

It is widely acknowledged that EM of macromolecules is entering a new phase of productivity (for review see Nogales and Grigorieff 2001). It is less well known that comparable innovations are afoot in cellular EM. Studies of biological “ultra-structure” were of key importance in the early years of cell biology, but more recently their importance has waned significantly. This change has been largely because much of what could be seen by classical techniques for EM has long since been described; it has also derived from the cost of EM in both time and money and the innovations that have recently revolutionized light microscopy, making it ever more powerful for the study of living cells. However, recent improvements in both methods and instrumentation for EM are now allowing the structure of organelles and cellular subsystems to be characterized with unprecedented detail and reliability. Thanks to tomography, the three-dimensional (3-D) structure of cells can now be visualized with 5–8-nm resolution (Frank 1995; Baumeister et al. 1999). Thanks to high pressure freezing, cellular specimens of considerable size can now be well frozen, even without the addition of chemical cryoprotectants (Shimoni and Muller 1998). With this and other methods for rapid freezing, samples become solidified within milliseconds (Gilkey and Staehelin 1986), whereupon they are embedded in glass-like ice (Sartori et al. 1993). The cellular milieu is still aqueous, but rapid freezing has immobilized all the cell's constituents before significant rearrangement is possible. Under these conditions, biological structure is trapped in an essentially native state, and ice crystals, which would deform the physiological organization, have had little time to grow. More rapidly frozen specimens retain impressive preservation of intracellular detail (Heuser and Reese 1981).
Frozen-hydrated cells can be examined directly in the EM by using a low temperature specimen holder, or they can subsequently be fixed by “freeze-substitution,” during which cellular water is replaced at −80°C to −90°C by an organic solvent that contains chemical fixatives to stabilize the biological structure before it is embedded in a matrix suitable for microtomy. Freeze-substituted samples appear similar to material prepared for EM by conventional methods, but they have a greater likelihood of displaying structures in their native state (for review see Steinbrecht and Muller 1987; McDonald and Morphew 1993). This review will describe recent progress in the 3-D imaging of both frozen-hydrated cells and those that have been preserved by freeze-substitution. We will then evaluate the impact of the resulting data on our understanding of cellular mechanisms.
EM Images of Frozen-hydrated Specimens, Purified from Cells, Can Reveal Functionally Important Conformational Changes in Large Assemblies of Macromolecules
Several fields of cell biology have profited from information obtained by cryo-EM of specimens isolated from whole cells, spread as very thin layers on supporting films, and then frozen by plunging into a “cryogen,” such as liquid ethane. For example, our appreciation of microtubule dynamics was significantly advanced by the realization that polymer end morphology is different for assembling and disassembling microtubules (Mandelkow et al. 1991). Likewise, the stroke-cycles of motor enzymes, bound to either microfilaments or microtubules in the presence of different nucleotides, have been defined by cryo-EM (Hirose et al. 1995; Whittaker et al. 1995). The resulting images have allowed both a correlation with atomic resolution information from x-ray diffraction (e.g., Hoenger et al. 1998) and an exploration of the structural changes that accompany changes of the bound nucleotide (Rice et al. 1999; Vale and Milligan 2000).
Much of this imaging has used isolated cellular subfractions that are small, stable, and symmetric enough to allow image averaging to facilitate high-resolution EM. The beam sensitivity of a frozen-hydrated sample requires that the total exposure to the electron beam be kept low (e.g., <10 electrons/A2 for a resolution of <1 nm). At this dose, the quality of a single image is severely limited by “shot noise,” i.e., the poor statistics with which the electron-scattering properties of the specimen are sampled. Symmetry in the structure allows a simple image averaging to increase the image signal-to-noise ratio, even when the dose per unit area is kept low. Image quality improves with the square root of the number of areas averaged, and several thousand can be used. Alternatively, a large number of images of identical, individual particles can be collected, the images oriented relative to one another, and their averages computed to accomplish the same task with particles that possess no symmetry. Image averaging based on symmetry or on galleries of distinct but identical particles (or in the case of viruses and some other objects, on the use of both approaches together) has made it possible to improve signal-to-noise ratios to the point that resolution has become as good or better than 1 nm (e.g., Bottcher et al. 1997; Matadeen et al. 1999; Gabashvili et al. 2000). With this much image detail, it has been possible to integrate information from x-ray crystallography with images from cryo-EM and build up models for changes in the structure of complex molecular assemblies that accompany their biological function (Agrawal et al. 2000; Stark et al. 2000; for review see Nogales and Grigorieff 2001). This important field of structural biology is rapidly expanding our knowledge of complex molecular mechanisms.
Images of Frozen-hydrated Cells Are Revealing New Aspects of Cell Structure but Pose Important Challenges to the Microscopist
The study of frozen-hydrated cells raises two issues that are not encountered during microscopy of isolated, subcellular assemblies: (a) cells are large and tremendously complex, so a huge amount of information is necessary to characterize them, and (b) no two cells, or even two large organelles like mitochondria, are sufficiently similar that their images can be simply averaged to enhance image signal-to-noise. Thus, the methods for detailed characterization of cellular structure differ from those that are appropriate for macromolecular assemblies. To tackle the problem of cell size, one must either select tiny cells or find a way to collect appropriately small pieces of the cell of choice. Such pieces of a frozen-hydrated cell must also be thin enough for view in the EM without too many of the beam's electrons suffering multiple scattering events, which seriously degrades image quality (less than ∼0.5 μm thick for 300 KeV electrons). One approach for the preparation of such specimens has been direct thin sectioning of frozen cells or tissues (e.g., McDowell et al. 1989). However, it has been very difficult to cut useful sections of samples that have simply been frozen. The embedding ice tends to crack during sectioning (Richter et al. 1991), and sample compression is usually significant, though improvements may be on the horizon (Studer and Gnaegi 2000).
When thinking about cryomicrotomy it is natural to recall the widespread success that has been achieved by cutting cryosections from samples that have first been chemically fixed and infiltrated with a cryoprotectant (usually sucrose) and then frozen (Tokuyasu 1980). This method is commonly used to prepare samples for immunolabeling, but it fails to take advantage of the power of rapid freezing to preserve cell structure with the greatest possible fidelity. Thus, in spite of its value for antigen localization, the approach is not an ideal way to study cell morphology. Its reliance on both chemical fixation and infiltration with an osmotically active cryoprotectant reduces the likelihood that the resulting images will reflect the condition in a living cell. Current work in the lab of Jan Slot (University of Utrecht, Utrecht, Netherlands) is directed at improving this situation (Malide et al. 2000), but much remains to be done.
Since sectioning frozen-hydrated samples has been difficult, several scientists have sought cellular samples that can be viewed without it. For example, the thin margin of a cell spread on an electron-transparent substrate has been imaged directly by high voltage electron microscopy using samples maintained at ∼−165°C (O'Toole et al. 1993; Fig. 1). The resulting images show how much detail is preserved in a rapidly frozen cell, but with a single projection of so much 3-D structure, the images are hard to interpret (Fig. 1 A). Even thin processes that extend from the cell's margin are complex, though in such a region microfilaments can readily be identified (Fig. 1 B).
Figure 1.

A platelet from human peripheral blood, frozen by plunging into liquid ethane and imaged at −165°C, using 1,000 KeV electrons, as described in O'Toole et al. 1993. A shows platelet granules (G) and mitochondria (M) whose boundaries are clearly defined by membranes. Nonetheless, the density of organelles is sufficiently high that positional relationships among them are hard to discern. B shows a slender projection from the margin of such a platelet. Its component microfilaments are evident (arrows). Images were provided by Eileen O'Toole (University of Colorado, Boulder, CO). Bars: (A) 200 nm; (B) 100 nm.
The problem of superposed detail in frozen hydrated samples has been tackled by using multiple tilted views and the mathematics of tomography to build 3-D images of small cells and isolated organelles (Grimm et al. 1998; for review see Baumeister et al. 1999; Nicastro et al. 2000). Fig. 2A and Fig. B show slices extracted from a tomographic reconstruction of the archaebacterium, Pyrodictium abyssi. Here, automation of data collection has been used to keep the electron dose-per-view to a minimum, and the principle of dose-fractionation (McEwen et al. 1995) has allowed multiple tilted views to be assembled into a single 3-D volume whose signal-to-noise ratio is defined approximately by the total dose for the series, rather than the dose/image (subject to the constraint that each image must show enough detail to permit its accurate alignment with other tilted views). This work shows tremendous promise, given both the nearly native state of the cells at the time they are imaged and the 3-D detail that is visible. Objects separated by only 6–8 nm are visible in the best of these cryotomograms. At this resolution some large protein complexes can be recognized by their structure alone, suggesting that cryotomography followed by appropriate image analysis may provide information about the localization of specific proteins (Fig. 2C and Fig. D). However, note that the promising results in the latter two figures have so far been achieved with frozen protein solutions, not with cytoplasm itself (Boehm et al. 2000).
Figure 2.


Electron tomographic reconstruction of frozen-hydrated specimens. A and B are two 12-nm slices extracted from a reconstruction of the archaebacterium, Pyrodictium abyssi (Baumeister et al. 1999). The distance between the slices is 90 nm. The boundary of the cell is evident, including both the plasma membrane and the “S-layer,” which is composed of ordered protein subunits. There are also crystalline structures in the cytoplasm (square arrays in A and B). Vesicles within the cell are evident. The dark circle in each image is a 250-nm latex sphere, which is apparently being internalized by endocytosis. Reprinted with permission from the author and Trends in Cell Biology. C displays a slice from a tomographic reconstruction of isolated, frozen-hydrated thermosomes, which are chaperonins with eightfold rotational symmetry. The proteins are arranged in a top view orientation. Image processing methods have been used to search for sites of correlations between the tomogram shown in C and a template generated from high resolution cryo-EM micrographs of the thermosome. Positions of strong correlation are marked by white crosses in D. This method for feature recognition holds promise as a way to identify the location of specific proteins in tomographic images of cytoplasm. Reprinted with permission of the authors and the Proceedings of the National Academy of Sciences. Bar, (B) 200 nm.
Technological Developments Have Opened New Possibilities, but There Are Still Many Challenges to Overcome
The emergence of EM tomography has depended on many technological advances. Examples include: (a) microscopes that operate at higher than conventional voltages. These reduce the deleterious impact of multiply scattered electrons on the quality of images from specimens thick enough to be suitable for tomography. (b) Field emission guns that are bright sources of comparatively coherent electrons. These increase the contrast and signal-to-noise in images of unstained specimens. (c) Eucentric stages that allow smooth tilting between images; these ease and speed the process of data collection. (d) Combinations of hardware and computer-facilitated controls that allow automated collection of the large numbers of tilted views necessary for tomographic reconstruction. These devices also minimize the beam damage to the region being recorded (Koster et al. 1997). Finally, there are indications that an electron energy loss filter, developed by several companies to remove electrons that were inelastically scattered by the specimen before an image is formed, can enhance the quality of images from thick, frozen-hydrated specimens (Grimm et al. 1997).
Although these technologies have led to significant progress in imaging frozen cells, there are still formidable problems to overcome before this approach will yield the most useful results. Both microscopes and cameras will have to be optimized, so every imaging electron is detected with high efficiency and negligible noise. This is a particular problem with higher electron energies (e.g., 300 KeV), where electron scattering and x-ray generation commonly reduce a camera's detector quantum efficiency (Downing and Hendrickson 1999). Even with improvements, though, the inherent radiation sensitivity of frozen hydrated samples will make it hard to push the resolution of cellular cryotomography as low as 2 nm. Specimen holders that maintain liquid helium temperatures and permit tilting about two orthogonal axes may provide improved data, but such big improvements in resolution are unlikely to be straight-forward. Nonetheless, this goal is worth significant effort, because at ∼2 nm one should be able to recognize the shape of many macromolecules, building on the pioneering work from the Baumeister laboratory (Max Plank Institute for Biochemistry, Martinsreid, Germany) (Fig. 2) to “dock” atomic resolution structures determined by crystallography or other methods into the lower resolution images available from EM (e.g., Wriggers et al. 2000). Such an advance might ultimately obviate labeling, e.g., by immuno-EM, because at least some large proteins could be recognized directly by their shapes. However, identifying small proteins will require improved imaging methods and probably novel ways of comparing cellular and molecular structures.
In another vein, cryotomography is now applicable only to cells that are small or thin. Cryomicrotomy of frozen hydrated cells will have to be improved, so specimen thickness can be controlled by the experimenter, not by the nature of the sample. Several laboratories are working on this and related issues (Studer and Gnaegi 2000; Dubochet and Sartori Blanc 2001), so progress can be expected. In addition, there are limitations to current methods for examining significant volumes from large frozen-hydrated cells. Even assuming that good cryosections can be cut, getting good SERIAL cryosections will present an additional hurdle. Moreover, the radiation sensitivity of frozen specimens, combined with current limitations on the size of the images that can practically be recorded, make it difficult to sample large areas from any one section. Thus, there will be serious difficulties in reconstructing volumes that correspond to a significant fraction of an entire frozen-hydrated, eukaryotic cell.
Images of Rapidly Frozen, Freeze-substituted Cells Reveal the Substructure of Extensive Cytoplasmic Volumes at ∼6-nm Resolution in 3-D
Given the complexities of cellular cryotomography, several labs have sought practical alternatives that would permit 3-D imaging of well-preserved samples that are structurally as close as possible to the living state, even if they are not frozen-hydrated. For such work, one would like samples that permit convenient microtomy for serial sections, exhibit good contrast, and are sufficiently robust in the electron beam that large sample areas can be imaged. Specimens prepared by rapid freezing followed by freeze-substitution fixation and embedding in plastic (RFFSE), fulfill these requirements and provide specimens suitable for detailed study of cells in 3-D.
3-D reconstruction of cellular samples has been accomplished for several years by tomography of chemically fixed, plastic-embedded samples stained with heavy metals (e.g., Olins et al. 1983; McEwen et al. 1986; Woodcock et al. 1991; Ladinsky et al. 1994; Moritz et al. 1995; Mannella et al. 1997; Martone et al. 1999; Harlow et al. 2001). However, the well-known limitations of chemical fixation for preserving cellular ultrastructure have limited the information that can be derived from such work. These problems have recently been addressed by combining the power of RFFSE with tomography to obtain 3-D views of cellular structures that should closely resemble the living state. The accuracy of these specimen preparation methods has been tested in three ways: (a) significant fractions (1/20–1/5) of rapidly frozen yeast cells are viable upon rewarming, showing that rapid freezing itself preserves the native structure of cells very well. (b) RFFSE samples have been compared with equivalent ones prepared by freeze-fracture EM, a method that visualizes replicas of fractured cellular surfaces formed by metal shadowing at very low temperatures. These studies have found no significant difference between the structures seen by the two methods (e.g., Linder and Staehelin 1979). (c) The images from samples prepared by RFFSE conform to expectations that are based on a broad range of knowledge about subcellular components: cytoskeletal fibers usually run straight; membrane profiles of the ER and of Golgi cisternae appear turgid and smooth; most vesicles are round; cytomatrix is even; even chromatin appears structured (McDonald and Morphew 1993; Ladinsky et al. 1999; Muller et al. 2000). Thus, studies that have melded RFFSE with tomography are likely to be showing us cellular structures that are essentially native, revealing organelle morphology in its normal context and in a situation where the parameters of cell physiology, like cell cycle stage, can be manipulated by the experimenter. Indeed, it is possible to freeze cells or parts of cells at well-defined times after a stimulus, allowing 3-D snapshots of cellular processes, even fast ones, like muscle contraction (Taylor et al. 1999).
This composite technology has been used to examine the architecture of mitotic kinetochores in mammalian cells (Mastronarde 1997; McEwen et al. 1998), the centrosomes of budding yeast (O'Toole et al. 1999), and the Golgi apparatus and associated organelles in mammalian cells (Ladinsky et al. 1999; Marsh et al. 2001). The results from mitotic systems show that the methods preserve not only spindle microtubules, but also the fine structure of microtubule ends and the organelles that initiate them (Fig. 3). These tomograms contain an impressive amount of 3-D detail. They even allow investigators to examine a given structure sliced computationally in several different orientations to get the most informative views for answering each specific question (Fig. 3 A). In addition, a highly asymmetric structure, like a spindle microtubule, can be sampled with a tomographic slice that exactly contains its axis, but that is only a fraction of the structure's thickness (Fig. 3, B–D). Such a feat is currently impossible by microtomy.
Figure 3.

A spindle pole body and its associated microtubules from the budding yeast, Saccharomyces cerevisiae (O'Toole et al. 1999). A shows a 3-nm slice from a tomographic reconstruction of one end of a forming mitotic spindle. Six layers of the spindle pole body are indicated by numbers (1, outer plaque [OP]; 2, inner layer 1 [IL1]; 3, inner layer 2 [IL2]; 4, central plaque [CP]; 5, inner plaque [IP]; 6, the minus ends of the spindle microtubules [MT]). The six small panels to the right of A are slices cut from the tomogram perpendicular to the slice shown on the left, and the placement of the slices corresponds to the positions of the labeled lines in A on the left. Order is quite evident in the arrangement of subunits in IL2. However, the shape of the plaque is distorted by the collapse of the section under the influence of the electron beam (see O'Toole et al. 1999 for details). B and C show minus ends of microtubules at IP and OP respectively (arrows). Both spindle and cytoplasmic microtubules are capped, though the shapes of the caps are not the same. D shows the plus ends of two spindle microtubules, revealing their flared shape (arrows). Reprinted with modifications and with permission from the authors and Molecular Biology of the Cell. Bars: (A) 50 nm; (D) 25 nm.
Application of the same techniques to the study of membranes in the Golgi apparatus has revealed several features of cisternal organization that had escaped detection by conventional EM (Fig. 4). For example, tomographic reconstruction of four serial, 1/4-μm sections has provided enough cellular volume to reveal continuity among all of the Golgi elements that bud clathrin; these elements lie at the transmost side of a Golgi stack (red object in Fig. 4 D; Ladinsky et al. 1999). Tubular projections, which are rarely seen in conventionally fixed and imaged material, form from the edges of several cisternae; these extend significant distances in either the cis- or transdirection. Such reconstructions have also revealed the pleomorphism of the membranous compartments that lie in noncompact regions of the Golgi region. Moreover, they have provided quantitative information about the numbers of different types of vesicles in particular cytoplasmic domains and on the proximity of membrane-bounded organelles with each other and with elements of the cytoskeleton.
Figure 4.
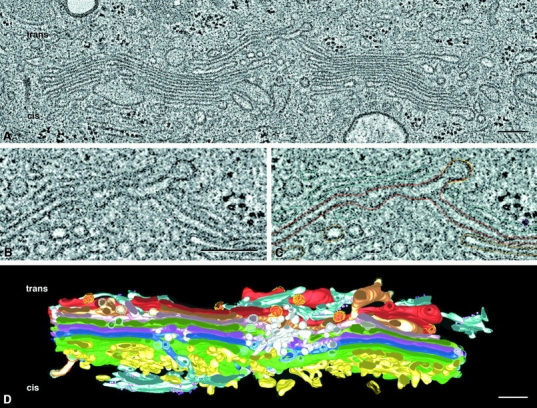
Tomographic reconstruction of a portion of the Golgi ribbon from a cultured normal rat kidney cell, fixed by plunge freezing and freeze substitution (Ladinsky et al. 1999). A shows one tomographic slice ∼4 nm thick from the 257 that comprise the entire reconstruction. The area shown is ∼3.8 × 1.9 μm, and the thickness of the whole tomogram (assembled from four serial 250-nm sections) is ∼1 μm. This volume is large enough that many features of Golgi morphology can be seen with greater clarity and detail than had previously been possible. B shows a small region from A, providing enough magnification to show that the two leaflets of a unit membrane can be resolved (∼6 nm). C shows the same area with graphic objects laid on to mark the positions of membranes in that slice. Similar modeling on every slice, followed by triangulation and surface rendering, permitted the construction of the model shown in D. The trans side of the Golgi apparatus is at the top, the cis at the bottom, and a noncompact region lies at the middle of the reconstruction. For details, see Ladinsky et al. 1999. Reprinted with permission from the authors and the Journal of Cell Biology. Bars, 250 nm.
An additional advantage of EM tomography is that many cellular structures are visualized at once with a single imaging technology. This is a significant difference from fluorescence light microscopy, where one sees only that which has been made fluorescent. Indeed, it is this feature of immunofluorescence that allows one to be satisfied with fixation protocols that preserve some cellular details very badly (Melan and Sluder 1992). EM tomography of well-fixed cells, on the other hand, reveals the relationships among diverse cellular structures at comparatively high resolution (Fig. 5). The stereo images presented here show a 1.2-μm slab of cytoplasm from a cultured mammalian cell. The cisternae of the Golgi region and the ER are displayed with all the nearby vesicles, microtubules, and ribosomes (Marsh et al. 2001). This view reveals just how crowded cytoplasm really is. However, note that even though the model in Fig. 5 looks densely packed with organelles, fully 65% of the volume modeled lies outside of organelles, allowing plenty of space for cytosol.
Figure 5.
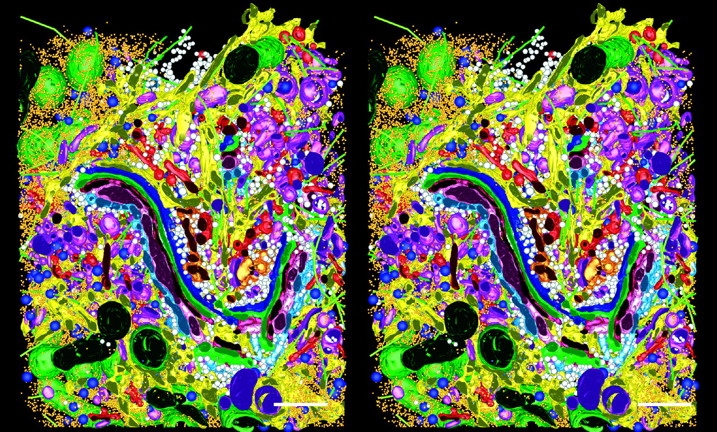
Stereo views of a model generated by tomographic reconstruction of a slab of cytoplasm from a cultured cell, strain HIT-T15. Three serial 400-nm sections were reconstructed by dual axis tomography, and the IMOD software package was used to model all visible objects within a volume 3.1 × 3.2 × 1.2 μm3 (see Marsh et al. 2001 for details). The model displays the Golgi complex in the context of surrounding organelles: ER, yellow; membrane-bound ribosomes, blue; free ribosomes, orange; MTs, bright green; dense core vesicles, bright blue; clathrin-negative vesicles, white; clathrin-positive compartments and vesicles, bright red; clathrin-negative compartments and vesicles, purple; mitochondria, dark green. Reprinted with permission from the authors and the Proceedings of the National Academy of Sciences. Bars, 500 nm.
Tomography of Cryofixed, Freeze-substituted, and Stained Specimens Is Currently Subject to Its Own Share of Limitations
For all its strengths, the composite method described above has limitations of its own that need to be considered. Plastic sections collapse under the action of an electron beam (Luther et al. 1988). Shrinkage in the section's plane is modest (5–10%), but along the direction of the beam it is 40–50%. Reduced temperatures provide some protection against this effect (Braunfeld et al. 1994), but in our hands there is some rapid initial collapse, even at low temperatures. Ice does not show this shrinkage, so if cryotomography can be made practical for large cells, this kind of sample deformation by the beam should be eliminated. Until that time, either we must correct for beam-induced distortions (Ladinsky et al. 1999; O'Toole et al. 1999) or simply live with them. There are also limitations to the resolution that can be expected from stained, plastic-embedded samples. Currently, these are ∼6 nm, probably set by the granularity of the stain after exposure to so much radiation and by the accuracy with which stain represents the positions and structures of underlying macromolecules. One fact that supports this resolution claim is that one can see two layers in a cytoplasmic membrane that is oriented appropriately relative to a tomographic slice (Fig. 4 B). However, one cannot resolve subunits in a microtubule wall, which are separated by 4–5 nm (Fig. 3 B). Improving on current resolution may require real breakthroughs in techniques for fixation, embedding, staining, and imaging at low dose.
Another current limitation of tomography is the difficulty of identifying particular macromolecules within the samples. When a protein assembles to form a characteristic shape, like tubulin or clathrin, it is easy to identify and localize in a tomogram. However, less idiosyncratic proteins are simply stain-binding regions of the sample. Postembedment antibody labeling of sections thick enough to merit tomography is unlikely to be useful, because immunoglobulins will not diffuse into the embedding plastic. Methods that remove the embedding resin tend to be harsh and therefore limit image resolution and reliability. Labeling before embedding usually requires cells to be “permeabilized” before antibody treatment, which results in some extraction of cytoplasm and the possibility of antigen movement. Therefore, such methods are intrinsically less reliable than ones that use optimal fixation (e.g., RFFSE). Developing methods that get identifying tags onto proteins or nucleic acids in samples suitable for tomography is a standing challenge.
Cellular Tomography Should Have Profound Effects on Future Directions of Cell Biology
It is encouraging how much can be seen with the new kinds of cellular EM. The cryotomograms of small cells are sufficiently promising to motivate a real attack on ways to get comparably thin samples from any frozen cell or tissue. The tomograms of RFFSE samples already show enough detail and quality to prompt serious study in their own right, and it is realistic to think about using these methods to view major cellular subsystems, like an entire nucleus or a Golgi complex. Results already obtained show that many structural details can be characterized, like the morphology of microtubule ends at their sites of cellular attachment. Future work should display changes in these structures as a function of interphase physiology or mitotic state. Volumes now under study comprise >10 μm3, permitting the description of extensive tubular projections from individual Golgi cisternae and of the endoplasmic reticulum as it penetrates the Golgi region and lines up with regular Golgi elements. Future work should elucidate the connectivity in 3-D of all the components of the endocytic pathway and their structural relationships with each Golgi compartment. One can even envision the reconstruction of an entire eukaryotic cell.
In spite of its far-reaching value, EM tomography is no longer oppressively time consuming. Using a modern microscope equipped with a digital camera, a good eucentric tilting stage, and software to control data collection, a skilled operator can collect the ∼140 images required for a complete tilt series of ∼70°C in <2 h. If one uses montages of digital data to enlarge the area per image, the time increases somewhat, and dual axis tomography (Mastronarde 1997) essentially doubles the time investment. Nonetheless, in our laboratory we are routinely collecting data for two and more tomograms per day. Comparatively modest computers, like a UNIX workstation or a fast PC, can align these images and calculate a tomogram in another few hours, so the preparation of a tomogram is now surprisingly fast. Using montages and serial thick sections, it should be feasible to image the entirety of a small eukaryote, like a budding yeast, in 3-D at ∼6-nm resolution (∼3 × 1010 bytes of image data) in <1 wk. However, our lab has concentrated on organelles and cellular subsystems in order to retain a functional focus for structural work.
Analyzing tomographic data is more time consuming than tomogram construction. Models like the ones shown in Fig. 4 and Fig. 5 have taken months to prepare, so there is significant room for improvement in the ways that features of interest in 3-D images are abstracted from their backgrounds (a process called segmentation). The coordination of computer-assisted feature recognition (e.g., Boehm et al. 2000) with hand–eye efforts by a trained biologist (e.g., Marsh et al. 2001) offer many avenues for productive work, and we anticipate that this aspect of structural cell biology will soon move much faster. Of more concern is the issue of recognizing macromolecules of interest within cellular tomograms. There is great promise in the approach under development by Baumeister's group, but the utility of this approach for smaller proteins in real cytoplasm remains to be demonstrated. Our lab and others are working to develop labeling methods that will be suitable for thick section EM reconstruction, but this work is still in its infancy. The concept of 3-D maps of specific antigens, localized at ∼6 nm in 3-D, is certainly attractive when thinking about data that could discriminate between competing models for complex cellular events, like mitosis, secretion, and endocytosis.
It seems likely that EM tomography will provide an important complement to the beautiful and informative images that are now emerging from work on live cells based on a range of fluorescent stains, including protein chimeras with green or other fluorescent proteins (Griffiths 2001). The spatial resolution of EM tomography is ∼40-fold better than most confocal microscopy, allowing the visualization of detail that is otherwise vague. When such methods are applied to cytoplasmic mechanisms, like endocytosis, they should reveal the connectivity among all relevant structural compartments. Applied to chromatin, they may identify aspects of higher order DNA structure that help to control gene expression. When used to view nuclear pore complexes in situ, aspects of protein and RNA traffic that have previously escaped our notice may become obvious. Host–pathogen relationships may also appear in a new light, and some cellular subtleties of developmental processes may emerge from the morass of molecular detail in which they are currently buried. We see a brave new world emerging from the combination of modern EM with the rapidly advancing methods for light microscopy. The resulting data should help us to understand the context for functional genomics and elucidate the continuum of structural order that bridges from inanimate molecules to the order of the living cell.
Acknowledgments
I thank Andrew Staehelin, Eileen O'Toole, and Daniela Nicastro for critical readings of this manuscript and members of the Boulder Laboratory for the 3-D structure and for many helpful discussions.
Work from our lab that is reviewed here was supported in part by grants from the National Institutes of Health (RR00592 and GM61306). J.R. McIntosh is a research professor of the American Cancer Society.
Footnotes
Abbreviations used in this paper: 3-D, three-dimensional; RFFSE, rapid freezing followed by freeze-substitution fixation and embedding in plastic.
References
- Agrawal R.K., Spahn C.M., Penczek P., Grassucci R.A., Nierhaus K.H., Frank J. Visualization of tRNA movements on the Escherichia coli 70S ribosome during the elongation cycle. J. Cell Biol. 2000;150:447–460. doi: 10.1083/jcb.150.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister W., Grimm R., Walz J. Electron tomography of molecules and cells. Trends Cell Biol. 1999;9:81–85. doi: 10.1016/s0962-8924(98)01423-8. [DOI] [PubMed] [Google Scholar]
- Boehm J., Frangakis A.S., Hegerl R., Nickell S., Typke D., Baumeister W. Toward detecting and identifying macromolecules in a cellular contexttemplate matching applied to electron tomograms. Proc. Natl. Acad. Sci. USA. 2000;97:14245–14250. doi: 10.1073/pnas.230282097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher B., Wynne S.A., Crowther R.A. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature. 1997;386:88–91. doi: 10.1038/386088a0. [DOI] [PubMed] [Google Scholar]
- Braunfeld M.B., Koster A.J., Sedat J.W., Agard D.A. Cryo automated electron tomographytowards high-resolution reconstructions of plastic-embedded structures. J. Microsc. 1994;174:75–84. doi: 10.1111/j.1365-2818.1994.tb03451.x. [DOI] [PubMed] [Google Scholar]
- Downing K.H., Hendrickson F.M. Performance of a 2k CCD camera designed for electron crystallography at 400 kV. Ultramicroscopy. 1999;75:215–233. doi: 10.1016/s0304-3991(98)00065-5. [DOI] [PubMed] [Google Scholar]
- Dubochet J., Sartori Blanc N. The cell in absence of aggregation artifacts. Micron. 2001;32:91–99. doi: 10.1016/s0968-4328(00)00026-3. [DOI] [PubMed] [Google Scholar]
- Frank J. Approaches to large-scale structures. Curr. Opin. Struct. Biol. 1995;5:194–201. doi: 10.1016/0959-440x(95)80075-1. [DOI] [PubMed] [Google Scholar]
- Gabashvili I.S., Agrawal R.K., Spahn C.M., Grassucci R.A., Svergun D.I., Frank J., Penczek P. Solution structure of the E. coli 70S ribosome at 11.5 A resolution. Cell. 2000;100:537–549. doi: 10.1016/s0092-8674(00)80690-x. [DOI] [PubMed] [Google Scholar]
- Gilkey J.C., Staehelin L.A. Advances in ultrarapid freezing for the preservation of cellular ultrastructure. J. Electron Microsc. Tech. 1986;3:177–210. [Google Scholar]
- Griffiths G. Bringing electron microscopy back into focus for cell biology. Trends Cell Biol. 2001;11:153–154. doi: 10.1016/s0962-8924(01)01949-3. [DOI] [PubMed] [Google Scholar]
- Grimm R., Barmann M., Hackl W., Typke D., Sackmann E., Baumeister W. Energy filtered electron tomography of ice-embedded actin and vesicles. Biophys. J. 1997;72:482–489. doi: 10.1016/S0006-3495(97)78689-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm R., Singh H., Rachel R., Typke D., Zillig W., Baumeister W. Electron tomography of ice-embedded prokaryotic cells. Biophys. J. 1998;74:1031–1042. doi: 10.1016/S0006-3495(98)74028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow M.L., Ress D., Stoschek A., Marshall R.M., McMahan U.J. The architecture of active zone material at the frog's neuromuscular junction. Nature. 2001;409:479–484. doi: 10.1038/35054000. [DOI] [PubMed] [Google Scholar]
- Heuser J.E., Reese T.S. Structural changes after transmitter release at the frog neuromuscular junction. J. Cell Biol. 1981;88:564–580. doi: 10.1083/jcb.88.3.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K., Lockhart A., Cross R.A., Amos L.A. Nucleotide-dependent angular change in kinesin motor domain bound to tubulin. Nature. 1995;376:277–279. doi: 10.1038/376277a0. [DOI] [PubMed] [Google Scholar]
- Hoenger A., Sack S., Thormahlen M., Marx A., Muller J., Gross H., Mandelkow E. Image reconstructions of microtubules decorated with monomeric and dimeric kinesinscomparison with x-ray structure and implications for motility. J. Cell Biol. 1998;141:419–430. doi: 10.1083/jcb.141.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster A.J., Grimm R., Typke D., Hegerl R., Stoschek A., Walz J., Baumeister W. Perspectives of molecular and cellular electron tomography. J. Struct. Biol. 1997;120:276–308. doi: 10.1006/jsbi.1997.3933. [DOI] [PubMed] [Google Scholar]
- Ladinsky M.S., Kremer J.R., Furcinitti P.S., McIntosh J.R., Howell K.E. HVEM tomography of the trans-Golgi networkstructural insights and identification of a lace-like vesicle coat. J. Cell Biol. 1994;127:29–38. doi: 10.1083/jcb.127.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky M.S., Mastronarde D.N., McIntosh J.R., Howell K.E., Staehelin L.A. Golgi structure in three dimensionsfunctional insights from the normal rat kidney cell. J. Cell Biol. 1999;144:1135–1149. doi: 10.1083/jcb.144.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder J.C., Staehelin L.A. A novel model for fluid secretion by the trypanosomatid contractile vacuole apparatus. J. Cell Biol. 1979;83:371–382. doi: 10.1083/jcb.83.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther P.K., Lawrence M.C., Crowther R.A. A method for monitoring the collapse of plastic sections as a function of electron dose. Ultramicroscopy. 1988;24:7–18. doi: 10.1016/0304-3991(88)90322-1. [DOI] [PubMed] [Google Scholar]
- Malide D., Ramm G., Cushman S.W., Slot J.W. Immunoelectron microscopic evidence that GLUT4 translocation explains the stimulation of glucose transport in isolated rat white adipose cells. J. Cell Sci. 2000;23:4203–4210. doi: 10.1242/jcs.113.23.4203. [DOI] [PubMed] [Google Scholar]
- Mandelkow E.M., Mandelkow E., Milligan R.A. Microtubule dynamics and microtubule capsa time-resolved cryo- electron microscopy study. J. Cell Biol. 1991;114:977–991. doi: 10.1083/jcb.114.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannella C.A., Marko M., Buttle K. Reconsidering mitochondrial structurenew views of an old organelle. Trends. Biochem. Sci. 1997;22:37–38. doi: 10.1016/s0968-0004(96)30050-9. [DOI] [PubMed] [Google Scholar]
- Marsh B.J., Mastronarde D.N., Buttle K.F., Havell K.E., McIntosh J.R. Organellar relationships in the Golgi region of the pancreatic beta cell line, HIT-T15, visualized by high resolution electron tomography. Proc. Natl. Acad. Sci. USA. 2001;98:2399–2406. doi: 10.1073/pnas.051631998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martone M.E., Jones Y.Z., Young S.J., Ellisman M.H., Zivin J.A., Hu B.R. Modification of postsynaptic densities after transient cerebral ischemiaa quantitative and three-dimensional ultrastructural study. J. Neurosci. 1999;19:1988–1997. doi: 10.1523/JNEUROSCI.19-06-01988.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde D.N. Dual-axis tomographyan approach with alignment methods that preserve resolution. J. Struct. Biol. 1997;120:343–352. doi: 10.1006/jsbi.1997.3919. [DOI] [PubMed] [Google Scholar]
- Matadeen R., Patwardhan A., Gowen B., Orlova E.V., Pape T., Cuff M., Mueller F., Brimacombe R., van Heel M. The Escherichia coli large ribosomal subunit at 7.5 A resolution. Structure Fold Des. 1999;7:1575–1583. doi: 10.1016/s0969-2126(00)88348-3. [DOI] [PubMed] [Google Scholar]
- McDonald K., Morphew M.K. Improved preservation of ultrastructure in difficult-to-fix organisms by high pressure freezing and freeze substitutionI. Drosophila melanogaster and Strongylocentrotus purpuratus embryos. Microsc. Res. Tech. 1993;24:465–473. doi: 10.1002/jemt.1070240603. [DOI] [PubMed] [Google Scholar]
- McDowell A., Gruenberg P., Remisch T., Griffiths G. The structure of organelles of the endocytic pathway in hydrated cryosections of cultured cells. Euro. J. Cell Biol. 1989;49:289–304. [PubMed] [Google Scholar]
- McEwen B.F., Radermacher M., Rieder C.L., Frank J. Tomographic three-dimensional reconstruction of cilia ultrastructure from thick sections. Proc. Natl. Acad. Sci. USA. 1986;83:9040–9044. doi: 10.1073/pnas.83.23.9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen B.F., Downing K.H., Glaeser R.M. The relevance of dose-fractionation in tomography of radiation- sensitive specimens. Ultramicroscopy. 1995;60:357–373. doi: 10.1016/0304-3991(95)00082-8. [DOI] [PubMed] [Google Scholar]
- McEwen B.F., Hsieh C.E., Mattheyses A.L., Rieder C.L. A new look at kinetochore structure in vertebrate somatic cells using high-pressure freezing and freeze substitution. Chromosoma. 1998;107:366–375. doi: 10.1007/s004120050320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melan M.S., Sluder G. Redistribution and differential extraction of soluble proteins in permeabilized cell culturesimplications for fluorescence microscopy. J. Cell Sci. 1992;101:731–743. doi: 10.1242/jcs.101.4.731. [DOI] [PubMed] [Google Scholar]
- Moritz M., Braunfeld M.B., Fung J.C., Sedat J.W., Alberts B.M., Agard D.A. Three-dimensional structural characterization of centrosomes from early Drosophila embryos. J. Cell Biol. 1995;130:1149–1159. doi: 10.1083/jcb.130.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W.H., Koster A.J., Humbel B.M., Ziese U., Verkleij A.J., van Aelst A.C., van der Krift T.P., Montijn R.C., Boekhout T. Automated electron tomography of the septal pore cap in Rhizoctonia solani . J. Struct. Biol. 2000;131:10–18. doi: 10.1006/jsbi.2000.4243. [DOI] [PubMed] [Google Scholar]
- Nicastro D., Frangakis A.S., Typke D., Baumeister W. Cryo-electron tomography of neurospora mitochondria. J. Struct. Biol. 2000;129:48–56. doi: 10.1006/jsbi.1999.4204. [DOI] [PubMed] [Google Scholar]
- Nogales E., Grigorieff N. Molecular machinesputting the pieces together. J. Cell Biol. 2001;52:F1–F10. doi: 10.1083/jcb.152.1.f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olins D.E., Olins A.L., Levy H.A., Durfee R.C., Margle S.M., Tinnel E.P., Dover S.D. Electron microscope tomographytranscription in three dimensions. Science. 1983;220:498–500. doi: 10.1126/science.6836293. [DOI] [PubMed] [Google Scholar]
- O'Toole E., Wray G., Kremer J., McIntosh J.R. High voltage cryomicroscopy of human blood platelets. J. Struct. Biol. 1993;110:55–66. doi: 10.1006/jsbi.1993.1004. [DOI] [PubMed] [Google Scholar]
- O'Toole E.T., Winey M., McIntosh J.R. High-voltage electron tomography of spindle pole bodies and early mitotic spindles in the yeast Saccharomyces cerevisiae . Mol. Biol. Cell. 1999;10:2017–2031. doi: 10.1091/mbc.10.6.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice S., Lin A.W., Safer D., Hart C.L., Naber N., Carragher B.O., Cain S.M., Pechatnikova E., Wilson-Kubalek E.M., Whittaker M. A structural change in the kinesin motor protein that drives motility. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- Richter K., Gnagi H., Dubochet J. A model for cryosectioning based on the morphology of vitrified ultrathin sections. J. Microsc. 1991;163:19–28. doi: 10.1111/j.1365-2818.1991.tb03156.x. [DOI] [PubMed] [Google Scholar]
- Sartori N., Richeter K., Dubochet J. Vitrification depth can be increased more than 10-fold by high-pressure freezing. J. Microsc. 1993;172:55–61. [Google Scholar]
- Shimoni E., Muller M. On optimizing high-pressure freezingfrom heat transfer theory to a new microbiopsy device. J. Microsc. 1998;192:236–247. doi: 10.1046/j.1365-2818.1998.00389.x. [DOI] [PubMed] [Google Scholar]
- Stark H., Rodnina M.V., Wieden H.J., van Heel M., Wintermeyer W. Large-scale movement of elongation factor G and extensive conformational change of the ribosome during translocation. Cell. 2000;100:301–309. doi: 10.1016/s0092-8674(00)80666-2. [DOI] [PubMed] [Google Scholar]
- Steinbrecht R.A., Muller M. Freeze-substitution and freeze drying. In: Steinbrecht R.A., Zierold K., editors. Cryotechniques in Biological Electron Microscopy. Springer-Verlag; Berlin: 1987. pp. 149–172. [Google Scholar]
- Studer D., Gnaegi H. Minimal compression of ultrathin sections with use of an oscillating diamond knife. J. Microsc. 2000;197:94–100. doi: 10.1046/j.1365-2818.2000.00638.x. [DOI] [PubMed] [Google Scholar]
- Taylor K.A., Schmitz H., Reedy M.C., Goldman Y.E., Franzini-Armstrong C., Sasaki H., Tregear R.T., Poole K., Lucaveche C., Edwards R.J. Tomographic 3D reconstruction of quick-frozen, Ca2+-activated contracting insect flight muscle. Cell. 1999;99:421–431. doi: 10.1016/s0092-8674(00)81528-7. [DOI] [PubMed] [Google Scholar]
- Tokuyasu K.T. Immunochemistry on ultrathin frozen sections. Histochem. J. 1980;12:381–403. doi: 10.1007/BF01011956. [DOI] [PubMed] [Google Scholar]
- Vale R.D., Milligan R.A. The way things movelooking under the hood of molecular motor proteins. Science. 2000;288:88–95. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- Whittaker M., Wilson-Kubalek E.M., Smith J.E., Faust L., Milligan R.A., Sweeney H.L. A 35-A movement of smooth muscle myosin on ADP release. Nature. 1995;378:748–751. doi: 10.1038/378748a0. [DOI] [PubMed] [Google Scholar]
- Woodcock C.L., McEwen B.F., Frank J. Ultrastructure of chromatin. II. Three-dimensional. J. Cell Sci. 1991;99:107–114. doi: 10.1242/jcs.99.1.107. [DOI] [PubMed] [Google Scholar]
- Wriggers W., Agrawal R.K., Drew D.L., McCammon A., Frank J. Domain motions of EF-G bound to the 70S ribosomeinsights from a hand- shaking between multi-resolution structures. Biophys. J. 2000;79:1670–1678. doi: 10.1016/S0006-3495(00)76416-2. [DOI] [PMC free article] [PubMed] [Google Scholar]