Karp PD. Using functional and organizational information to improve genome-wide computational prediction of transcription units on pathway-genome databases. Bioinformatics. 2004;20:709–717. doi: 10.1093/bioinformatics/btg471. [DOI] [PubMed] [Google Scholar]
- 16.Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BO. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007;3 doi: 10.1038/msb4100155. article 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun J, Tuncay K, Haidar AA, Ensman L, Stanley F, Trelinski M, Ortoleva P. Transcriptional regulatory network discovery via multiple method integration: application to E. coli K12. Algorithms Mol. Biol. 2007;2:2. doi: 10.1186/1748-7188-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ouzounis CA, Karp PD. Global properties of the metabolic map of Escherichia coli. Genome Res. 2000;10:568–576. doi: 10.1101/gr.10.4.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green ML, Karp PD. A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases. BMC Bioinformatics. 2004;5:76. doi: 10.1186/1471-2105-5-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reed JL, Vo TD, Schilling CH, Palsson BO. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR) Genome Biol. 2003;4:R54. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar VS, Dasika MS, Maranas CD. Optimization based automated curation of metabolic reconstructions. BMC Bioinformatics. 2007;8:212. doi: 10.1186/1471-2105-8-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guilloton MB, Lamblin AF, Kozliak EI, Gerami-Nejad M, Tu C, Silverman D, Anderson PM, Fuchs JA. A physiological role for cyanate-induced carbonic anhydrase in Escherichia coli. J. Bacteriol. 1993;175:1443–1451. doi: 10.1128/jb.175.5.1443-1451.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, et al. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhn RM, Karolchik D, Zweig AS, Trumbower H, Thomas DJ, Thakkapallayil A, Sugnet CW, Stanke M, Smith KE, et al. The UCSC genome browser database: update 2007. Nucleic Acids Res. 2007;35:D668–D673. doi: 10.1093/nar/gkl928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huerta AM, Collado-Vides J. Sigma70 promoters in Escherichia coli: specific transcription in dense regions of overlapping promoter-like signals. J. Mol. Biol. 2003;333:261–278. doi: 10.1016/j.jmb.2003.07.017. [DOI] [PubMed] [Google Scholar]
- 26.Rudd KE. EcoGene: a genome sequence database for Escherichia coli K-12. Nucleic Acids Res. 2000;28:60–64. doi: 10.1093/nar/28.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misra RV, Horler RS, Reindl W, Goryanin II, Thomas GH. EchoBASE: an integrated post-genomic database for Escherichia coli. Nucleic Acids Res. 2005;33:D329–D333. doi: 10.1093/nar/gki028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, et al. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jardine O, Gough J, Chothia C, Teichmann SA. Comparison of the small molecule metabolic enzymes of Escherichia coli and Saccharomyces cerevisiae. Genome Res. 2002;12:916–929. doi: 10.1101/gr.228002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 32.Ma HW, Kumar B, Ditges U, Gunzer F, Buer J, Zeng AP. An extended transcriptional regulatory network of Escherichia coli and analysis of its hierarchical structure and network motifs. Nucleic Acids Res. 2004;32:6643–9. doi: 10.1093/nar/gkh1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bowers PM, Pellegrini M, Thompson MJ, Fierro J, Yeates TO, Eisenberg D. Prolinks: a database of protein functional linkages derived from coevolution. Genome Biol. 2004;5:R35. doi: 10.1186/gb-2004-5-5-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Price MN, Huang KH, Alm EJ, Arkin AP. A novel method for accurate operon predictions in all sequenced prokaryotes. Nucleic Acids Res. 2005;33:880–892. doi: 10.1093/nar/gki232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weber J, Hoffmann F, Rinas U. Metabolic adaptation of Escherichia coli during temperature-induced recombinant protein production: 2. Redirection of metabolic fluxes. Biotechnol. Bioeng. 2002;80:320–330. doi: 10.1002/bit.10380. [DOI] [PubMed] [Google Scholar]
- 37.Karp PD. Pathway databases: a case study in computational symbolic theories. Science. 2001;293:2040–2044. doi: 10.1126/science.1064621. [DOI] [PubMed] [Google Scholar]