

Abstract
The microtubule-binding integral 63 kD cytoskeleton-linking membrane protein (CLIMP-63; former name, p63) of the rough endoplasmic reticulum (ER) is excluded from the nuclear envelope. We studied the mechanism underlying this ER subdomain–specific localization by mutagenesis and structural analysis. Deleting the luminal but not cytosolic segment of CLIMP-63 abrogated subdomain-specific localization, as visualized by confocal microscopy in living cells and by immunoelectron microscopy using ultrathin cryosections. Photobleaching/recovery analysis revealed that the luminal segment determines restricted diffusion and immobility of the protein. The recombinant full-length luminal segment of CLIMP-63 formed α-helical 91-nm long rod-like structures as evident by circular dichroism spectroscopy and electron microscopy. In the analytical ultracentrifuge, the luminal segment sedimented at 25.7 S, indicating large complexes. The complexes most likely arose by electrostatic interactions of individual highly charged coiled coils. The findings indicate that the luminal segment of CLIMP-63 is necessary and sufficient for oligomerization into α-helical complexes that prevent nuclear envelope localization. Concentration of CLIMP-63 into patches may enhance microtubule binding on the cytosolic side and contribute to ER morphology by the formation of a protein scaffold in the lumen of the ER.
Keywords: coiled coils, CLIMP-63 (p63), endoplasmic reticulum, green fluorescent protein, microtubule-binding membrane protein
Introduction
The characteristic reticular structure of the ER, with its interconnected tubules and cisternae, is unique among the membrane compartments in the cell. Classically, the ER is subdivided into the rough ER, including the nuclear envelope, and the less-defined smooth ER. The rough ER is predominantly involved in synthesis, modification, and degradation of proteins, and in lipid biogenesis in all cells, whereas the smooth ER is particularly prominent in cells dealing with steroid precursor synthesis or biotransformation of endogenous and exogenous lipophilic compounds. The rough ER can be further subdivided into a peripheral reticular domain and the nuclear envelope. Additional compartmentalization of the ER may exist (Deschuyteneer et al. 1988; Vertel et al. 1989; Ellinger and Pavelka 1992; Baumann 1998), but has been poorly explored (for reviews see Sitia and Meldolesi 1992; Hauri and Schweizer 1997).
Most rough ER membrane proteins are present in both the reticular part of the rough ER and the nuclear envelope. This is due to physical continuity of these two subdomains allowing for uninhibited protein diffusion. However, there are exceptions to this rule. A few membrane proteins are exclusively localized to the nuclear envelope (Bastos et al. 1995). They are either associated with the nuclear pores or with lamins (Stuurman et al. 1998). Nuclear envelope–restricted proteins are primarily involved in bidirectional traffic between cytoplasm and nucleoplasm (Mattaj and Englmeier 1998; Adam 1999; Talcott and Moore 1999), and the maintenance of the architecture of the nucleoplasm (Mancini et al. 1996). Other types of proteins may function in nuclear envelope–specific signaling (Csermely et al. 1995), sterol synthesis (Silve et al. 1998), unfolded protein response, and ER overload response (Chapman et al. 1998; Sidrauski et al. 1998; Pahl 1999).
In marked contrast to these examples, the ER 63 kD cytoskeleton-linking membrane protein (CLIMP-63; previous name, p63) is restricted to the reticular part of the rough ER and excluded from the nuclear envelope (Klopfenstein et al. 1998). CLIMP-63 is a 63 kD nonglycosylated type II integral ER membrane protein with an extracytoplasmic segment of 474 amino acids and an NH2-terminal cytoplasmic segment of 106 amino acids (Schweizer et al. 1993a,Schweizer et al. 1993b, Schweizer et al. 1994, Schweizer et al. 1995b). The cytoplasmic segment binds to microtubules (Klopfenstein et al. 1998). CLIMP-63 is reversibly palmitoylated during mitosis and under conditions that interfere with intracellular protein transport (Mundy and Warren 1992; Schweizer et al. 1993b; Mundy 1995). Upon overexpression, CLIMP-63 can arrange the ER into bundled membrane tubules (Schweizer et al. 1993a, Schweizer et al. 1995a). This morphological change occurs in the absence of a functional microtubule-binding cytoplasmic segment, suggesting that the rearrangement is due to other parts of CLIMP-63. The molecular basis for the rearrangement may lie in the known feature of CLIMP-63 to form aggregates in vitro (Schweizer et al. 1994).
In this study we have tested the possibility that the restricted localization of CLIMP-63 in the ER is mediated by its luminal segment, which is predicted to fold into an extensive coiled coil. The α-helical coiled coil is the most frequently encountered motif involved in subunit oligomerization of multimeric proteins. In the secretory pathway, for example, coiled coil interactions play a major role in vesicular transport by mediating the formation of SNARE complexes (Skehel and Wiley 1998) and vesicle tethering (Barr et al. 1998; Waters and Pfeffer 1999). Coiled coils typically consist of two to five right-handed amphipatic α-helices which coil around each other to form a slightly left-handed supercoil (Lupas 1996a; Kammerer 1997; Kohn et al. 1997). The portion of the protein involved in coiled coil interaction is typically characterized by heptad repeats of seven amino acids designated a–g in alphabetical order (Sodek et al. 1972; McLachlan and Stewart 1975). The so-called 3,4-hydrophobic heptad repeats display apolar residues in positions a and d, whereas polar and charged residues are frequently found at the other positions. Although many of the predicted coiled coil sequences are relatively short in length (20–60 amino acids), longer heptad repeat segments are found in structural proteins, such as myosin or tropomyosin, in the microtubule-based motor kinesin (Lupas 1996a) and in vesicle-tethering Golgi proteins such as giantin (Linstedt and Hauri 1993; Sönnichsen et al. 1998) and GM130 (Shorter and Warren 1999).
To probe for a possible function of the luminal segment of CLIMP-63 in subdomain localization, we generated several green fluorescent protein (GFP)-tagged mutants and studied their localization, diffusion, and mobility in living cells. This analysis was complemented by in vitro studies on the structure and oligomerization state of the luminal segment. The results suggest that the luminal segment immobilizes CLIMP-63 in the reticular subdomain of the ER by coiled coil–based homooligomerization into large complexes. These CLIMP-63 complexes may facilitate microtubule binding and contribute to ER morphology.
Materials and Methods
Antibodies
The following primary antibodies were used: mAb G1/296 against CLIMP-63 (Schweizer et al. 1993a), an mAb against the ER marker B cell antigen receptor–associated protein of M-31 (BAP31) (Klumperman et al. 1998), an mAb against GFP (Boehringer), and a rabbit antibody against GFP (provided by Graham Warren, Yale University, New Haven, CT). Secondary antibodies were: TRITC goat anti–mouse (ICN Biomedicals) and HRP rabbit anti–mouse (Dako).
Cell Culture and Transfection
COS cells were cultured in DME supplemented with 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 μg/ml fungizone at 37°C according to standard techniques. The cells were transiently transfected using the DEAE-dextran method (Cullen 1987).
Recombinant DNAs and Expression of Recombinant Protein
CLIMP-63 cDNA was mutated using standard PCR protocols. GFP (CLONTECH Laboratories, Inc.) was fused to wild-type (wt)-pECE-CLIMP-63 at the COOH terminus by ligating GFP cDNA, followed by a TAA stop codon into an additionally inserted NheI-ClaI site of the sequence 5′-GTA TCG ATG GAA CTA GCT AGC TAA-3′ at nucleotide position 1,888, thereby coding for an extra serine between CLIMP-63 and GFP. ΔLuminal-GFP was constructed by insertion of a PCR GFP cDNA fragment followed by TAA stop codon into the same linker sequence at position 486. CLONTECH Laboratories, Inc. vector pEGFP-C1 was used for the constructs in which the cytoplasmic segment was replaced by GFP. A PCR-amplified cDNA fragment encoding for amino acids 101–601, followed by a TAA stop codon, were ligated as a BglII-EcoRI insert into the polylinker site of the vector, resulting in the construct GFP-Δcytoplasmic. For the construction of the luminal deletion mutants, GFP-Δcytoplasmic was used as a template. Unique restrictions sites BamHI, HindIII, ApaLI, and BssHII were used in double digests with XbaI to cut out respective cDNA fragments and replaced by hybridized linker oligonucleotides of the sequence 5′-G ATC CAT TAA TAA GAT CTT-3′ for Δ218–601 (BamHI), 5′-AG CTT TAA TAA GAT CTT-3′ for Δ354–601 (HindIII), 5′-C CTG TAA TAA GAT CTT-3′ for Δ428–601 (BssHII), and 5′-CG CGC TAA TAA GAT CTT-3′ for Δ511–601 (ApaLI). Δ296–492 was constructed by using two in-frame SacI sites to remove the cDNA fragment corresponding to amino acids 246–492 and religation of the vector. The double luminal segment construct was obtained by inserting a cDNA fragment encoding amino acids 101–601 and an in-frame EcoRI site coding for E602F603 into the polylinker site of the vector. A second insert encoding the luminal segment with a COOH-terminal TAA stop codon was ligated into the EcoRI site. Construct Δ132–354 was obtained by inserting a HindIII site at nucleotide position 478 by the Quickchange kit (Stratagene), removing the corresponding fragment 132–354 with HindIII, and self-ligation of the vector. GFP fusions to cytochrome P450, Sec61β, and vesicular stomatitis virus glycoprotein (VSV-G) were provided by Byron Kemper (University of Illinois, Urbana, IL), Jamie White, and Rainer Pepperkok (both from the European Molecular Biology Laboratory, Heidelberg, Germany), respectively. A His6-tagged construct encompassing amino acids 127–601, 127–469, 127–268, and 268–459 of the luminal segment of CLIMP-63 was obtained by PCR amplification of the corresponding cDNA sequence using an upstream primer with a BglII and a downstream primer with two TAA stop codons, followed by an EcoRI restriction site. All of the constructs were confirmed by DNA sequencing. The resulting fragments were cloned into the BamHI-EcoRI site of the Escherichia coli expression vector pHis based on the vector pET-15b (Novagen, Inc.). The recombinant His6-tagged polypeptide was expressed in E. coli strain BL21 (DE3) and purified under denaturing conditions by Ni2 +-agarose affinity chromatography according to the manufacturer (QIAGEN). The thrombin cleavage site was used to remove the His6 tag, leaving two additional amino acids, Gly-Ser, at the NH2 terminus.
In Vitro Aggregation Assay
In vitro aggregation was analyzed as described in Schweizer et al. 1994.
EM
Purified proteins encompassing the luminal segment of CLIMP-63 were adsorbed to carbon-coated parlodion films that were mounted on electron microscope grids and rendered hydrophilic by glow discharge at low pressure in air. After washing with H2O, the samples were negatively stained with 0.75% uranyl formate, pH 4.2. Images were recorded on film (SO-163; Kodak) at a nominal magnification of 20,000 or 50,000× under low dose conditions using an LEO 910 transmission electron microscope operated at 80 kV. For immuno-EM cells were fixed in a mixture of 2% formaldehyde and 0.2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. Preparation of ultrathin cryosections and subsequent immunogold labeling according to the protein A method was performed as described (Slot et al. 1991; Klumperman et al. 1998).
For the calculation of labeling densities, grids were randomly scanned for transfected cells having a visible nucleus. Two or three pictures were taken per cell at a magnification of 15,000×. For each transfectant, 10 cells were sampled as follows. The pictures were printed at a magnification of 40,500× and overlayed with a transparent foil having horizontal and vertical lines at a distance of 1 cm. Gold particles were then counted over the nuclear envelope and the ER. All gold particles within a distance of 20 nm from the membrane were ascribed to that membrane. The length of the sampled membranes was established by counting the number of intersections with the lines on the overlay (Weibel 1979). For the nuclear envelope, both the outer and the inner membranes were taken into account. Counting was performed in a blind experiment, with the identity of the cells under study unknown.
Confocal Laser Scanning Microscopy (CLSM) and FRAP
Conventional CLSM images were acquired on a microscope (TCS NT; Leica). Transiently transfected COS cells were trypsinized 24 h after transfection and seeded into 8-well multi chamber slides. 19–20 h later the cells were fixed with 3% paraformaldehyde in PBS, permeabilized with PBS/0.1% saponin, and processed for immunofluorescence microscopy (Schweizer et al. 1993a). The specimens were examined with 0.28-μm optical sections. Corrections of bleed through in double-labeling experiments were done according to the recommendations of the manufacturer and were tested in single-labeling experiments. FRAP was imaged on a confocal microscope (LSM510; ZEISS). For FRAP experiments (Lippincott-Schwartz et al. 1999), transiently transfected COS cells have been grown on 15-mm coverslips and were examined 40–64 h after transfection. Cells were imaged with a 63× objective (NA 1.4). With an excitation wavelength of 488 nm and a long-pass LP505 filter, images were recorded with a laser intensity of 200–1,000-fold lower than the maximal laser power available for the photobleach. The region of interest (a stripe of w = 2 μm width spanning the entire cell) was bleached for 2–4 s and images during the recovery phase were taken every 0.5–4 s. An overall loss of fluorescence due to the bleaching of the cell did not exceed 20%. Control paraformaldehyde fixed cells showed no recovery in the bleached region of interest. The mean pixel values in the region of interest were background subtracted and fluorescence relative to the prebleach intensities was fitted to the following formula for one-dimensional diffusion (Lippincott-Schwartz et al. 1999) using the computer program Mathematica 3.0:
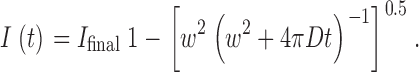 |
1 |
Diffusion coefficient (D) and the amount of mobile fraction (I final) can be directly obtained from the fit. I is a percentage of fluorescent intensity versus the prebleach intensity; w, stripe width in μm; t, time in seconds.
Circular Dichroism (CD) Spectroscopy
Far-UV CD spectroscopy and thermal unfolding profiles were recorded on a Cary 61 spectropolarimeter equipped with a temperature-controlled quartz cell of 0.1-cm path length as described by (Kammerer et al. 1998). A ramping rate of 1°C/min was used for all experiments.
Analytical Ultracentrifugation (AUC)
Purified polypeptides (1 mg/ml) were analyzed by AUC in 20 mM Tris, pH 8.0, 500 mM NaCl, and 1–3 M urea depending on the experiment. Sedimentation velocity and sedimentation equilibrium experiments were carried out in an analytical ultracentrifuge (model E; Beckman Coulter) equipped with interference and schlieren optics and in an ultracentrifuge (model XLA; Beckman Coulter) equipped with absorption optics. Sedimentation velocity was performed at 24,000 rpm at 20°C in a single sector 12-mm cell with schlieren optics. Equilibrium sedimentation was performed using interference optics (5,200 rpm at 20°C in a 12-mm double sector cell) as well as absorption optics (scanned at 4,500 rpm at 20°C at 283 nm). Results of equilibrium sedimentation were analyzed by using a floating baseline computer program with adjusted baseline absorbance (or zero fringe) to obtain lnA versus r2 (A, the absorbance or the fringe shift in equilibrium sedimentation performed with absorption optics or interference optics, respectively; r, the radial distance). A partial specific volume of 0.73 cm3/g and a solution density of 1.03 g/cm3 were used; for the conversion of s′ to s20w, the solution viscosity was taken as 1.31 centipoise.
Secondary Structure Prediction
Secondary structure prediction was done on a DEC α-computer (UNIX v4.0) using the program COILS2 (Lupas et al. 1991; Lupas 1996b) or COILSCAN of the Wisconsin Package Version 10.0 (Genetics Computer Group).
Results
The Luminal Segment of CLIMP-63 Is Required for Exclusion from the Nuclear Envelope and Confers Immobility in the ER
To explore the function of the luminal segment of CLIMP-63, a mutant lacking this part was generated and compared with the wild-type protein. For reasons of detection, a GFP tag was fused to the COOH terminus of the truncated and wild-type CLIMP-63, resulting in the constructs Δlumen-GFP and wt-GFP, respectively (Fig. 1 A). Because CLIMP-63 is a type II membrane protein, the GFP tag of these constructs is exposed on the luminal side of the ER. Immunofluorescence microscopy of COS cells transfected with wt-GFP showed ER-like staining. No fluorescent ring was seen around the nucleus, indicating that wt-GFP was excluded from the nuclear envelope (Fig. 2, A–C) in the same way as endogenous CLIMP-63 (Klopfenstein et al. 1998). In marked contrast, the Δlumen-GFP showed classical ER staining, including ring-like fluorescence around the nucleus (Fig. 2, D–F), as confirmed by costaining with an antibody against the ER membrane protein BAP31 (Klumperman et al. 1998; Fig. 2B and Fig. E). To establish these staining patterns at the ultrastructural level, immuno-EM was performed using anti-GFP antibodies (Fig. 3) and the data were quantified (Table ). This analysis unequivocally showed that wt-GFP was excluded from the nuclear envelope, whereas high labeling densities were obtained in ER cisternae (Fig. 3 A). Quantification showed that the labeling of the nuclear envelope was not different from background labeling. The labeling ratio of nuclear envelope to ER cisternae was 1:34. Δlumen-GFP, on the other hand, was associated with both ER domains (Fig. 3 B and Table ). The labeling ratio of the nuclear envelope to ER cisternae was 1:4. These morphological results indicate that the luminal segment of CLIMP-63 is required for its subdomain-specific localization in the peripheral ER.
Figure 1.

CLIMP-63 constructs. Schematic representation of CLIMP-63 constructs. (A) GFP constructs used for in vivo dynamics and in vitro aggregation assays. (B) Fragments of the luminal segment recombinantly expressed in E. coli and used for biochemical and structural analysis.
Figure 2.


The luminal segment of CLIMP-63 is required for nuclear envelope exclusion and mobility. COS cells were transiently transfected with wt-GFP (A–C) or Δlumen-GFP (D–F). 48 h after transfection, the cells were fixed with 3% paraformaldehyde, stained with monoclonal antibodies against the ER marker BAP31 (B and E) and analyzed by CLSM. Note that the construct lacking the luminal segment is no longer excluded from the nuclear envelope (D and F). (G) Quantification of FRAP experiments comparing the diffusion and mobility wt-GFP (•) with Δlumen-GFP (○). A representative, transiently transfected cell was photobleached over a 2-μm wide strip covering the ER. Mean fluorescent intensities in the strip were plotted versus time. The prebleach intensity was normalized arbitrarily to 100%. The least square fits to are indicated as lines. Note the excellent correlation between experimental data and fitting.
Figure 3.
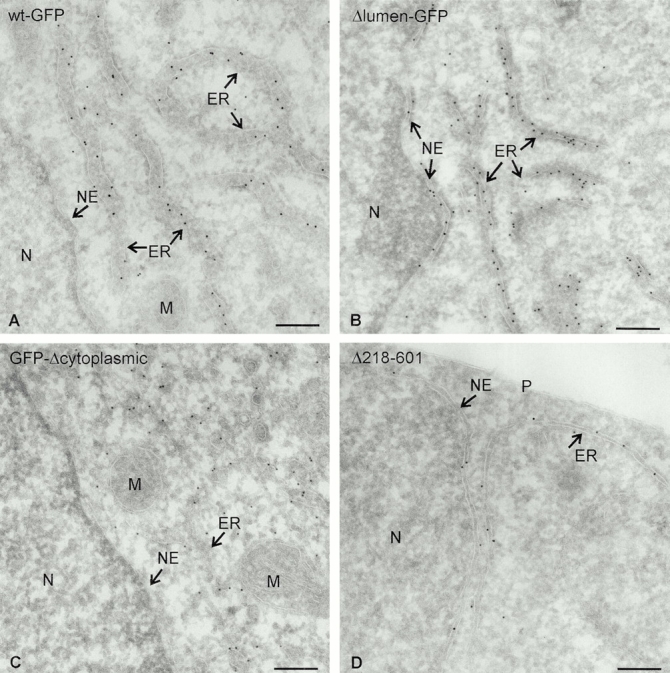
Localization of CLIMP-63 constructs by immuno-EM. Ultrathin cryosections of COS cells expressing different CLIMP-63 constructs were labeled with an antibody against GFP followed by protein A–gold. Note that wt-GFP (A) and GFP-Δcytoplasmic (C), but not Δlumen-GFP (B) and Δ218–601 (D), are excluded from the nuclear envelope (NE). N, nucleus; M, mitochondrion; P, plasma membrane. Bars, 200 nm.
Table 1.
Quantification of Immunogold Labeling of Ultrathin Cryosections of COS Cells Transfected with GFP-tagged CLIMP-63 Constructs
| Gold particles per membrane length (arbitrary units) ± SD | |||
|---|---|---|---|
| Nuclear envelope | ER | Nuclear envelope:ER | |
| wt-GFP | 0.06 ± 0.07 | 1.31 ± 0.52 | 1:34 |
| Δlumen-GFP | 0.29 ± 0.18 | 0.94 ± 0.36 | 1:4 |
| GFP-Δcytoplasmic | 0.06 ± 0.06 | 0.54 ± 0.39 | 1:12 |
| Δ218–601 | 0.15 ± 0.15 | 0.62 ± 0.39 | 1:9 |
Labeling densities represent the means of 10 cells. Membrane lengths were measured by counting the number of intersections with a squared line lattice of 1 cm2 at a magnification of 40,500×. Nuclear envelope/ER ratios were calculated per cell and represent the means of 10 cells.
Previous biochemical studies have shown that CLIMP-63 forms large homooligomeric aggregates that are largely insoluble in Triton X-100. It has been proposed that due to these aggregates, CLIMP-63 is retained as an ER-resident protein (Schweizer et al. 1994). Therefore, we considered the possibility that oligomer formation mediated by the luminal segment may immobilize CLIMP-63 in the lipid bilayer of the peripheral ER. This was tested by photobleaching experiments in living cells. Diffusion of CLIMP-63–GFP constructs in the reticular regions of the ER was examined using the FRAP technique. In FRAP experiments, a region of interest was selected (typically a stripe of 2-μm diameter covering peripheral ER over the entire width of a cell) and bleached with a high-intensity laser beam. The bleached area was smaller than 1/16 of the total area of the cell. Recovery of fluorescence in the bleached area was then measured over time. wt-GFP was found to have a very low recovery rate (Fig. 2 G, filled circles); even after a long observation time of 5 min, only 40–50% of the initial fluorescence intensity was recovered. By contrast, the recovery rate of the Δlumen-GFP was rapid. Fluorescence nearly reached prebleach intensity levels with this construct (Fig. 2 G, open circles). The diffusion coefficients were Deff of 1.5 × 10−10 cm2s−1 for the wt-GFP and 8.5 × 10−10 cm2s−1 for Δlumen-GFP as calculated according to Lippincott-Schwartz et al. 1999. Interestingly, there was a difference not only in diffusion but also in mobility. Mobility is indicated by the percentage of maximal fluorescence recovery after photobleaching. Mobility was >90% for the Δlumen-GFP and ∼50% for wt-GFP. The data suggest that both diffusion and mobility of CLIMP-63 are dependent on the luminal segment. The difference in mobility suggests a role for the luminal segment in mediating the formation of large, slowly diffusing complexes.
Subdomain-specific ER Localization Requires the Entire Luminal Segment
To test if the entire luminal segment is required for nuclear envelope exclusion and immobilization, we made deletions in the luminal part of CLIMP-63 (Fig. 1 A) and tested the constructs in FRAP experiments. In these constructs the cytosolic segment of CLIMP-63 was replaced by GFP. This replacement was required to exclude a possible interference by the cytosolic microtubule–binding segment. COS cells transfected with the GFP-Δcytoplasmic construct that carries the entire luminal segment of CLIMP-63 (Fig. 1 A) showed reticular ER staining without a nuclear ring (Fig. 4 A) comparable to wt-GFP. In addition to reticular ER staining, ∼25% of the transfected cells (n = 864) exhibited highly fluorescent perinuclear areas (Fig. 4 D). Other ER markers, such as BAP31 (Klumperman et al. 1998), colocalized with these areas (Fig. 4 E, merger in F). Analysis by immuno-EM showed that these areas of intense fluorescence correspond to regions of densely packed ER elements (Fig. 5). The labeling for CLIMP-63 in clustered and nonclustered ER elements was comparable. Obviously, CLIMP-63 can concentrate ER membranes by its luminal segment in the absence of the cytoplasmic segment. The exclusion of the GFP-Δcytoplasmic construct from the nuclear membrane was confirmed by immuno-EM (Fig. 3 C and Table ). Unlike GFP-Δcytoplasmic, all luminal deletion constructs (Fig. 1 A) had access to the nuclear envelop (Fig. 4 and Fig. 7). One of these constructs, Δ218–601, was also studied by immuno-EM, which confirmed its dual localization in both subdomains of the ER (Fig. 3 D and Table ). These results suggest that exclusion of CLIMP-63 from the nuclear envelope depends on an intact luminal segment.
Figure 4.
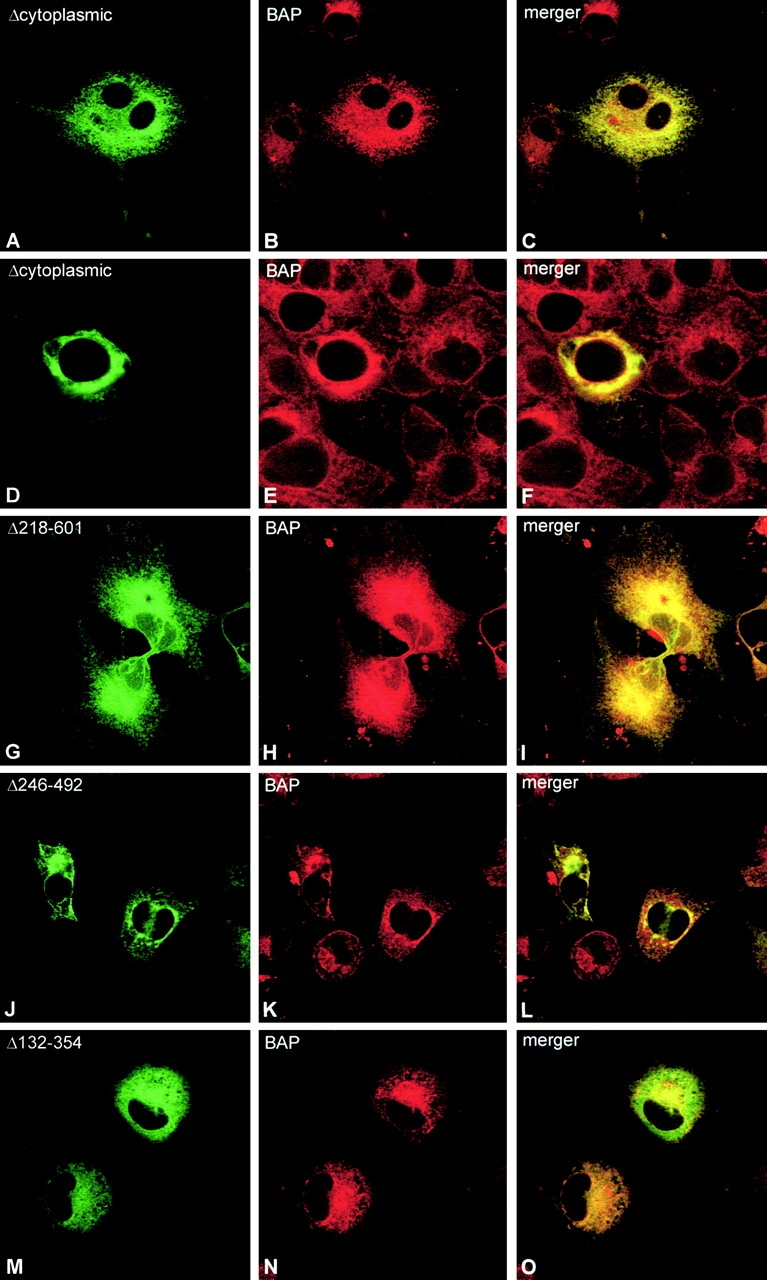
Luminal deletion constructs are no longer excluded from the nuclear envelope. COS cells were transiently transfected with GFP-Δcytoplasmic (A–F), GFP-Δcytoplasmic-Δ218–601 (G–I), GFP-Δcytoplasmic-Δ246–492 (J–L), and GFP-Δcytoplasmic-Δ132–354 (M–O). 48 h after transfection, the cells were fixed and stained with mAb against the ER marker BAP31 (B, E, H, K, and N). Colocalization of the GFP-fusions with BAP31 is apparent in the merger images (C, F, I, L, and O). Note the concentrated ER in (D–F) and (J–L).
Figure 5.

Overexpression of CLIMP-63 results in formation of ER clusters in restricted areas of the cytoplasm. COS cells were transiently transfected with wt-GFP and subjected to ultrathin cryosectioning followed by immunolabeling with anti-GFP and protein A–gold. (A) The cell at the left expresses high levels of wt-GFP as judged by the high density of gold particles and displays clustered ER (arrows). The cell at the right expresses no or undetectable levels of wt-GFP and shows nonclustered ER (arrows). (B) Another example of an ER cluster in a cell with high expression of wt-GFP. N, nucleus; P, plasma membrane. Bars: (A) 1 μm; (B) 500 nm.
Figure 7.

Transient expression of luminal mutants show clustering of ER membranes. COS cells were transiently transfected with GFP-Δcytoplasmic (A), GFP-Δcytoplasmic double luminal (B), GFP-Δcytoplasmic-Δ511–601 (C), and GFP-Δcytoplasmic-Δ218–601 (D). 48 h after transfection the cells were fixed and analyzed by CLSM. Note that the luminal deletion constructs (C and D) are not excluded from the nuclear envelope.
We noticed differences among the luminal deletion constructs regarding the extent of perinuclear concentration of ER elements. Although cells expressing the COOH-terminal deletion constructs Δ246–492 (Fig. 4, J–L) and Δ218–601 (Fig. 7 D) showed a concentration phenotype with a similar frequency to GFP-Δcytoplasmic (Fig. 4, D–F), cells expressing the NH2-terminal deletion construct Δ132–354 more rarely had concentrated ER (Fig. 4, M–O; 11% of cells with concentrated ER, n = 599). The extent of ER cluster formation was not due to overall expression levels, which were comparable for all constructs (not shown). These results indicate that the intact luminal segment has the highest propensity to cluster ER elements.
To determine whether the deletion mutants differ in protein mobility, we performed FRAP experiments in living cells. A strip of 2-μm width covering the reticular ER over the entire cell was bleached for 2 s and fluorescence recovery was quantified. Fig. 6 A shows that the recovery rate was faster for the luminal deletion construct than for GFP-Δcytoplasmic, whereas the differences among the deletion constructs were rather minimal (Table ). As a reference, we also included the Δlumen-GFP construct which showed high mobility and diffusion in the same range as the ER membrane enzyme cytochrome P450, Sec61β, and VSV-G protein (Table II; Cole et al. 1996; Szczesna-Skorupa et al. 1998; Rolls et al. 1999). The fraction of mobile protein was dependent on the presence of the luminal segment. The calculated diffusion coefficients ranging from 1.5–17.8 × 10−10cm2s−1 are shown in Table . The rate of fluorescence recovery in the nuclear envelope and in the reticular domain of the ER was comparable as judged from visual inspection. However, there was a considerable difference in mobility of a given construct if reticular and condensed ER areas were compared. Condensed ER regions of cells expressing the constructs Δcytoplasmic (Fig. 6 B, left) and ΔcytoplasmicΔ246–492 (right) displayed lower mobility than the corresponding reticular ER. These results suggest that molecular clustering renders the constructs nearly immobile and prevents them from mixing with the mobile reticular pool.
Figure 6.

FRAP shows fast diffusion for luminal deletion mutants. Quantitative FRAP experiments with transiently transfected COS cells expressing CLIMP-63–GFP constructs. (A) The recovery profiles of five constructs are shown as the mean value of intensity in the region of interest as a function of time. (B) GFP-Δcytoplasmic (left) and GFP-Δcytoplasmic-Δ246–492 (right) show differences in diffusion comparing mobile, reticular ER (○) with immobile, clustered ER (•).
Table 2.
Diffusion Coefficients and Mobility of CLIMP-63 Constructs
| Constructs | D | Mobility | n |
|---|---|---|---|
| 10−10 cm2s−1 | |||
| wt-GFP | 1.5 ± 0.4 | + | 5 |
| Δlumen-GFP | 8.6 ± 3.3 | +++ | 6 |
| Δ24–101-GFP | 3.1 ± 2.1 | + | 12 |
| Δ24–101Δlumen-GFP | 13.7 ± 2.2 | +++ | 4 |
| GFP-Δcytoplasmic | 8.9 ± 2.4 | ++ | 9 |
| GFP-Δcytoplasmic double luminal | 2.7 ± 1.2 | − | 4 |
| GFP-Δcytoplasmic-Δ511–601 | 7.8 ± 4.2 | ++ | 5 |
| GFP-Δcytoplasmic-Δ428–601 | 16.4 ± 8.2 | +++ | 4 |
| GFP-Δcytoplasmic-Δ354–601 | 17.8 ± 2.7 | +++ | 5 |
| GFP-Δcytoplasmic-Δ218–601 | 17.8 ± 4.9 | +++ | 5 |
| GFP-Δcytoplasmic-Δ246–492 | 6.7 ± 2.5 | ++ | 8 |
| GFP-Δcytoplasmic-Δ132–354 | 10.8 ± 5.5 | +++ | 6 |
| P450 2C2-GFP | 13.1 ± 6.6 | +++ | 6 |
| Sec61β-GFP | 7.8 ± 1.2 | +++ | 6 |
| VSV-G-GFP | 5.9 ± 1.3 | +++ | 5 |
FRAP analysis of GFP fusion constructs reveal strong influence of the luminal segment on diffusion. Deletion of the luminal segment increases diffusion and mobility (Δlumen-GFP) in contrast to deletion of the microtubule binding domain (Δ24–101-GFP), which increases diffusion but not mobility. Note that deletions of the COOH-terminal part have a higher diffusion coefficient. D, diffusion coefficient; n, number of independent experiments. Mobility is measured as a percentage of initial fluorescence: 0–25%, −; 25–50%,+; 50–75%, ++; 75–100%, +++.
That the luminal segment promotes clustering of ER membranes led us to test if this phenomenon can be enhanced by expressing a construct with a tandemly repeated luminal segment (Fig. 1 A, double luminal). A double luminal segment in fact increased the percentage of COS cells exhibiting condensed ER and the extent of concentration was also increased (compare Fig. 7A and Fig. B). 77% of the transfected cells (n = 318) showed areas with concentrated ER, and in FRAP experiments the apparent mobility was further decreased in comparison to the construct with the single luminal segment (Fig. 6 A). This further confirms an important role of the luminal segment in restricting mobility of CLIMP-63. Overall, these mutagenesis experiments reveal that the entire luminal segment is required for the subdomain-specific localization of CLIMP-63.
The Luminal Segment of CLIMP-63 Consists of Heptad Repeats and Forms α-Helical Rods
To gain further insight into the mechanisms underlying domain-specific localization and aggregation, we studied structural aspects of CLIMP-63. Amino acid sequence analysis identified stretches of heptad repeats in the luminal segment of CLIMP-63 using the COILS2 program (Lupas et al. 1991; Lupas 1996b; Fig. 8 A), suggesting that assemblies of CLIMP-63 may be mediated by a coiled coil oligomerization domain. To test this assumption, we expressed a His6-tagged peptide fragment encompassing the luminal sequence of CLIMP-63 (Fig. 1 B) in E. coli. The fragment was affinity purified by Ni2+-agarose chromatography and analyzed by transmission EM after negative staining. Fig. 8 B shows that the purified luminal segment forms rod-shaped particles with a mean length of 91 ± 24 nm. This is somewhat longer than the calculated length of 69 nm for a two-stranded, α-helical coiled coil consisting of 70 heptad repeats (with the assumption that the axial rise per residue corresponds to 1.485 Å; Fraser et al. 1964).
Figure 8.
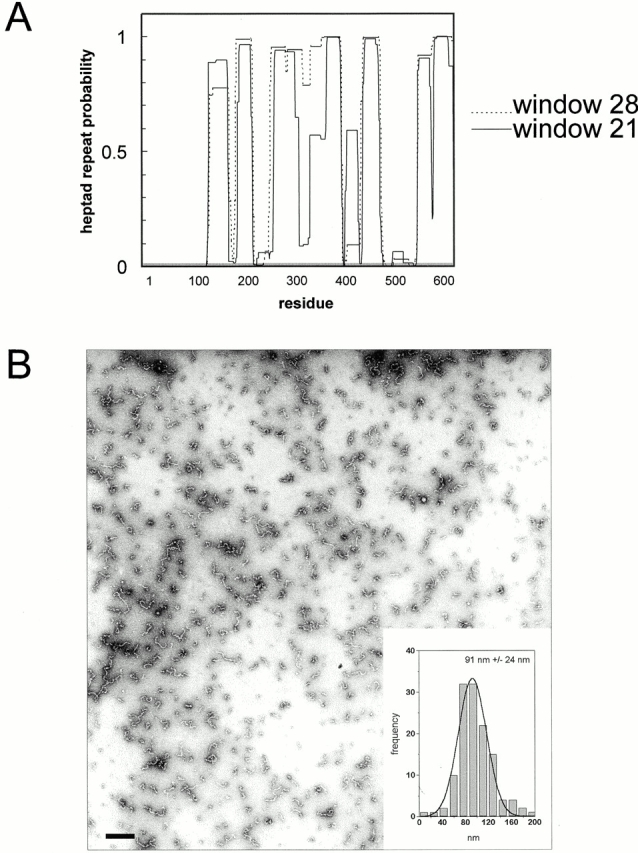
The luminal segment forms rod-shaped molecules. (A) Structure prediction based on the COILSCAN program (GCG package) reveals high probability for coiled coil formation in the luminal segment of CLIMP-63. Window sizes of 21 or 28 were used. (B) Electron micrograph of the purified luminal segment showing rod-shaped molecules of 91 nm average length. (Inset) A single Gaussian fit for the distribution of the molecular length and mean length ± 1 SD (n = 132). Bar, 100 nm.
Next, far ultraviolet CD spectroscopy was applied for secondary structure analysis of the luminal part. At 5°C and a protein concentration of 19 μM, the CD spectrum was characteristic for an α-helical structure exhibiting minima near 208 and 222 nm (not shown). Based on a [Θ]222 value of −32,000 deg.cm2/dmol, a helical content of 80–90% was estimated assuming that a value of 37,400 deg.cm2/dmol would correspond to a helicity of 100% for a polypeptide chain of this size (Chen et al. 1974). The stability of the recombinant luminal peptide was analyzed by thermal unfolding profiles monitored by CD at [Θ]222. At a concentration of 19 μM the luminal peptide showed a sigmoidal unfolding profile with a Tm at ∼45°C (not shown). The thermal unfolding was monophasic and reversible with >90% recovery of the starting signal after cooling. These results suggest that the recombinant luminal segment of CLIMP-63 can fold into an α-helical coiled coil structure.
The Luminal Segment Can Form Large Molecular Complexes
To determine the oligomerization state of the rod-shaped particles, AUC was used. At 20°C in 10 mM Tris, 150 mM NaCl, 1 M urea, pH 8.0, a sedimentation equilibrium of the recombinant luminal segment yielded an apparent molecular mass of 2,547 kD and a sedimentation coefficient of 25.7 S (Table ). This indicates that the luminal segment forms large aggregates in solution. Because CLIMP-63 is less soluble at an acidic pH, the sedimentation behavior of the luminal segment was also analyzed at pH 6.0. Lowering the pH by two units resulted in an increase in particle size, as indicated by the sedimentation coefficient of 171 S (Table ). The difference in sedimentation velocity correlated with a change in particle shape observed in EM. Whereas particles prepared at pH 8.0 appeared mainly as single rods, assembly rods in networks were observed at pH 6.0 in addition to single particles (not shown). Next, we sought to test the stability of the aggregates and dissociate the large complexes formed at pH 8.0 into smaller subunits by increasing the concentration of urea or NaCl. In the absence of urea, the solubility of the luminal segment was salt-dependent. NaCl concentrations ≥0.5 M prevented precipitation, which was observed with lower concentrations of NaCl. The sedimentation properties of a protein sample in buffer with 0.5–3 M NaCl showed a large sedimentation coefficient of 25 S (not shown), which was not different from that of a sample in buffer with 1 M urea (Table ). At urea concentrations of ≥3 M the luminal segment was apparently in an unfolded monomeric state, whereas no intermediate-sized complexes were observed at concentrations between 1–3 M urea as judged from CD and AUC analyses. These data suggest that the recombinant wild-type luminal segment forms large complexes in vitro that are probably stabilized by electrostatic interactions.
Table 3.
AUC, CD Spectroscopy Data and Melting Curves
| Condition | ||||||
|---|---|---|---|---|---|---|
| Fragments | Quantity of urea | pH level | [Θ]222 | Tm | Sedimentation coefficient s20, w | Molecular mass |
| M | 10−3 deg.cm2/dmol | °C | S | kD | ||
| 127–601 | 1 | 8.0 | −32.6 | 45 | 25.7 | 2547 (55) |
| 127–601 | 1.5 | 8.0 | −20.3 | ND | 80% 26:20% 2 | ND |
| 127–601 | 2 | 8.0 | −13.7 | ND | 50% 26:50% 2 | ND |
| 127–601 | 2.5 | 8.0 | −5.8 | ND | 20% 26:80% 2 | ND |
| 127–601 | 3 | 8.0 | −4.5 | ND | 2.1 | 58 (55) |
| 127–601 | 1 | 6.0 | −33.2 | 45 | 177.0 | ND |
| 127–459 | 1 | 8.0 | −31.6 | 45 | >20 | ND |
| 127–268 | 1 | 8.0 | −14.6 | 45 | >20 | ND |
| 268–459 | 1 | 8.0 | ND | ND | ND | 40 (21) |
Mean molar ellipcities at 222 nm, thermal melting temperatures at the midpoint of transition, sedimentation coefficients, and molecular masses of various CLIMP-63 constructs at different conditions.
To determine whether fragments of the luminal segment still oligomerize into large complexes, deletion constructs were recombinantly expressed, purified, and subjected to biophysical analysis. The constructs used are shown in Fig. 1 B. CD spectra consistently showed an α-helicity of >80% for the constructs tested except 127–268, which showed 50% α-helicity (Table ). In AUC, large complexes were observed for all fragments, with the exception of 268–459, that was also present as dimers (Table ). Analysis of these constructs by EM revealed rods organized into network-like structures (not shown). These assemblies most likely represent filaments generated by staggered alignment of individual molecules. These biochemical and morphological data are consistent with a high propensity of the luminal domain of CLIMP-63 to form large molecular complexes.
Discussion
Role of the Luminal Segment for Subdomain-specific Localization in the ER
Our mutagenesis approach, in conjunction with confocal microscopy and immuno-EM, revealed that without its luminal segment, CLIMP-63 is no longer excluded from the nuclear envelope, whereas absence of the microtubule-binding cytosolic segment had no effect on the localization of CLIMP-63. This indicates that the luminal segment is required for subdomain-specific localization of CLIMP-63 in the reticular part of the ER.
Exclusion from the nuclear membrane is paralleled by CLIMP-63's immobility in the plane of the bilayer membrane as assessed by FRAP experiments in living cells. Deleting the entire luminal segment considerably increased the mobility. By contrast, doubling the luminal segment reduced the mobility of CLIMP-63, confirming an important role of the luminal segment for restricting mobility. In general, the mobility was inversely proportional to the length of the luminal segment: the shorter the luminal segment the higher the mobility of the construct.
Interestingly, the mobility of intact CLIMP-63 is considerably lower than that of two other ER membrane proteins, cytochrome P450 and Sec61β in the ER, as well as that of VSV-G protein in transit through the Golgi region. The diffusion constants we measured for these proteins were similar to published values (Cole et al. 1996; Szczesna-Skorupa et al. 1998; Rolls et al. 1999). In contrast to CLIMP-63, we observed no restriction in mobility of cytochrome P450, Sec61b, and VSV-G protein in FRAP experiments, arguing that these proteins do not form large, immobile complexes. For cytochrome P450, these results are consistent with the finding that partial deletions in the protein had little effect on its mobility (Szczesna-Skorupa et al. 1998).
The different GFP-tagged constructs showed areas of bright fluorescence in the vicinity of the nucleus to various extents. Analysis of wt-GFP by immuno-EM showed that these areas correspond to sites of concentrated ER membranes. At the immunofluorescence level, the extent of this ER clustering correlated with the length of the luminal segment. The strongest effect was seen for the double luminal construct with extensive clustering and the weakest for the GFP-Δlumen, which only rarely exhibited such clusters. Photobleaching of the clusters revealed a remarkable decrease in mobility in comparison to areas with a reticular ER in the same cell. This suggests that CLIMP-63 can occur in two states of mobility, dynamic and immobile. An analogous duality was reported for lamin B receptor with an immobile nuclear envelope fraction and a mobile reticular ER fraction (Ellenberg et al. 1997). However, immobility of lamin B receptor is mediated by cytosolic interaction, whereas immobility of CLIMP-63 is mediated by the luminal segment.
It is important to note that ER cluster formation is different from ER tubulation reported previously (Klopfenstein et al. 1998). Overexpression of wt-CLIMP-63 can lead to ER tubules extending into the cell periphery. If the central part of the cytoplasmic segment containing the microtubule-binding domain (i.e., amino acids 24–101) is deleted, short, apparently retracted ER tubules appear (Klopfenstein et al. 1998) that are morphologically different from ER clusters reported here for luminal deletion constructs. This comparison indicates that both the luminal and the cytoplasmic segments contribute to the ER rearrangement induced by CLIMP-63, but they do so in a characteristically different way.
Loss of subdomain-specific localization of CLIMP-63 constructs is also accompanied by increased solubility in detergents (not shown). Most likely, the reduced mobility in vivo and the reduced detergent solubility in vitro are a reflection of the remarkable feature of CLIMP-63 to form large homooligomers.
The Luminal Segment Forms α-Helical Structures that Assemble into Large Complexes
CD spectroscopy showed that the recombinant luminal segment has a high content of α-helicity. Due to the aggregation properties of the luminal segment, AUC studies could only resolve large oligomeric structures. Since most extracellular coiled coil domains form trimers (Kammerer 1997), we assume that the basic unit of CLIMP-63 aggregates are trimeric α-helical bundles, but this remains to be verified. In agreement with a coiled coil structure, EM data revealed rod-shaped luminal molecules. Their length of 91 ± 24 nm exceeds 69 nm, a value calculated for a parallel 474–amino acid long coiled coil (Fraser et al. 1964). It is possible that the rod-shaped molecules are composed of staggered monomers into parallel or antiparallel structures, which would also explain the high variance toward longer molecules (Fig. 8 B, inset). A staggered assembly of monomers into filaments is well known for intermediate filament proteins (Abumuhor et al. 1998).
Aggregation observed by AUC is likely to occur via hydrophilic interactions since increasing concentrations of NaCl renders the protein more soluble. Consistent with this, the luminal segment carries a large number of charged amino acids, including 33 lysine, 31 arginine, 29 aspartic acid, 11 histidine, and 61 glutamic acid residues, totaling 165 charged residues over the entire 474–amino acid long luminal segment. The charged residues are predominantly found in position b, c, and f that are facing the solvent-exposed side of the helix. It is conceivable that the shielding of these charged residues by high salt concentrations keeps the protein in a less aggregated state. Based on our structural analysis, we propose that the luminal segment of CLIMP-63 forms coiled coils that can form higher order oligomers, presumably stabilized by electrostatic intermolecular forces.
Functions of CLIMP-63
Our previous studies have established a role for CLIMP-63 in linking the ER to microtubules (Klopfenstein et al. 1998). The minimal requirement to fulfill such a function is an ER membrane anchor at one end of the molecule and a microtubule binding domain at the other end. However, our present studies suggest a more complex scenario involving the luminal segment of CLIMP-63 as well. The luminal segment is largely responsible for coiled coil–based oligomerization that results in the exclusion of CLIMP-63 from the nuclear envelope. Although the cell nucleus can bind to MTs in vitro (Reinsch and Karsenti 1997) it is conceivable that in vivo such a binding is undesirable for a large spherical organelle, or at least unnecessary. Microtubules are more important for dynamic, tubule-forming organelles such as reticular ER, ERGIC, Golgi apparatus, or endosomes.
The simplest way to restrict a CLIMP to the reticular domain of the ER would be transmembrane domain–mediated aggregation without the need of a large luminal domain. The fact that CLIMP-63 has a large luminal domain may therefore indicate functions that go beyond microtubule binding. The 91-nm long rod-shaped luminal segment may play a morphogenetic role in the ER. Assuming a luminal width of ER cisternae of 60–120 nm (Gotow and Hashimoto 1989; Senda and Yoshinaga-Hirabayashi 1998), CLIMP-63 can be envisaged to assemble in different ways which would determine different functions. CLIMP-63 may aggregate into patches and thereby form MT binding sites of high avidity. In addition, these aggregates may form a large luminal scaffold that plays a structural role for ER morphology. Alternatively, the luminal segments of CLIMP-63 molecules of opposing cisternal membranes may bind to each other in an antiparallel or parallel manner. Such an arrangement would help to keep ER cisternae flat. These possibilities are not mutually exclusive. It remains even possible that the oligomeric assemblies of CLIMP-63 contain additional proteins that regulate the oligomerization of the heptad repeats or operate as additional linkers in the formation of a luminal protein scaffold. Further studies are required to test these putative additional functions of CLIMP-63.
Acknowledgments
We thank Käthy Bucher for expert technical assistance, Therese Schulthess for assisting with the CD spectrometer, Andreas Hefti for providing some electron micrographs, Rainer Pepperkok for discussions and providing the advanced light microscopy facilities at the European Molecular Biology Laboratory in Heidelberg, Jan Ellenberg and Jamie White for helpful suggestions regarding the FRAP experiments, Byron Kemper for providing the cytochrome P450 cDNA, and Graham Warren for providing antibodies to GFP.
This work was supported by the University of Basel and by a grant from the Swiss National Science Foundation (to H.-P. Hauri).
Footnotes
Abbreviations used in this paper: AUC, analytical ultracentrifugation; BAP31, B cell antigen receptor–associated protein of M-31; CD, circular dichroism; CLIMP-63, 63 kD cytoskeleton-linking membrane protein; CLSM, confocal laser scanning microscopy; GFP, green fluorescent protein; VSV-G, vesicular stomatitis virus glycoprotein G; wt, wild-type.
References
- Abumuhor I.A., Spencer P.H., Cohlberg J.A. The pathway of assembly of intermediate filaments from recombinant alpha-internexin. J. Struct. Biol. 1998;123:187–198. doi: 10.1006/jsbi.1998.4040. [DOI] [PubMed] [Google Scholar]
- Adam S.A. Transport pathways of macromolecules between the nucleus and the cytoplasm. Curr. Opin. Cell Biol. 1999;11:402–406. doi: 10.1016/S0955-0674(99)80056-8. [DOI] [PubMed] [Google Scholar]
- Barr F.A., Nakamura N., Warren G. Mapping the interaction between GRASP65 and GM130, components of a protein complex involved in the stacking of Golgi cisternae. EMBO J. 1998;17:3258–3268. doi: 10.1093/emboj/17.12.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos R., Pante N., Burke B. Nuclear pore complex proteins. Int. Rev. Cytol. 1995;162:257–302. doi: 10.1016/s0074-7696(08)62619-4. [DOI] [PubMed] [Google Scholar]
- Baumann O. Association of spectrin with a subcompartment of the endoplasmic reticulum in honeybee photoreceptor cells. Cell Motil. Cytoskeleton. 1998;41:74–86. doi: 10.1002/(SICI)1097-0169(1998)41:1<74::AID-CM7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Chapman R., Sidrauski C., Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus. Annu. Rev. Cell Dev. Biol. 1998;14:459–485. doi: 10.1146/annurev.cellbio.14.1.459. [DOI] [PubMed] [Google Scholar]
- Chen Y.H., Yang J.T., Chau K.H. Determination of the helix and beta-form of proteins in aqueous solution by circular dichroism. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- Cole N.B., Smith C.L., Sciaky N., Terasaki M., Edidin M., Lippincott-Schwartz J. Diffusional mobility of Golgi proteins in membranes of living cells. Science. 1996;273:797–801. doi: 10.1126/science.273.5276.797. [DOI] [PubMed] [Google Scholar]
- Csermely P., Schnaider T., Szanto I. Signaling and transport through the nuclear membrane. Biochim. Biophys. Acta. 1995;1241:425–451. doi: 10.1016/0304-4157(95)00015-1. [DOI] [PubMed] [Google Scholar]
- Cullen B.R. DEAE-Dextran-mediated transfection of COS cells. Methods Enzymol. 1987;152:692–693. [Google Scholar]
- Deschuyteneer M., Eckhardt A.E., Roth J., Hill R.L. The subcellular localization of apomucin and nonreducing terminal N-acetylgalactosamine in porcine submaxillary glands. J. Biol. Chem. 1988;263:2452–2459. [PubMed] [Google Scholar]
- Ellenberg J., Siggia E.D., Moreira J.E., Smith C.L., Presley J.F., Worman H.J., Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cellstargeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinger A., Pavelka M. Subdomains of the rough endoplasmic reticulum in colon goblet cells of the ratlectin-cytochemical characterization. J. Histochem. Cytochem. 1992;40:919–930. doi: 10.1177/40.7.1607641. [DOI] [PubMed] [Google Scholar]
- Fraser R.D.B., MacRae T.P., Miller A. The coiled-coil model of alpha-keratin structure. J. Mol. Biol. 1964;10:147–156. doi: 10.1016/s0022-2836(64)80034-6. [DOI] [PubMed] [Google Scholar]
- Gotow T., Hashimoto P.H. Substructure of cisternal organelles of neuronal perikarya in immature rat brains revealed by quick-freeze and deep-etch techniques. Cell Tissue Res. 1989;256:53–64. doi: 10.1007/BF00224718. [DOI] [PubMed] [Google Scholar]
- Hauri H.-P., Schweizer A. The ER–Golgi membrane systemcompartmental organization and protein traffic. In: Hoffman J.F., Jamieson J.D., editors. Handbook of Physiology, Section 14Cell Physiology. Oxford University Press; New York: 1997. pp. 605–647. [Google Scholar]
- Kammerer R.A. Alpha-helical coiled-coil oligomerization domains in extracellular proteins. Matrix Biol. 1997;15:555–565. doi: 10.1016/s0945-053x(97)90031-7. [DOI] [PubMed] [Google Scholar]
- Kammerer R.A., Schulthess T., Landwehr R., Lustig A., Fischer D., Engel J. Tenascin-C hexabrachion assembly is a sequential two-step process initiated by coiled-coil alpha-helices. J. Biol. Chem. 1998;274:10602–10608. doi: 10.1074/jbc.273.17.10602. [DOI] [PubMed] [Google Scholar]
- Klopfenstein D.R., Kappeler F., Hauri H.P. A novel direct interaction of endoplasmic reticulum with microtubules. EMBO J. 1998;17:6168–6177. doi: 10.1093/emboj/17.21.6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J., Schweizer A., Clausen H., Tang B.L., Hong W., Oorschot V., Hauri H.-P. The recycling pathway of protein ERGIC-53 and dynamics of the ER-Golgi intermediate compartment. J. Cell Sci. 1998;111:3411–3425. doi: 10.1242/jcs.111.22.3411. [DOI] [PubMed] [Google Scholar]
- Kohn W.D., Kay C.M., Hodges R.S. Salt effects on protein stabilitytwo-stranded alpha-helical coiled-coils containing inter- or intrahelical ion pairs. J. Mol. Biol. 1997;267:1039–1052. doi: 10.1006/jmbi.1997.0930. [DOI] [PubMed] [Google Scholar]
- Linstedt A.D., Hauri H.P. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell. 1993;4:679–693. doi: 10.1091/mbc.4.7.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Presley J.F., Zaal K.J., Hirschberg K., Miller C.D., Ellenberg J. Monitoring the dynamics and mobility of membrane proteins tagged with green fluorescent protein. Methods Cell Biol. 1999;58:261–281. doi: 10.1016/s0091-679x(08)61960-3. [DOI] [PubMed] [Google Scholar]
- Lupas A. Coiled coilsnew structures and new functions Trends Biochem. Sci 21 1996. 375 382a [PubMed] [Google Scholar]
- Lupas A. Prediction and analysis of coiled-coil structures Methods Enzymol 266 1996. 513 525b [DOI] [PubMed] [Google Scholar]
- Lupas A., Van Dyke M., Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Mattaj I.W., Englmeier L. Nucleocytoplasmic transportthe soluble phase. Annu. Rev. Biochem. 1998;67:265–306. doi: 10.1146/annurev.biochem.67.1.265. [DOI] [PubMed] [Google Scholar]
- Mancini M.A., He D., Ouspenski I.I., Brinkley B.R. Dynamic continuity of nuclear and mitotic matrix proteins in the cell cycle. J. Cell Biochem. 1996;62:158–164. doi: 10.1002/(SICI)1097-4644(199608)62:2%3C158::AID-JCB3%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- McLachlan A.D., Stewart M. Tropomyosin coiled-coil interactionsevidence for an unstaggered structure. J. Mol. Biol. 1975;293:293–304. doi: 10.1016/s0022-2836(75)80119-7. [DOI] [PubMed] [Google Scholar]
- Mundy D.I. Protein palmitoylation in membrane trafficking. Biochem. Soc. Trans. 1995;23:572–576. doi: 10.1042/bst0230572. [DOI] [PubMed] [Google Scholar]
- Mundy D.I., Warren G. Mitosis and inhibition of intracellular transport stimulate palmitoylation of a 62-kD protein. J. Cell Biol. 1992;116:135–146. doi: 10.1083/jcb.116.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl H.L. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol. Rev. 1999;79:683–701. doi: 10.1152/physrev.1999.79.3.683. [DOI] [PubMed] [Google Scholar]
- Reinsch S., Karsenti E. Movement of nuclei along microtubules in Xenopus egg extracts. Curr. Biol. 1997;7:211–214. doi: 10.1016/s0960-9822(97)70092-7. [DOI] [PubMed] [Google Scholar]
- Rolls M.M., Stein P.A., Taylor S.S., Ha E., McKeon F., Rapoport T.A. A visual screen of a GFP-fusion library identifies a new type of nuclear envelope membrane protein. J. Cell Biol. 1999;146:29–44. doi: 10.1083/jcb.146.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer A., Ericsson M., Bachi T., Griffiths G., Hauri H.P. Characterization of a novel 63 kDa membrane protein. Implications for the organization of the ER-to-Golgi pathway J. Cell Sci 104 1993. 671 683a [DOI] [PubMed] [Google Scholar]
- Schweizer A., Rohrer J., Jeno P., DeMaio A., Buchman T.G., Hauri H.P. A reversibly palmitoylated resident protein (p63) of an ER-Golgi intermediate compartment is related to a circulatory shock resuscitation protein J. Cell Sci 104 1993. 685 694b [DOI] [PubMed] [Google Scholar]
- Schweizer A., Rohrer J., Hauri H.P., Kornfeld S. Retention of p63 in an ER-Golgi intermediate compartment depends on the presence of all three of its domains and on its ability to form oligomers. J. Cell Biol. 1994;126:25–39. doi: 10.1083/jcb.126.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer A., Rohrer J., Kornfeld S. Determination of the structural requirements for palmitoylation of p63 J. Biol. Chem 270 1995. 9638 9644a [DOI] [PubMed] [Google Scholar]
- Schweizer A., Rohrer J., Slot J.W., Geuze H.J., Kornfeld S. Reassessment of the subcellular localization of p63 J. Cell Sci 108 1995. 2477 2485b [DOI] [PubMed] [Google Scholar]
- Senda T., Yoshinaga-Hirabayashi T. Intermembrane bridges within membrane organelles revealed by quick-freeze deep-etch electron microscopy. Anat. Rec. 1998;251:339–345. doi: 10.1002/(SICI)1097-0185(199807)251:3<339::AID-AR9>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Shorter J., Warren G. A role for the vesicle tethering protein, p115, in the post-mitotic stacking of reassembling Golgi cisternae in a cell-free system. J. Cell Biol. 1999;146:57–70. doi: 10.1083/jcb.146.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C., Chapman R., Walter P. The unfolded protein responsean intracellular signaling pathway with many surprising features. Trends Cell Biol. 1998;8:245–249. doi: 10.1016/s0962-8924(98)01267-7. [DOI] [PubMed] [Google Scholar]
- Silve S., Dupuy P.H., Ferrara P., Loison G. Human lamin B receptor exhibits sterol C14-reductase activity in Saccharomyces cerevisiae . Biochim. Biophys. Acta. 1998;1392:233–244. doi: 10.1016/s0005-2760(98)00041-1. [DOI] [PubMed] [Google Scholar]
- Sitia R., Meldolesi J. Endoplasmic reticuluma dynamic patchwork of specialized subregions. Mol. Biol. Cell. 1992;3:1067–1072. doi: 10.1091/mbc.3.10.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C. Coiled coils in both intracellular vesicle and viral membrane fusion. Cell. 1998;95:871–874. doi: 10.1016/s0092-8674(00)81710-9. [DOI] [PubMed] [Google Scholar]
- Slot J.W., Geuze H.J., Gigengack S., Lienhard G.E., James J.E. Immunolocalization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodek J., Hodges R.S., Smillie L.B., Jurasek L. Amino-acid sequence of rabbit skeletal tropomyosin and its coiled-coil structure. Proc. Natl. Acad. Sci. USA. 1972;69:3800–3804. doi: 10.1073/pnas.69.12.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sönnichsen B., Lowe M., Levine T., Jamsa E., Dirac-Svejstrup B., Warren G. A role for giantin in docking COPI vesicles to Golgi membranes. J. Cell Biol. 1998;140:1013–1021. doi: 10.1083/jcb.140.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuurman N., Heins S., Aebi U. Nuclear laminstheir structure, assembly, and interactions. J. Struct. Biol. 1998;122:42–66. doi: 10.1006/jsbi.1998.3987. [DOI] [PubMed] [Google Scholar]
- Szczesna-Skorupa E., Chen C.D., Rogers S., Kemper B. Mobility of cytochrome P450 in the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA. 1998;95:14793–14798. doi: 10.1073/pnas.95.25.14793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talcott B., Moore M.S. Getting across the nuclear complex. Trends Cell Biol. 1999;9:312–318. doi: 10.1016/s0962-8924(99)01608-6. [DOI] [PubMed] [Google Scholar]
- Vertel B.M., Velasco A., LaFrance S., Walters L., Kaczman-Daniel K. Precursors of chondroitin sulfate proteoglycan are segregated within a subcompartment of the chondrocyte endoplasmic reticulum. J. Cell Biol. 1989;109:1827–1836. doi: 10.1083/jcb.109.4.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters M.G., Pfeffer S.R. Membrane tethering in intracellular transport. Curr. Opin. Cell Biol. 1999;11:453–459. doi: 10.1016/s0955-0674(99)80065-9. [DOI] [PubMed] [Google Scholar]
- Weibel E.R. Stereological Methods. I. Practical methods for biological morphometry. Academic Press, Inc; London: 1979. [Google Scholar]