

Abstract
The [URE3] prion (infectious protein) of yeast is a self-propagating, altered form of Ure2p that cannot carry out its normal function in nitrogen regulation. Previous data have shown that Ure2p can form protease-resistant amyloid filaments in vitro, and that it is aggregated in cells carrying the [URE3] prion. Here we show by electron microscopy that [URE3] cells overexpressing Ure2p contain distinctive, filamentous networks in their cytoplasm, and demonstrate by immunolabeling that these networks contain Ure2p. In contrast, overexpressing wild-type cells show a variety of Ure2p distributions: usually, the protein is dispersed sparsely throughout the cytoplasm, although occasionally it is found in multiple small, focal aggregates. However, these distributions do not resemble the single, large networks seen in [URE3] cells, nor do the control cells exhibit cytoplasmic filaments. In [URE3] cell extracts, Ure2p is present in aggregates that are only partially solubilized by boiling in SDS and urea. In these aggregates, the NH2-terminal prion domain is inaccessible to antibodies, whereas the COOH-terminal nitrogen regulation domain is accessible. This finding is consistent with the proposal that the prion domains stack to form the filament backbone, which is surrounded by the COOH-terminal domains. These observations support and further specify the concept of the [URE3] prion as a self-propagating amyloid.
Keywords: amyloid, yeast prion, immunoelectron microscopy, protease resistance, Ure2p
Introduction
The [URE3] prion (infectious protein) concept arose in studies of the transmissible spongiform encephalopathies (TSEs) of mammals (Alper et al. 1967; Griffith 1967; Prusiner 1982). The remarkable UV radiation resistance of the scrapie agent (Alper et al. 1967) led to speculation that the infectious agent might not involve nucleic acids, and might instead be a sort of nucleation phenomenon (Griffith 1967), precisely the present concept of this disease. Sinc, a chromosomal gene controlling the disease (Dickinson et al. 1968) encodes the major protein component (PrP) of infectious material (Bolton et al. 1982; Chesebro et al. 1985; Oesch et al. 1985; Carlson et al. 1986). Considerable evidence has now accumulated in support of the idea that scrapie is an infectious form of the PrP protein, although attempts to induce infectious scrapie de novo using recombinant PrP protein or transgenes have not yet succeeded.
[URE3] and [PSI+] are nonchromosomal genetic elements of Saccharomyces cerevisiae (Cox 1965; Lacroute 1971). Stringent genetic criteria were formulated to distinguish prions from nucleic acid replicons or other epigenetic phenomena, and it was shown that [URE3] and [PSI+] satisfy these criteria as prions of Ure2p and Sup35p, respectively (Wickner 1994). Ure2p normally regulates nitrogen catabolism, but on rare occasions can change to an inactive form that is able to convert the normal (active) form into the same inactive state. The Sup35 protein is a subunit of the translation termination factor, and in the same way, a self-propagating, inactive form of Sup35p was identified as the [PSI] nonchromosomal genetic element (for review see Wickner et al. 2000). Although the agents of TSEs kill infected cells, and [URE3] or [PSI+] at most only retard growth, these infections are believed to have similar mechanisms, i.e., self-propagating amyloids. TSEs are often associated with amyloid deposition in infected tissues, and both Sup35p and Ure2p have been shown to form amyloids in vitro (Glover et al. 1997; King et al. 1997; Taylor et al. 1999).
Both Ure2p and Sup35p have Asn–Gln-rich NH2-terminal “prion domains” (Tuite 2000). Deletion analysis showed that the Ure2p prion domain, residues 1–80, is necessary for prion-based inactivation of Ure2p, and sufficient to induce the de novo appearance of [URE3] when overexpressed in a normal cell (Masison and Wickner 1995, Maddelein and Wickner 1999). Moreover, the peptide covering residues 1–65 spontaneously forms amyloid filaments in vitro (Taylor et al. 1999). The COOH-terminal portion, residues 81–354, is sufficient to carry out the nitrogen regulation function (Coschigano and Magasanik 1991; Masison and Wickner 1995). Full-length native Ure2p is a stable, soluble dimer, but forms cofilaments when the prion domain peptide is added. This in vitro amyloid formation is highly specific and selfpropagating, thus providing a possible explanation for the [URE3] prion (Taylor et al. 1999).
We have sought to clarify this hypothesis by examining the state of Ure2p in [URE3] cells by electron microsopy. Intracellular aggregation has been shown by fluorescence light microscopy of green fluorescent protein (GFP) fusions with Sup35p (Patino et al. 1996) and Ure2p (Edskes et al. 1999). In the latter case, Ure2p was aggregated in [URE3] cells, but evenly distributed through the cytoplasm of normal cells, and aggregation was induced only in molecules containing the prion domain. However, intracellular amyloid filaments have not been demonstrated for any yeast prion. Here we report visualization of such filaments in [URE3] cells in thin sections and immunolabeling studies with antibodies specific to both the COOH- and NH2-terminal domains of Ure2p. To complement these observations, we extracted Ure2p from [URE3] cells and found it to be in large aggregates that resemble in vitro–produced Ure2p amyloid, insofar as its insolubility in denaturants and resistance to proteinase K digestion (Masison and Wickner 1995). Taken together, these observations establish a link between the in vivo and in vitro data that support the concept of the [URE3] prion as a self-propagating amyloid.
Materials and Methods
Antibody Production
A Ure2p (64–339)–His6 fusion was constructed by PCR, and expressed in Escherichia coli K38 using the pT7.7 vector (gift of Dr. Herbert Tabor, National Institutes of Health). The fusion protein was purified by NiNTA-agarose chromatography and 12% SDS-PAGE. The Coomassie blue–stained band was used to generate the rabbit polyclonal antiserum (Babco), Ure2-3C. The AminoLink Plus Immobilization Kit (Pierce Chemical Co.) and recombinant His6–Ure2p were used for affinity purification of the antibody. The antibody specific for the NH2-terminal domain has been described (Wickner 1994; Taylor et al. 1999).
Thin Section Electron Microscopy
Cells of the isogenic strains YHE134 (MAT a/MATα ura2/ura2 leu2/leu2 trp1/+ +/his −pH14 [URE3]) and YHE129 (MAT a/MATα ura2/ura2 leu2/leu2 trp1/+ +/his − pH14 [ure-o]) were grown in leucine dropout medium to exponential phase. For ultrastructural analysis, they were processed according to Byers and Goetsch 1991, except that 0.1 mg/ml zymolase 20T (Seikagaku Corp.) was used in addition to 10% vol/vol glusulase (Dupont; NEN Life Science Products) to remove the cell wall. After fixation with 2% glutaraldehyde on ice overnight, cells were treated with the enzymes for 2 h at 30°C, postfixed with 2% osmium tetroxide, treated en bloc with 1% uranyl acetate, dehydrated, and embedded in Spurr resin. Silver/gray sections were stained with 2% uranyl acetate followed by Reynolds lead citrate. All grids, including those with immunolabeled specimens described below, were examined in a ZEISS EM902 at 80 kV using the energy-filtering system in “zero-loss” mode.
Immunogold Labeling
Cryosections were prepared according to van Tuinen 1996. Thawed cryosections were blocked on drops of 5% BSA/0.1% cold water fish skin gelatin in PBS for 30 min, incubated for 40 min on the affinity-purified COOH-terminal antibody, washed, and transferred for 40 min to a drop of protein A–gold (10 nm; Sigma-Aldrich) diluted to A520 = 0.05. Both the primary antibody and protein A–gold were diluted in PBS containing 0.1% BSA-c (Aurion). In the absence of the antibody, the protein A–gold conjugate did not react with the cell sections. After extensive washing with PBS, sections were fixed with 2% glutaraldehyde for 10 min and contrasted with methylcellulose–uranyl acetate (Griffiths et al. 1984).
To quantitate the incidence of aggregates in [URE3] cells, micrographs of 127 randomly chosen cell sections were recorded at a magnification of 12,000. 54 of these were eliminated as either too small (tangential sections) or poorly preserved. Of the remaining 73, 4 had aggregates of 3–6 μm in diameter, 3 had aggregates of 0.8–1.5 μm, 3 had aggregates of 0.4–0.8 μm, and 6 had short lines of 3–5 closely spaced gold particles. In comparison, no such alignments were seen in >300 examples of the majority kind of control [ure-o] sections (see Fig. 3 A), whereas the two other kinds of [ure-o] labeling patterns (see Fig. 3B and Fig. C) were qualitatively different, rare, and readily recognizable. To assess the average densities of dispersed gold particles, we measured the areas and counted the gold particles of 20 [URE3] sections (minus areas with aggregates) and 15 [ure-o] sections.
Figure 3.
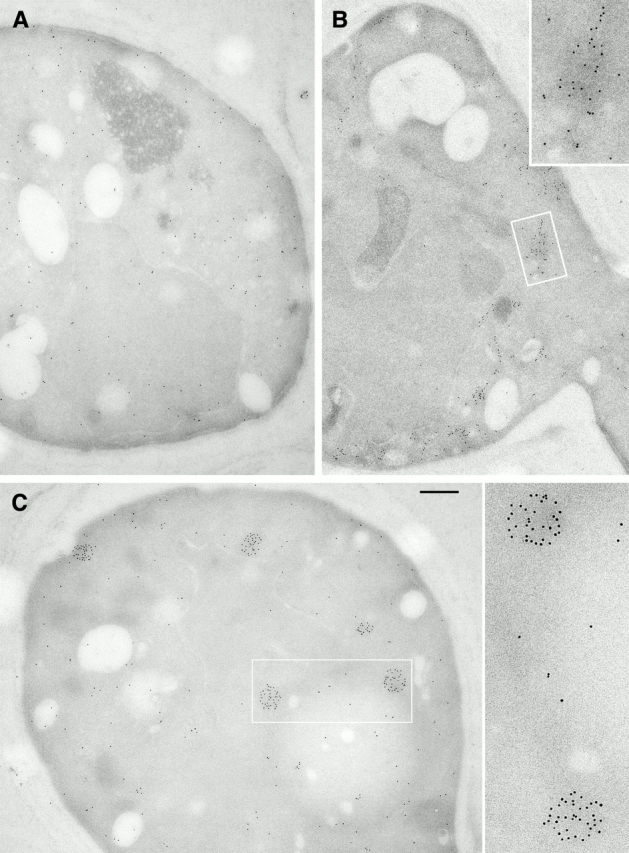
Immunoelectron microscopy of control [ure-o] cells. Most labeled cryosections (>80%) exhibit a sparse distribution of gold particles all over the cell (e.g., A). Approximately 5% of sections show concentrations of label at the cell periphery (B). Another 5–10% show one or more focal clusters within which the gold particles appear to be distributed randomly (C), in addition to diffuse labeling elsewhere. Bar, 0.5 μm.
Proteinase K Assays
In 320 μl, 45 μg of proteinase K was added to 800 μg of cell extract protein in 50 mM Tris-HCl (pH 7.5) containing 0.15 M NaCl, and incubated at 37°C. At 0, 1, 2, 5, 10, 15, and 30 min, 32 μl of the reaction was quenched by the addition of 34 μl of 8 M urea and 14 μl of SDS-PAGE loading buffer containing 2 mM PMSF. Samples were boiled for 5 min and 30-μl aliquots were loaded onto a 10–20% SDS-PAGE. The gels were blotted on to polyvinylidene difluoride membranes, and immunoblots were probed with an antibody specific for the NH2-terminal region of Ure2p (Wickner 1994; Taylor et al. 1999).
Results and Discussion
Aggregates of Ure2p-containing Filaments in [URE3] Cells
Yeast cells with the [URE3] prion and control [ure-o] cells were thin sectioned after fixation and embedding in an epoxy resin. Distinctive filamentous aggregates were seen in the cytoplasm of [URE3] cells of a strain that overexpresses Ure2p (Fig. 1). Large aggregates, at least 3 μm across, were seen in 5–10% of cell profiles, with smaller aggregates (or peripheral sections of large aggregates) being observed with a similar frequency (see below). These structures were never seen in control sections of [ure-o] cells. In sections showing these structures, there was typically one such aggregate, which was approximately globular in outline. They contain irregularly associated filaments ∼25 nm in diameter. In sections of [URE3] cells expressing normal levels of Ure2p, we observed a few candidate filamentous features but not frequently enough to allow conclusive identification (data not shown).
Figure 1.
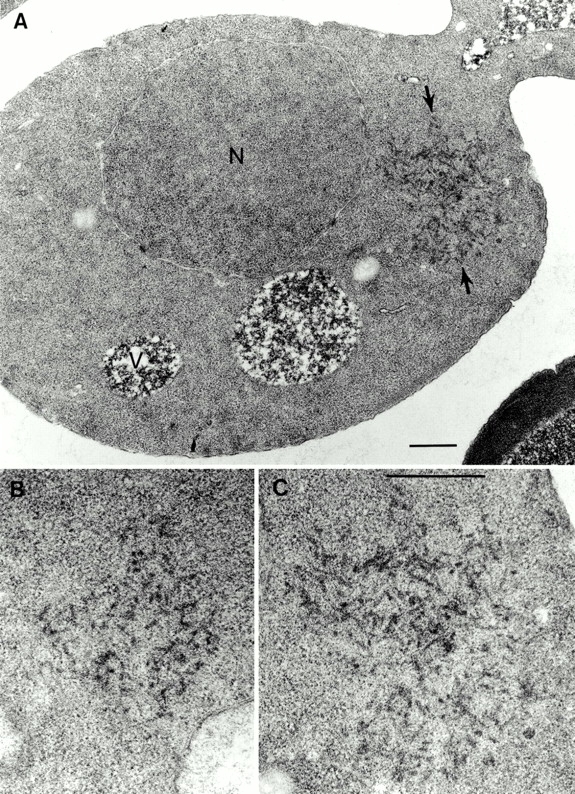
Filamentous aggregates in the cytoplasm of [URE3] cells overexpressing Ure2p. (A) Electron micrograph of a thin section through a [URE3] cell showing a cytoplasmic aggregate of 20–30 nm fibrils (arrows). (B and C) Enlarged views of aggregates from A (C) and from another cell (B). N, nucleus; V, vacuole. Bar, 0.5 μm.
Because these aggregates were found only in [URE3] cells, and filaments of similar diameter have been assembled from Ure2p in vitro (Taylor et al. 1999; Schlumpberger et al. 2000), we suspected that they contain Ure2p. To test this hypothesis, we prepared cryosections and labeled them with affinity-purified polyclonal antibodies raised against the COOH-terminal moiety of Ure2p (residues 64–339, Materials and Methods). Heavy and very specific labeling of the aggregates was observed (Fig. 2). The location and dimensions of the labeled regions matched those of the networks seen in the epoxy sections, as did their incidence. 5–10% of sections had a large aggregate and, when present, there was usually only one aggregate per cell section. Cryosections being less efficacious for conveying substructure, the filamentous substructure was less apparent in these micrographs, but the aggregates tended to stain more heavily than the surrounding cytoplasm. However, many of the gold particles were organized in linear formations (Fig. 2), attesting to filamentous organization of the antigen. Qualitatively similar but weaker and less specific labeling was observed with an antibody against the NH2-terminal prion domain (data not shown).
Figure 2.
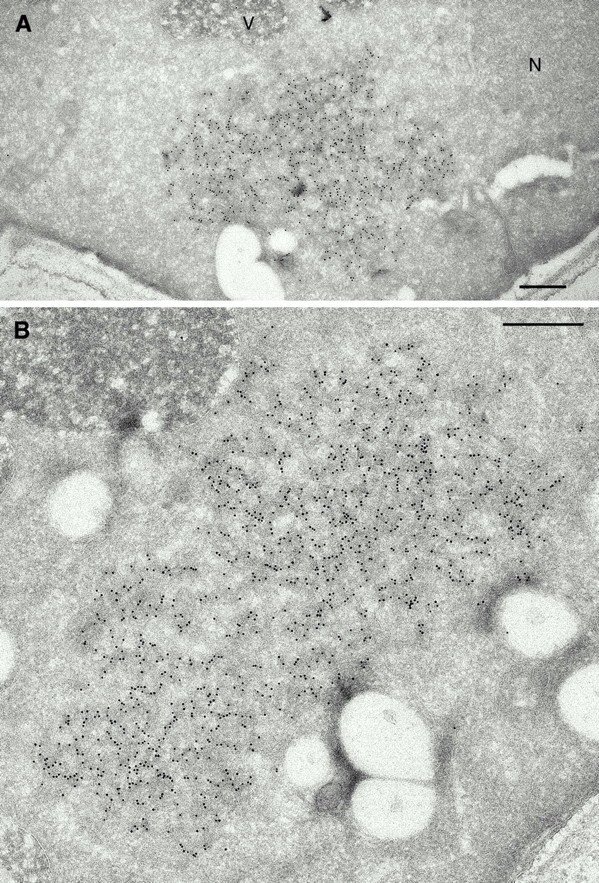
Cytoplasmic aggregates in [URE3] cells are labeled by anti-Ure2p antibodies. Cryosections were incubated with an affinity-purified antibody against the COOH-terminal domain of Ure2p. 10 nm gold particles show highly specific binding of the antibodies to cytoplasmic aggregates (A and B). Note the almost complete absence of label elsewhere in the cell. In the aggregates, gold particles are often locally organized in linear strands, suggesting multiple labeling of filaments. N, nucleus; V, vacuole. Bar, 0.5 μm.
As a control, we performed immunolabeling on [ure-o] cells overexpressing Ure2p at approximately the same level, as judged by protein detected with the COOH-terminal antibody. Several labeling patterns were observed. Most sections (>80%) showed sparse labeling throughout the cell (Fig. 3 A), occasionally with multiple small, focal concentrations of gold particles (Fig. 3 C). The remainder showed less regular (i.e., less round) concentrations of gold particles (Fig. 3 B), as if Ure2p were associated with an intracellular membrane, or no labeling, as if the plasmid had been lost. All these patterns were quite distinct from the labeling seen on [URE3] sections. Because different patterns were encountered on adjacent areas of the same [ure-o] section, their diversity does not reflect variations in procedure. Rather, we speculate that it may represent fortuitously different levels of Ure2p expression or differing stages of the cell cycle. Importantly, filaments were never observed in epoxy sections of these cells, and we have no reason to suppose that they harbor any Ure2p-containing filaments.
Fraction of Cells Containing Filamentous Aggregates
As noted above, we reproducibly saw a large aggregate in 5–10% of sections of [URE3] cells overexpressing Ure2p. This is quite a small fraction and raises the question of how many cells actually contain filaments. The compact nature of the aggregates implies that a considerably higher percentage of cells harbor such a structure as many random sections will miss the aggregate. To estimate this effect quantitatively, we first included all sections that showed smaller aggregates or even single filaments. Although we could not identify them in epoxy sections, we could detect filaments with the immunogold method as alignments of at least three closely spaced gold particles. This identification was possible because of the low level and random nature of background labeling obtained with this antibody (Materials and Methods). In this analysis, a total of 22%, i.e., 16 of 73 randomly chosen sections, scored positive for aggregates or at least one filament.
To put this number in context, we measured the fraction of cell sections that showed a nucleus and obtained a value of 29%, although every cell should have one. Comparing this number with the 22% of sections that show aggregates or filaments, and bearing in mind that the latter structures, being generally smaller than nuclei, are more likely to be missed, there is no discrepancy between these data and the proposition that essentially all of these cells contain filaments, aggregated or otherwise. Moreover, the density of single, randomly dispersed gold particles on [URE3] sections was very low, essentially at background level, and markedly less than on [ure-o] sections (0.8 vs. 14 gold particles per μm2, respectively). This observation suggests that almost all of the Ure2p in the [URE3] cells was in aggregates.
We also attempted to visualize filaments in normally expressing [URE3] cells by these methods, but did not obtain conclusive results. Because the level of Ure2p expression was >20-fold lower, the frequency of observing the structures of interest was presumably reduced to a prohibitively low level. Nevertheless, we suppose that a similar mechanism is in effect in these cells, i.e., amyloid filaments are present which sequester and inactivate Ure2p, but have not accumulated in amounts large enough to be seen in our sections.
Masking of NH2-terminal Epitope in Ure2p-containing Aggregates from [URE3] Cells
To complement these in situ observations, we examined Ure2p aggregates in extracts from prion-infected cells. Cells were suspended in buffer in 8 M urea and broken with glass beads. The whole cell extract was analyzed by SDS-PAGE after boiling in SDS-containing buffer, and probed by Western blotting with antibody specific for either the NH2 terminus or the COOH terminus of Ure2p (Fig. 4, left). In [URE3] extracts, the COOH-terminal–specific antibody detected at least 75% of Ure2p-containing material as aggregates that were retained in the stacking gel (Fig. 4, left, upper arrow). No such material was detected in [ure-o] extracts, in which Ure2p ran as a monomer. The [URE3] extracts also yielded some monomer, but in very low amounts if the concentration of urea was reduced (data not shown). These observations indicate that most Ure2p is aggregated in [URE3] cells, and that the minority of monomers most likely derive from small aggregates solubilized by the urea–SDS treatment. Interestingly, the [URE3]-derived aggregates were not detected by the NH2-terminal antibody, which readily detected the monomeric species (Fig. 4, left). We conclude that its epitope is masked in the aggregates.
Figure 4.

Solubility, antibody reactivity, and protease resistance of Ure2p in extracts of [URE3] and [ure-o] cells. [ure-o] (YHE129) and [URE3] (YHE134) cells carrying pH14 were suspended in 8 M urea, 100 mM Tris-HCl, pH 8.0, and were broken using a bead beater (2 × 3 min). (Left) Extract protein (15 μg) was boiled for 5 min in a total of 24 μl of SDS-PAGE loading buffer containing a final concentration of 3 M urea and 2% SDS. Samples were run on a 10–20% SDS-PAGE and immunoblotted with anti-Ure2p antibodies specific to either the NH2 or COOH terminus. (Right) Protease resistance of Ure2p in extracts of [URE3] cells (see Materials and Methods).
Ure2p in extracts from [URE3] cells also differs from the protein from [ure-o] cells in terms of resistance to proteinase K digestion. Whereas the latter material rapidly disappeared over the time course of the experiment, resistant fragments with apparent molecular masses of ∼30 and 14 kD accumulated in the [URE3] digestion (Fig. 4, right). Fragments with these molecular weights were detected previously in digestion of in vitro–assembled amyloid filaments (Taylor et al. 1999).
A Molecular Model for Ure2p Amyloid
The above observations support the proposal (Taylor et al. 1999) whereby the NH2-terminal domains stack axially to form the filament backbone from which the COOH-terminal domains protrude (Fig. 5). Sequestration of the NH2 termini in this way explains their inaccessibility to the NH2-terminal antibody. It also accounts for the lower reactivity of the NH2-terminal antibody with cryo-sections, although as expected, some labeling was observed as cross-cutting should expose some NH2-terminal epitopes.
Figure 5.
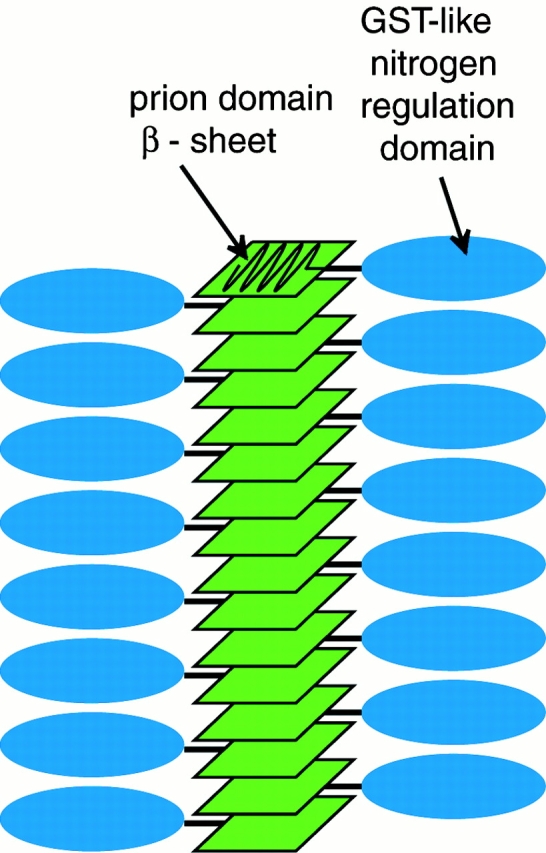
Molecular model of [URE3] amyloid filament composed of Ure2p. Polymerized prion domains (red) form the protease-resistant core of the filament and are inaccessible to antibodies because they are masked by the nitrogen regulation domain (blue), and perhaps also because of their compact structure. β Strands in the prion domain are perpendicular to the long axis of the filament. The nitrogen regulation domain is exposed both to antibody binding and to protease digestion.
A central tenet of the prion scenario is that Ure2p is inactivated in the polymerized form. Because the COOH-terminal domains carry its regulatory activity and are externally exposed, they are potentially accessible to reaction partners. Accordingly, their inactivation may be accomplished by at least partial unfolding upon becoming incorporated into the filament, or by steric inhibition of their functional sites (but not the whole domain), as packed in filaments. The former explanation is favored by the observed enhancement of β-sheet content when Ure2p enters its filamentous state (Taylor et al. 1999; Schlumpberger et al. 2000).
Curing and Induction of [URE3] as Expression-dependent Phenomena
Although genetic and biochemical evidence for yeast prions is now strong (reviewed by Wickner et al. 2000), the mechanism of prion propagation is not as well understood. Both Ure2p and Sup35p have Asn–Gln-rich NH2-terminal domains that are essential for prion generation and propagation (TerAvanesyan et al. 1994; Masison and Wickner 1995, Maddelein and Wickner 1999; DePace et al. 1998). Aggregation of Ure2p and Sup35p is an important feature of [URE3] and [PSI+], respectively (Patino et al. 1996; Paushkin et al. 1996; Edskes et al. 1999), but the nature of this aggregation has not been clear. GFP fusions with Ure2p form globular aggregates in [URE3] cells (Edskes et al. 1999). At first sight, the nonfilamentous nature of these aggregates might be considered as counter to the amyloid filament hypothesis (although some “filiform” aggregates have been reported in cells expressing the fusion of a mutant Ure2p with GFP [Fernandez-Bellot et al. 2000]). The present results account for this observation in that they reveal that the globular aggregates in [URE3] cells are tangles of filaments that are not resolved as such at the resolution of light microscopy.
Key to the concept of the [URE3] prion as a self-propagating amyloid is the provision that when cells divide, each daughter cell should receive at least one amyloid particle (a filament or aggregate of filaments) that then recruits newly expressed Ure2p and so maintains the [URE3] phenotype. In this context, it appears paradoxical that overexpression of the Ure2p prion domain should induce [URE3] in an initially [ure-o] cell, but the same overexpression of the same prion domain fragment in an initially [URE3] cell should cure it. The former phenomenon is readily explained by the observed efficacy of the prion domain in inducing cofilament assembly in vitro. The latter phenomenon has been discussed in terms of a poisoning of the prion “crystal” growth by incorporation of heterologous components (Edskes et al. 1999). Peptides of PrP inhibiting the propagation of scrapie in tissue culture cells have been similarly interpreted (Chabry et al. 1999). The observation that the amyloid filaments coalesce into single large aggregates in overexpressing [URE3] cells suggests an alternative curing mechanism. A larger aggregate may be more likely to soak up all the amyloid filaments, so that one of the daughter cells receives that aggregate and the other receives no amyloid and hence is cured. We observed more than one network within the same cell section only rarely, but cannot say that following or preceding sections of the same cell would not reveal another network (and Ure2p–GFP fusion proteins often show more than one aggregate per cell [Edskes et al. 1999]). However, it is clear that the number of aggregates per cell is small, and with two or three aggregates the chances of cosegregation would still be high enough to support this curing mechanism.
Both [PSI+] and [URE3] require the chaperone Hsp104 for their stable propagation (Chernoff et al. 1995; Moriyama et al. 2000). It was suggested that Hsp104 may help in the conversion of normal Sup35p and Ure2p to their altered forms (Chernoff et al. 1995). However, the disaggregating ability of Hsp104 (Parsell et al. 1994) and the finding that Sup35p is aggregated in [PSI+] extracts led Paushkin et al. 1996 to suggest that Hsp104's role may be to split up Sup35p aggregates to ensure equal distribution of the prion form to daughter cells in cell division. This segregational role of Hsp104 is also possible in [URE3], and is suggested by our finding that we mainly observe Ure2p in overexpressing [URE3] cells to be present in single large aggregates rather than dispersed populations of filaments. Without a disaggregating function, it is likely that one daughter cell would receive no filaments and thus be cured.
Acknowledgments
We thank Drs. D. Winkler and N. Cheng for providing electron microscopy facilities.
Footnotes
Abbreviations used in this paper: GFP, green fluorescent protein; TSE, transmissible spongiform encephalopathy.
References
- Alper T., Cramp W.A., Haig D.A., Clarke M.C. Does the agent of scrapie replicate without nucleic acid? Nature. 1967;214:764–766. doi: 10.1038/214764a0. [DOI] [PubMed] [Google Scholar]
- Bolton D.C., McKinley M.P., Prusiner S.B. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309–1311. doi: 10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- Byers B., Goetsch L. Preparation of yeast cells for thin-section electron microscopy. Methods Enzymol. 1991;194:602–608. doi: 10.1016/0076-6879(91)94044-d. [DOI] [PubMed] [Google Scholar]
- Carlson G.A., Kingsbury D.T., Goodman P.A., Coleman S., Marshall S.T., DeArmond S., Westaway D., Prusiner S.B. Linkage of prion protein and scrapie incubation time genes. Cell. 1986;46:503–511. doi: 10.1016/0092-8674(86)90875-5. [DOI] [PubMed] [Google Scholar]
- Chabry J., Priola S.A., Wehrly K., Nishio J., Hope J., Chesebro B. Species-independent inhibition of abnormal prion protein (PrP) formation by a peptide containing a conserved PrP sequence. J. Virol. 1999;73:6245–6250. doi: 10.1128/jvi.73.8.6245-6250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff Y.O., Lindquist S.L., Ono B.-I., Inge-Vechtomov S.G., Liebman S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- Chesebro B., Race R., Wehrly K., Nishio J., Bloom M., Lechner D., Bergstrom S., Robbins K., Mayer L., Keith J.M. Identification of scrapie prion protein-specific mRNA in scrapie-infected brain. Nature. 1985;315:331–333. doi: 10.1038/315331a0. [DOI] [PubMed] [Google Scholar]
- Coschigano P.W., Magasanik B. The URE2 gene product of Saccharomyces cerevisiae plays an important role in the cellular response to the nitrogen source and has homology to glutathione S-transferases. Mol. Cell. Biol. 1991;11:822–832. doi: 10.1128/mcb.11.2.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox B.S. PSI, a cytoplasmic suppressor of super-suppressor in yeast. Heredity. 1965;20:505–521. [Google Scholar]
- DePace A.H., Santoso A., Hillner P., Weissman J.S. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell. 1998;93:1241–1252. doi: 10.1016/s0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- Dickinson A.G., Meikle V.M.H., Fraser H. Identification of a gene which controls the incubation period of some strains of scrapie in mice. J. Comp. Path. 1968;78:293–299. doi: 10.1016/0021-9975(68)90005-4. [DOI] [PubMed] [Google Scholar]
- Edskes H.K., Gray V.T., Wickner R.B. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc. Natl. Acad. Sci. USA. 1999;96:1498–1503. doi: 10.1073/pnas.96.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Bellot E., Guillemet E., Cullin C. The yeast prion [URE3] can be greatly induced by a functional mutated URE2 allele. EMBO J. 2000;19:3215–3222. doi: 10.1093/emboj/19.13.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover J.R., Kowal A.S., Shirmer E.C., Patino M.M., Liu J.-J., Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae . Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- Griffith J.S. Self-replication and scrapie. Nature. 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- Griffiths G., McDowall A., Back R., Dubochet J. On the preparation of cryosections for immunocytochemistry. J. Ultrastruct. Res. 1984;89:65–78. doi: 10.1016/s0022-5320(84)80024-6. [DOI] [PubMed] [Google Scholar]
- King C.-Y., Tittmann P., Gross H., Gebert R., Aebi M., Wuthrich K. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc. Natl. Acad. Sci. USA. 1997;94:6618–6622. doi: 10.1073/pnas.94.13.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroute F. Non-mendelian mutation allowing ureidosuccinic acid uptake in yeast. J. Bacteriol. 1971;106:519–522. doi: 10.1128/jb.106.2.519-522.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddelein M.-L., Wickner R.B. Two prion-inducing regions of Ure2p are non-overlapping. Mol. Cell. Biol. 1999;19:4516–4524. doi: 10.1128/mcb.19.6.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masison D.C., Wickner R.B. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science. 1995;270:93–95. doi: 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- Moriyama H., Edskes H.K., Wickner R.B. prion propagation in Saccharomyces cerevisiaerequirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p Mol. Cell. Biol. 20 2000. 8916 8922[URE3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesch B., Westaway D., Walchli M., McKinley M.P., Kent S.B., Aebersold R., Barry R.A., Tempst P., Templow D.B., Hood L.E. A cellular gene encodes scrapie PrP 27-30 protein. Cell. 1985;40:735–746. doi: 10.1016/0092-8674(85)90333-2. [DOI] [PubMed] [Google Scholar]
- Parsell D.A., Kowal A.S., Singer M.A., Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- Patino M.M., Liu J.-J., Glover J.R., Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- Paushkin S.V., Kushnirov V.V., Smirnov V.N., Ter-Avanesyan M.D. Propagation of the yeast prion-like [psi +] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- Prusiner S.B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Schlumpberger M., Willie H., Baldwin M.A., Butler D.A., Herskowitz I., Prusiner S.B. The prion domain of yeast Ure2p induces autocatalytic formation of amyloid fibers by a recombinant fusion protein. Protein Sci. 2000;9:440–451. doi: 10.1110/ps.9.3.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K.L., Cheng N., Williams R.W., Steven A.C., Wickner R.B. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science. 1999;283:1339–1343. doi: 10.1126/science.283.5406.1339. [DOI] [PubMed] [Google Scholar]
- TerAvanesyan A., Dagkesamanskaya A.R., Kushnirov V.V., Smirnov V.N. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae . Genetics. 1994;137:671–676. doi: 10.1093/genetics/137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuite M.F. Yeast prions and their prion-forming domain. Cell. 2000;100:289–292. doi: 10.1016/s0092-8674(00)80663-7. [DOI] [PubMed] [Google Scholar]
- van Tuinen E.-J. Immunoelectron microscopy. Methods Mol. Biol. 1996;53:407–420. doi: 10.1385/0-89603-319-8:407. [DOI] [PubMed] [Google Scholar]
- Wickner R.B. as an altered URE2 proteinevidence for a prion analog in Saccharomyces cerevisiae Science. 264 1994. 566 569[URE3] [DOI] [PubMed] [Google Scholar]
- Wickner R.B., Taylor K.L., Edskes H.K., Maddelein M.-L., Moriyama H., Roberts B.T. Prions of yeast as heritable amyloidoses. J. Struct. Biol. 2000;130:310–322. doi: 10.1006/jsbi.2000.4250. [DOI] [PubMed] [Google Scholar]