

Abstract
Xist RNA expression, methylation of CpG islands, and hypoacetylation of histone H4 are distinguishing features of inactive X chromatin. Here, we show that these silencing mechanisms act synergistically to maintain the inactive state. Xist RNA has been shown to be essential for initiation of X inactivation, but not required for maintenance. We have developed a system in which the reactivation frequency of individual X-linked genes can be assessed quantitatively. Using a conditional mutant Xist allele, we provide direct evidence for that loss of Xist RNA destabilizes the inactive state in somatic cells, leading to an increased reactivation frequency of an X-linked GFP transgene and of the endogenous hypoxanthine phosphoribosyl transferase (Hprt) gene in mouse embryonic fibroblasts. Demethylation of DNA, using 5-azadC or by introducing a mutation in Dnmt1, and inhibition of histone hypoacetylation using trichostatin A further increases reactivation in Xist mutant fibroblasts, indicating a synergistic interaction of X chromosome silencing mechanisms.
Keywords: X chromosome, Xist gene, DNA methylation, histone deacetylase, gene silencing
Introduction
In mammals, equal X-linked gene dosage between the sexes is achieved by X chromosome inactivation in females. The inactivated X chromosome resembles constitutive heterochromatin in that it is condensed in interphase (Barr and Carr 1962), hypoacetylated on histone H4 (Jeppesen and Turner 1993), and replicates late in S phase (Priest et al. 1967). It is also methylated on CpG islands of housekeeping genes (Norris et al. 1991) and is enriched in histone macroH2A1, a histone H2A variant with a large nonhistone domain (Costanzi and Pehrson 1998). The inactive X (Xi) expresses Xist (Borsani et al. 1991; Brockdorff et al. 1991; Brown et al. 1991), a nuclear untranslated RNA (Brockdorff et al. 1992) that coats the chromosome in cis (Clemson et al. 1996). Both X chromosomes of an undifferentiated embryonic female cell are active, and X inactivation is initiated at the time of differentiation in vitro or in vivo (Monk and Harper 1979). Once an X chromosome is inactivated in a cell, the inactive state of the chromosome is clonally inherited through many rounds of cell division.
The most remarkable feature of Xi chromatin is its stability with respect to reactivation. Overall experimental reactivation of the entire chromosome in somatic cells has not been observed. Reactivation of one or few genes on the Xi has been seen, but the rate of reactivation is low, on the order of 10−5 to 10−4, in cultured somatic cells (Mohandas et al. 1981; Graves 1982). Many lines of evidence indicate that multiple molecular mechanisms are responsible for the high fidelity of maintenance of X inactivation (for review see Migeon 1994), but the contribution of individual mechanisms to silencing and their complex relationship remains to be elucidated.
Methylation of CpG dinucleotides in the promoter region of repressed genes has long been thought of as a mechanism stabilizing X chromosome inactivation. 5-azadC, an inhibitor of DNA methyltransferase 1 (Dnmt1), the major enzyme responsible for maintaining genomic methylation patterns, has been used to derepress several Xi-linked genes, providing the experimental evidence for the importance of methylation in X inactivation (Mohandas et al. 1981; Graves 1982). Methylation of the CpG island of hypoxanthine phosphoribosyl transferase (Hprt) follows X inactivation by several days, implying that methylation plays a maintenance role (Lock et al. 1987). An in vivo demonstration of the importance of CpG methylation in X inactivation maintenance is the instability of silencing on the Xi of ICF (immunodeficiency centromeric instability facial anomalies) patients. The Xi of patients suffering from ICF syndrome is hypomethylated at all CpG islands analyzed (Hansen et al. 2000). The syndrome is caused by a defect in the DNA methyltransferase DNMT3B (Hansen et al. 1999; Okano et al. 1999; Xu et al. 1999), suggesting that the enzyme may be responsible for establishing CpG methylation on Xi. X inactivation in the virtual absence of CpG methylation is less stable, as evidenced by reactivation of some loci in cells from these patients (Hansen et al. 2000).
Underacetylation of NH2-terminal lysine residues on histones H3 and H4 is another feature of Xi chromatin (Jeppesen and Turner 1993). It is well established that transcriptional activity of genes is regulated by histone acetylation. Histone deacetylation generally correlates with transcriptional silencing and high levels of acetylation with transcriptional activity (Cheung et al. 2000). However, reactivation of X-inactivated genes by altering histone acetylation levels has not been reported. The appearance of a hypoacetylated X chromosome is a late event during differentiation and X inactivation, suggesting that deacetylation of histones is a maintenance rather than establishment mechanism (Keohane et al. 1996).
Xist RNA is essential for the initiation of X chromosome inactivation in cis (Penny et al. 1996; Marahrens et al. 1997). However, several lines of evidence indicate that after X inactivation has been established, Xist is no longer required for maintenance. In studies using mouse–human somatic cell hybrids, a human Xi chromosome fragment that lacked the XIST gene remained transcriptionally silent and its sensitivity to reactivation by 5-azadC did not increase (Brown and Willard 1994). Similarly, in human leukemia cells, an Xi-derived isodicentric chromosome maintained its inactive state despite missing the XIST gene (Rack et al. 1994). To directly address the role of continued Xist RNA expression in karyotypically normal somatic cells, we have previously generated a conditional Xist allele (Csankovszki et al. 1999) using the Cre-loxP system (Sauer and Henderson 1988). After Cre-mediated deletion of Xist in primary mouse embryonic fibroblasts, the Xi remained silent (Csankovszki et al. 1999), again arguing that, in the absence of Xist RNA, other silencing mechanisms are sufficient to keep Xi silent. Another study used embryonic stem cells expressing an inducible Xist cDNA transgene, in which the timing of expression can be experimentally manipulated (Wutz and Jaenisch 2000). This approach defined an initial differentiation time window in which X inactivation is reversible and Xist dependent, followed by irreversible and Xist-independent X inactivation in fully differentiated cells (Wutz and Jaenisch 2000).
Nevertheless, continued transcription of Xist RNA and its close association with Xi throughout the lifetime of the female mammal suggests a role for Xist in somatic cells (Clemson et al. 1996). Indeed, in rodent–human somatic cell hybrids where the human XIST RNA does not localize correctly to Xi (Clemson et al. 1998; Hansen et al. 1998), the stability of silencing is greatly reduced. It has been speculated that reduced efficiency of silencing in hybrid cells is due to the absence of correctly functioning XIST RNA (Clemson et al. 1998; Hansen et al. 1998). Furthermore, deletion of Xist in mouse embryonic fibroblasts disrupts preferential localization of histone macroH2A1 to Xi, and this alteration of chromatin may also lead to decreased stability of silencing (Csankovszki et al. 1999).
To assess the possible role of Xist RNA in X inactivation maintenance and to study the relative contribution of Xist, DNA methylation, and histone hypoacetylation, we developed a system in which even low levels of reactivation can be quantitatively measured. Reactivation of two markers on Xi were studied, a green fluorescent protein (GFP) transgene, GFP (Hadjantonakis et al. 1998), and the endogenous Hprt gene. Using a conditional deletion of Xist (Csankovszki et al. 1999), we provide direct evidence for the first time that Xist RNA contributes to silencing in somatic cells. Additionally, using the drugs 5-azadC and trichostatin A (TSA) and by introducing a mutation in the Dnmt1 gene (Jackson-Grusby et al. 2001), we show that Xist RNA, DNA methylation, and histone hypoacetylation act synergistically to achieve a highly stable inactive state.
Materials and Methods
Mice and Preparation of Mouse Embryonic Fibroblasts
The Xist2lox(Csankovszki et al. 1999), the Xist Δ (Marahrens et al. 1997), the GFP mice (Hadjantonakis et al. 1998), and the Hprt Δ mice (Hooper et al. 1987) have been described elsewhere. To obtain mice carrying the GFP transgene and the Xist2lox allele in cis, germline recombinants were generated from double heterozygous females. Recombination frequency was 40% (n = 115). Similarly, we obtained mice with the Hprt Δ and Xist Δ alleles in cis from mice heterozygous for both mutations in trans (27% recombination frequency, n = 81). Finally, Xist Δ ;Hprt Δ/Xist + ;Hprt Δ females were mated to Xist2lox;Hprt + ;GFP/Y males to generate Xist conditional mutant fibroblasts and Xist + ;Hprt + ;GFP/Y males to generate controls. The Dnmt2lox(Jackson-Grusby et al. 2001) and DnmtS(Lei et al. 1996) mutations were bred into the colony to create Xist, Dnmt1 double conditional knockout fibroblasts.
Primary mouse embryonic fibroblasts were derived from dissociation and trypsinization of embryonic day 14 embryos, cultured, and when necessary immortalized with SV-40 T-antigen (Jat et al. 1986). For Cre-mediated recombination in fibroblasts, cells were infected with an adenovirus vector carrying the gene for Cre recombinase (Anton and Graham 1995). Infection was carried out in monolayer culture in DME with 2% fetal calf serum for 2 h. The lowest possible multiplicity of infection that yielded 100% recombination without cytotoxic effects was experimentally determined. Uninfected cells were treated identically, without the addition of virus. After infection, cells were grown in DME with 15% fetal calf serum with antibiotics. When appropriate, 5-azadC (Sigma-Aldrich) was added to the cultures to a final concentration of 0.3 μM, and TSA (Sigma-Aldrich) to final concentration of 500 nM.
Southern Blotting
To analyze the efficiency of Cre-mediated Xist deletion, genomic DNA of infected cells was digested with XbaI, blotted, and hybridized with probe 7, a 1.1-kb EcoRI-XbaI fragment at the beginning of exon 7. To analyze recombination at the Dnmt1 locus, genomic DNA was digested with SpeI and hybridized with the HV probe (Jackson-Grusby et al. 2001). To analyze demethylation of genomic DNA, an HpaII digest was performed, and the blot was hybridized with a probe covering the gag coding region of intracisternal A particle (IAP) element (nucleotides 1570–1899, sequence data available from GenBank/EMBL/DDBJ under accession no. M17551) (Walsh et al. 1998).
FACS® Analysis
Fibroblast cultures were trypsinized to obtain single cell suspension and resuspended in complete medium. Propidium iodide was added to 1 μg/ml. Viable cells were gated using scatter properties and exclusion of propidium iodide. 100,000–500,000 cells were analyzed for each bulk sample and 10,000 cells for poorly growing hypoxanthine/aminopterin/thymidine (HAT)–resistant clones. To isolate GFP-positive clones, single GFP-positive cells were sorted into wells of a 96-well plate containing DME with 15% fetal calf serum. After 2 wk, 20–30% of the wells contained fibroblast clones, of which ∼30% were GFP-positive or contained a high percentage of GFP-positive cells.
HAT Selection and Calculation of Reactivation Frequencies
For selection of clones carrying a reactivated Hprt gene, the appropriate number of cells were plated and selected in 1× ESQ HAT (Stratagene) containing media for 14 d. After selection, HAT-resistant clones were picked into regular media. For counting the number of HAT-resistant clones, plates were fixed in methanol/acetic acid (3:1) and stained with Giemsa. For Luria-Delbrück fluctuation analysis (Luria and Delbrück 1943), small independent fibroblast cultures were expanded to the appropriate size, plated at a density equivalent of a 1:9 to 1:6 split in HAT containing medium, and selected, fixed, and stained as above. Reactivation rates were calculated according to the P0 method of Luria and Delbrück 1943 (rate = −ln[proportion of negative cultures]/average culture size).
Analysis of Replication Timing
Cells were grown in the presence of 30 μM BrdU for the last 5 h before fixation. Colcemid (Sigma-Aldrich) was added for the last hour. Cells were fixed in methanol/acetic acid (3:1) and dropped onto slides. Slides were denatured in 70% formamide/2× SSC at 70–74°C for 2 min. BrdU signal was detected using a monoclonal anti-BrdU antibody (Becton Dickinson) followed by fluorescein-conjugated anti–mouse antibody (Vector Laboratories) in blocking buffer (1× PBS, 5% goat serum, 0.2% Tween, 0.2% fish skin gelatin). Slides were washed in 1× PBS/0.2% Tween, dehydrated, and used for DNA FISH without further denaturation of the chromosomes. The Xi was identified with a directly labeled GFP/Pgk-Puro probe. The inserts of EGFP-N1 plasmid (CLONTECH Laboratories, Inc.), and pPGKPuro were isolated, pooled, and labeled with Cy3-dCTP (Amersham Pharmacia Biotech) using random priming. Using only the GFP insert as a probe yielded identical results, but the signals were weaker. Only results obtained with the GFP/Pgk-Puro probe are shown. Hybridization of the probe and washing were performed as described (Panning and Jaenisch 1996).
Results
Generation of Xist Mutant Mouse Embryonic Fibroblasts
To study the stability of silencing on Xi, we generated fibroblasts with two Xi-linked markers, where reactivation was designed to be detectable even at low frequencies. One marker was an X-linked GFP transgene that is subject to X inactivation (Hadjantonakis et al. 1998). When the GFP transgene was located on Xi, cells were GFP negative and reactivation could be monitored using FACS® analysis. The insertion site of the transgene was determined by DNA FISH, and GFP was mapped to a position near the centromere (Fig. 1 A, and see Fig. 5 A). The second marker was the endogenous X-linked Hprt gene, the activity of which is required in order to survive in HAT containing medium. When cells carrying a wild-type Hprt allele on Xi and a mutant allele, Hprt Δ (Hooper et al. 1987), on the active X (Xa) are subjected to HAT selection, even a few reactivants in a large population can be isolated. To generate a homogeneous population of cells with defined Xa and Xi chromosomes, a null mutation in the Xist gene, Xist Δ (Marahrens et al. 1997), was introduced onto the chromosome carrying Hprt Δ. Since the Xist Δ allele cannot be chosen for X inactivation (Marahrens et al. 1998), cells with genotype Hprt + ;GFP/Xist Δ ;Hprt Δ carry the GFP transgene and the Hprt + allele on Xi.
Figure 1.

Generation of Xist conditional mutant fibroblasts with X-inactivated GFP and Hprt genes. (A) Map of the X chromosome with approximate genetic distances between genes. (B) Genotypes of Xist conditional mutant and control fibroblasts. Both cell types are phenotypically GFP negative and HAT sensitive, as the GFP transgene and the only functional Hprt allele are on the Xi and are inactivated (i). Conditional mutant cells carry the Xist2lox allele on Xi, whereas controls carry a wild-type Xist allele. (C) Cre-mediated deletion of Xist after adenovirus-Cre infection. Southern blotting of XbaI-digested DNA hybridized with probe 7 (pr7) indicates 100% recombination in primary (1°) and SV-40 T-antigen–immortalized (Tag) cells.
Figure 5.

Late-replicating Xi chromosomes in clones with reactivated GFP and Hprt. (A) Mapping of the GFP transgene insertion site by DNA FISH. A Cy3-labeled GFP/Pgk-Puro probe (red) was hybridized to denatured chromosomes (DAPI, blue). Enlargement of a single X chromosome is shown with the centromere staining brighter with DAPI than the rest of the chromosome. The arrow shows the site of transgene integration near the centromere. The doublet signal corresponds to sister chromatids. (B) Analysis of replication timing of the inactive chromosome. BrdU incorporation into late-replicating regions of the genome was detected using a monoclonal anti-BrdU antibody and fluorescein-anti–mouse antibody (green) on DAPI-stained metaphase chromosome spreads (blue). Xi (arrow) was identified using the GFP/Pgk-Puro probe (red). In all clones analyzed, the Xi with reactivated genes was late replicating. (C) An enlargement of a single late-replicating Xi.
To study the effect of deletion of Xist from Xi, a conditional allele of the gene Xist2lox (Csankovszki et al. 1999) was introduced onto the chromosome. Cells with genotype Xist2lox;Hprt + ;GFP/Xist Δ ;Hprt Δ will be referred to as conditional mutants. Control cells with genotype Xist + ;Hprt + ;GFP/Xist Δ ;Hprt Δ lack the Xist2lox allele, and therefore cannot delete the Xist gene (Fig. 1 B). To induce Cre-mediated deletion of Xist2lox, we infected cells with an adenovirus carrying the gene encoding Cre recombinase (Anton and Graham 1995). An Xist2lox to Xist1lox recombination was observed in 100% of the cells, as assayed by Southern blotting (Fig. 1 C). Conditional mutants and controls were treated identically, and infections and all further analyses were done in triplicate. Cre-mediated deletion of Xist took place over a period of 2–3 d, and Xist RNA levels were undetectable by day 4 (Csankovszki et al. 1999; data not shown). In a previous study, we showed that deletion of Xist in mouse embryonic fibroblasts does not interfere with late replication timing and underacetylation of histone H4 residues, but it disrupts preferential localization of histone macroH2A1 to Xi (Csankovszki et al. 1999).
Deletion of Xist Leads to Increased Reactivation of GFP and Hprt
GFP expression was analyzed using FACS® in Xist mutant and control cells that were infected with adenovirus-Cre or left untreated. GFP fluorescence was compared with autofluorescence, and cells showing greater GFP fluorescence than autofluorescence were counted as positive. We found that the number of cells expressing GFP was dependent on cell density and both the number of cells expressing GFP and the intensity of fluorescence decreased upon long term culture (data not shown). Therefore, care was taken to ensure that an equal number of cells were plated for each sample and cells from the same early passage were used for each experiment. Results of a typical experiment are shown in Fig. 2. Fibroblasts not containing the GFP transgene (genotype +/Y) were negative, whereas over 99.9% of cells with GFP on Xa (GFP/Y) were positive. The majority of cells with GFP on Xi were also GFP negative with a small number of positives, ∼10–20 in 100,000, representing the spontaneous reactivation frequency (Fig. 2 A). In primary Xist conditional mutant fibroblasts after Cre-mediated deletion of Xist, the number of GFP-positive cells increased about twofold (Fig. 2 B). This increase cannot be attributed to the effect of adenovirus infection, as the number of GFP-positive cells did not increase in control cells after viral infection. Although the effect is small, we consider it significant as we consistently detected a twofold difference in GFP expressing cells between mutant and control cells in five independent repetitions of the assay.
Figure 2.

Reactivation of GFP in Xist mutant fibroblasts. FACS® analysis of cells. (A) Live cells were gated, and their GFP fluorescence was plotted against autofluorescence. Dots to the right of the diagonal represent cells in which GFP fluorescence is greater than autofluorescence and therefore are considered GFP positive. GFP-negative (+/Y) and GFP-positive (GFP/Y) populations are shown for control. Xist conditional mutant cell populations, before Cre-mediated deletion of Xist (−cre), contained a small number of GFP-positive cells. After Cre-mediated deletion of Xist (+cre), the number of GFP-positive cells increased. (B) Number of GFP-positive cells in primary conditional mutants and in controls with or without adenovirus-Cre infection on day 7 after infection. In Xist conditional mutants, Cre-mediated deletion of Xist led to a twofold increase in GFP-positive cells, whereas controls remained unchanged. (C) Long-term culture of SV-40 T-antigen–transformed conditional mutant cells. FACS® analysis was performed at various time points after adenovirus-Cre infection. Although initially we observed an increase in the number of GFP-positive cells after Cre-mediated deletion of Xist, the number decreased after another week in culture to reach the level of spontaneous reactivation and remained at that level for the duration of the experiment.
We considered the possibility that allowing the cells to go through many rounds of cell division in the absence of Xist will lead to an increased number of cells reactivating GFP. To test this hypothesis, we derived permanent cell lines by SV-40 T-antigen transformation and cultured the cells for over 2 mo. GFP expression was analyzed at various time points (Fig. 2 C). 7 d after adenoviral infection, the transformed cells behaved similarly to primary cells: the conditional mutants exhibited a two- to threefold increase in the number of GFP-positive cells after Cre-mediated deletion of Xist, whereas there was no difference between infected and uninfected control cells. However, by day 14 after infection, the difference between the infected and uninfected mutant cells disappeared, and the proportion of GFP-positive cells remained unchanged for >2 mo. Reactivated GFP alleles are likely subject to resilencing, consistent with our observations that GFP expression declines over time and with earlier studies that showed that this transgene is subject to nonspecific silencing, even when carried on Xa (Eggan et al. 2000). In addition, it is possible that reactivation of X-inactivated genes confers selective disadvantage on the cells leading to a decrease in the number of GFP-positive cells over time (see below).
Next, we analyzed reactivation of an endogenous X-inactivated gene, Hprt, by subjecting cells to selection in HAT medium. Since HAT-resistant colonies from rare reactivants can only be obtained in permanent cell lines, these experiments were performed in SV-40 T-antigen–immortalized cells. Two million cells were plated and selected in HAT medium for 2 wk, after which plates were fixed and stained to count the number of colonies (Fig. 3 A). Very few (<1/plate) colonies were seen on control plates and on plates containing conditional mutant fibroblasts before Cre-mediated Xist, indicating that the spontaneous reactivation frequency of Hprt is very low. However, after deletion of Xist, a small but significant number of cells reactivated Hprt, and Xist mutant cultures yielded ∼30–70 HAT-resistant colonies per plate. The proportion of HAT-resistant cell in the population increased for the first 4 wk in culture indicating ongoing reactivation of Hprt. Furthermore, HAT-resistant cells were detected even after 3 mo in culture (Fig. 3 B). Northern analysis of total RNA indicated that different clones transcribed different amounts of Hprt RNA (data not shown). We conclude that deletion of Xist leads to an increased, albeit low, frequency of reactivation of an endogenous X-inactivated gene, Hprt, indicating that Xist RNA contributes to stabilizing the inactive state in the maintenance phase of X inactivation.
Figure 3.
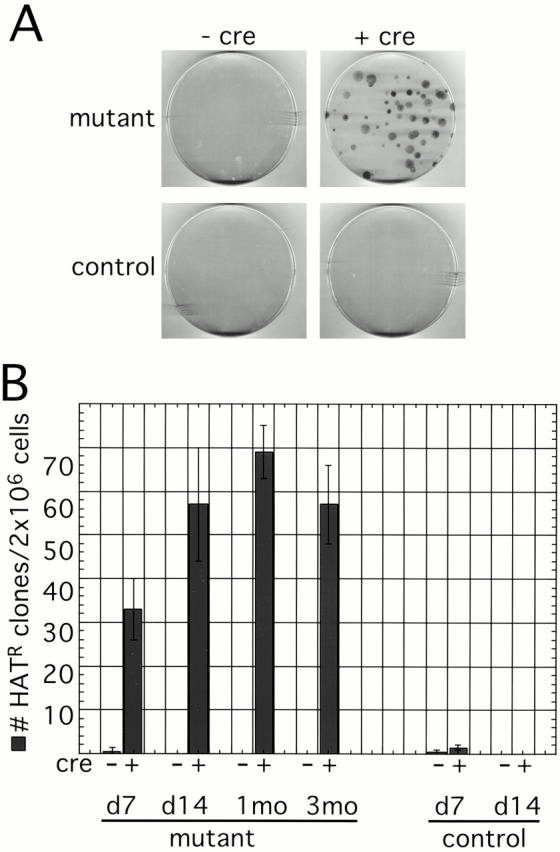
Reactivation of Hprt in Xist mutant fibroblasts. (A) HAT selection of Xist conditional mutant and control SV-40 T-antigen–immortalized fibroblasts with (+cre) or without (−cre) adenovirus-Cre infection. 2 × 106 cells were plated, selected in HAT media, and then fixed and stained. Cells that reactivated the Xi-linked Hprt gene were able to proliferate and form colonies. No or very few colonies were observed on control plates and plates containing conditional mutants before adenovirus-Cre infection. However, after Cre-mediated deletion of Xist in conditional mutants, HAT-resistant colonies were routinely detected. (B) The proportion of HAT-resistant cells in the culture does not change significantly after culturing cells for 7 and 14 d, and 1 and 3 mo after Cre-mediated deletion of Xist.
Patchy Reactivation of X-inactivated Genes in Xist Mutant Fibroblasts
As the nature of Xist RNA–mediated silencing has not been studied in detail in somatic cells, we next examined whether Xist RNA coordinately silences genes on Xi or whether genes are regulated independently of one another. First, we isolated HAT-resistant clones with a reactivated Hprt allele and asked whether these cells are also GFP-positive (Fig. 4). We analyzed two types of clones, fast-growing ones that grew almost as well as the unselected population (n = 6), and slow-growing ones that divided barely enough to yield sufficient cells for FACS® analysis (n = 7). In Xist mutant and Xist wild-type bulk populations before HAT selection, the number of GFP-positive cells was ∼1 in 10,000. Among fast-growing clones, the variation was large, but on average the clones contained 10–20 times more GFP-positive cells than the unselected populations. In slow-growing clones, the number of GFP-positive cells was on average another 10-fold higher (Fig. 4). Yet, even in the clones with the highest proportion of GFP-positive cells, over 98% of cells remained GFP negative. Therefore, most cells that reactivated Hprt generally did not also reactivate GFP. However, a chromosome that reactivated Hprt was more prone to reactivate GFP than a chromosome with an inactive Hprt gene.
Figure 4.

FACS® analysis of Hprt-positive clones. The number of GFP-positive cells is higher in HAT-resistant clones than in bulk unselected cultures. Slow-growing clones reactivated GFP in a higher proportion of cells than fast-growing ones.
It is interesting to note that HAT-resistant clones with a higher proportion of GFP-positive cells grew slower than those with fewer GFP-positive cells. This result is consistent with the conclusion that reactivation of X-inactivated genes confers selective disadvantage on the cells. In clones with a higher proportion of GFP-positive cells, the more extensive reactivation is more detrimental to cell growth.
Finally, we isolated GFP-positive clones by sorting and expanding individual GFP-positive cells, and tested whether these clones also reactivated Hprt. Similarly to the HAT-resistant clones, GFP-positive clones did not grow well, and most died under HAT selection without yielding a single colony. However, occasionally (2 of the 42 clones analyzed), the clone grew well in HAT-containing medium, indicating that most, if not all, cells of the clone were also Hprt positive. We conclude that most clones that reactivated one X-linked gene do not also reactivate another. Yet, there is a certain level of cooperativity in reactivation, as a chromosome that reactivated one gene on Xi, is more likely to reactivate another one than a chromosome that has not reactivated any gene.
X Chromosomes with Reactivated Genes Remain Late Replicating
An Xi chromosome after deletion of Xist remains late replicating when analyzed in bulk population with presumably very few of the cells containing reactivated genes (Csankovszki et al. 1999). We wished to see whether in the reactivated clones, Xi, or a cytologically visible portion of it, became early replicating. We analyzed two slow-growing HAT-resistant clones with a high proportion of GFP-positive cells and one GFP-positive clone that was also HAT resistant. As the rest of the clones grew too poorly to obtain sufficient numbers of cells for the assay, we pooled small HAT-resistant colonies and GFP-positive clones, and the assay was also performed on the pools. BrdU incorporation into late-replicating regions was detected using an anti-BrdU antibody and Xi was identified by DNA FISH using a probe that detects the GFP transgene. In all clones and pools analyzed, the Xi marked by the GFP transgene was late replicating (Fig. 5B and Fig. C). These results indicate that even after reactivating one or more genes on Xi, the chromosome as a whole remained inactive. We conclude that reactivation of Xi-linked genes after loss of Xist RNA occurs at one or a few loci and does not result in a detectable change in late replication of the chromosome.
Synergism of Xist RNA, DNA Methylation, and Histone Hypoacetylation in X Chromosome Silencing
Next, we examined the relationship between Xist RNA-mediated silencing and inactivation by other mechanisms. DNA methylation and hypoacetylation of core histones are believed to contribute to inactivation of X-linked genes (Cedar 1988; Keohane et al. 1998). A low frequency of reactivation of X-linked genes has been observed after demethylating cells using 5-azadC (Mohandas et al. 1981; Graves 1982). Transcriptional activation of silenced genes has also been seen after treating cells with TSA, a potent inhibitor of histone deacetylases (Yoshida et al. 1995). We wanted to see whether the Xist mutant cells with their already compromised ability to silence Xi are more sensitive to demethylation and/or inhibition of histone deacetylation than wild-type cells, leading to a further increase in the number of cells reactivating GFP and Hprt.
Adenovirus-Cre–infected and –uninfected Xist conditional mutant cells were treated with 5-azadC and/or TSA, and the number of cells that reactivated GFP was analyzed using FACS® (Fig. 6 A). Due to the toxicity of the drugs, the experiments were performed in SV-40 T-antigen–transformed cells. The cells were infected with adenovirus-Cre, allowed to recover, and then treated twice with 5-azadC on days 7 and 9 after infection. After allowing the cells to go through several rounds of cell division to achieve demethylation of DNA, half of the cultures were treated with TSA on day 12 after infection. FACS® analysis was performed on day 13. By day 13 after infection, the Xist deletion–induced increase in the number of GFP reactivants disappeared, and we could no longer observe a difference between Xist wild-type and Xist-deficient cells without drug treatment (Fig. 2 C). Inhibition of histone deacetylases by itself had no effect on the number of GFP-positive cells, whether or not Xist was deleted. 5-azadC–induced demethylation increased the number of GFP-positive cells by ∼20-fold in cells that did not delete Xist. However, the combined effect of Xist deletion and 5-azadC treatment was a 30–40-fold increase in GFP reactivants. 5-azadC treatment followed by TSA further increased the number of GFP-positive cells by another twofold in both Xist mutant and control cells. These results are summarized in Table . The ≤60-fold increase in the number of cells reactivating GFP indicates that the effects of Xist RNA deletion, 5-azadC, and TSA treatments are synergistic, rather than simply additive.
Figure 6.

Synergism of X chromosome silencing mechanism. (A) FACS® analysis of GFP reactivation in Xist mutant fibroblasts with or without Cre infection treated with 5-azadC and/or TSA. TSA by itself had no effect. However, 5-azadC by itself, or followed by TSA treatment, led to significant GFP reactivation. Xist deleted cells were more sensitive to the treatment than those that did not delete Xist. (B) GFP reactivation in Xist/Dnmt1 double conditional mutant fibroblasts. Control cells (Dnmt12lox/Sand Xist +/Δ) delete Dnmt1 upon adenovirus-Cre infection, whereas Xist mutant cells (Dnmt12lox/Sand Xist2lox/ Δ) delete both Dnmt1 and Xist. Both primary (1°) and SV-40 T-antigen–immortalized (Tag) cells were analyzed. Deletion of Dnmt1 alone led to reactivation of GFP in ≤7% of primary and ≤15% of T-antigen–transformed cells. Deletion of Xist also in addition to Dnmt1 increased the number of GFP-positive cells about twofold. (C) The number of HAT-resistant colonies in Xist conditional mutant treated with 5-azadC. Cre-mediated deletion of Xist had a more significant effect on the number of HAT-resistant cells than 5-azadC treatment. However, deletion of Xist followed by 5-azadC treatment reactivated Hprt in a much higher proportion of cells than either treatment alone. Note the use of log scale.
Table 1.
Synergistic Effect of X Chromosome Silencing Mechanisms on the Repression of GFP and Hprt
| Xist RNA | Histone macroH2A enrichment | DNA methylation | Histone hypoacetylation | Increase GFP | Increase Hprt |
|---|---|---|---|---|---|
| + | + | + | + | 1× | 1× |
| Δ | − | + | + | 2–3× | 100× |
| + | + | 5-azadC | + | 19× | 12× |
| + | + | + | TSA | 1× | ND |
| Δ | − | 5-azadC | + | 30× | 4,800× |
| + | + | 5-azadC | TSA | 29× | ND |
| Δ | − | 5-azadC | TSA | 60× | ND |
| + | + | Δ | + | 1,500× | ND |
| Δ | − | Δ | + | 2,500× | ND |
Fold increase in the number of GFP and Hprt expressing cells following deletion of Xist and/or Dnmt1, inhibition of DNA methylation using 5-azadC, and/or inhibition of histone deacetylation using TSA. Basal level of reactivation in untreated wild-type cells is designated as 1×. +, intact; Δ, a null mutation in the gene Xist or Dnmt1; 5-azadC, cells were demethylated using 5-azadC; TSA, histone deacetylation was inhibited.
5-azadC treatment leads to limited genomic demethylation (Fig. 7 A). More extensive demethylation can be achieved by deleting Dnmt1. To study how X inactivation is maintained in the absence of Xist RNA and DNA methylation, we bred a Dnmt1 conditional allele (Jackson-Grusby et al. 2001) into the Xist mutant colony and generated double conditional mutant fibroblasts. Fibroblast genotypes were Xist2lox;GFP/Xist Δ ,Dnmt1 2lox/S, for Xist conditional mutants, and Xist + ;GFP/Xist Δ ,Dnmt1 2lox/S, for control. The Dnmt1S allele is a constitutive null mutation of the gene (Lei et al. 1996). Upon adenovirus-Cre infection of these cells, both Xist and Dnmt1 were deleted in Xist mutant cells, whereas only Dnmt1 was deleted in the controls (Fig. 7 B). Deletion of Dnmt1 led to a more pronounced demethylation of bulk genomic DNA than 5-azadC treatment (Fig. 7). Primary Dnmt1 mutant fibroblasts arrested within 1 wk of adenovirus-Cre infection, whereas SV-40 T-antigen–transformed cells continued dividing, although at a much slower rate than wild-type cells (Jackson-Grusby et al. 2001). We analyzed both primary and transformed cells 7 d after infection. A much higher proportion of cells reactivated GFP in the Dnmt1 mutants than in 5-azadC–treated cells (Fig. 6 B), most likely as a result of the Dnmt1 mutants being much more demethylated. Transformed cells consistently yielded more GFP, reactivants possibly because of their increased ability to proliferate and further demethylate or as a result of decreased stability of silencing in transformed cells. Xist/Dnmt1 double mutants reactivated GFP in about twice as many cells as Dnmt1 single mutants, indicating again that Xist and DNA methylation cooperate to silence GFP on Xi. GFP was reactivated in ≥30% of Dnmt1/Xist double mutant cells, representing an almost 3,000-fold increase over controls (Table ). Interestingly, the Dnmt1 mutation, or even limited demethylation using 5-azadC, has a much more significant effect on the number of GFP reactivants than the Xist mutation, arguing that at least for the X-linked GFP transgene, DNA methylation is a more important contributor to silencing than Xist.
Figure 7.
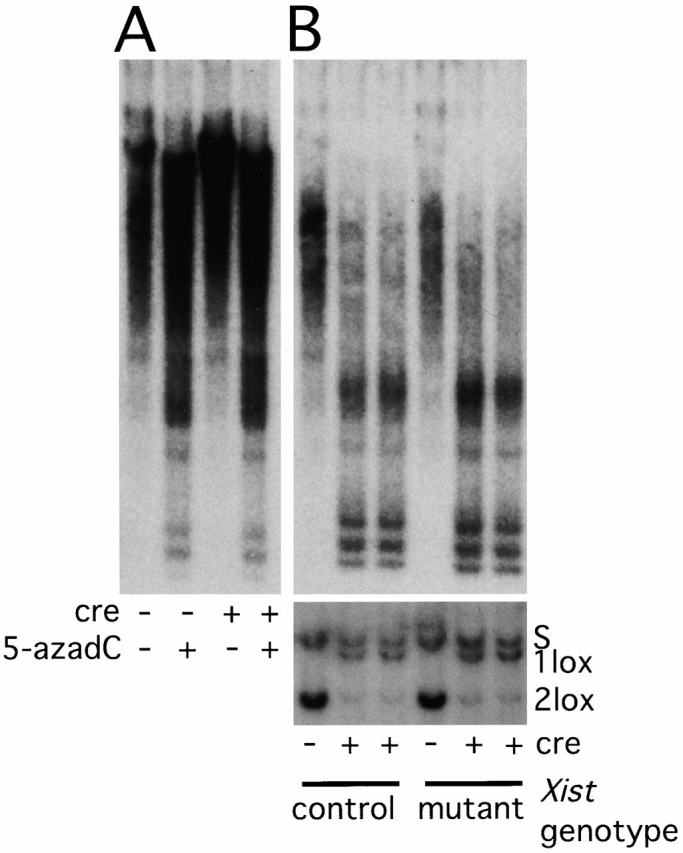
Demethylation of genomic DNA. (A) Demethylation of genomic DNA after 5-azadC treatment of cells. Adenovirus-Cre–infected or –uninfected Xist conditional mutant cells were treated with 5-azadC or were left untreated. Genomic DNA was isolated and digested with the methylation-sensitive restriction enzyme HpaII. The blotted DNA was hybridized with an IAP probe to analyze demethylation of bulk genomic DNA. Demethylation due to 5-azadC treatment is indicated by the appearance of low molecular weight bands. (B) Adenovirus-Cre–mediated recombination in Dnmt12lox/S; Xist +/Δ (control) and Dnmt12lox/S; Xist2lox/ Δ (mutant) cells. Only Dnmt1 recombination in SV-40 T-antigen–immortalized cells is shown on Southern blots of SpeI-digested DNA hybridized with the HV probe (lower blot). Nearly 100% recombination is seen in both controls and mutants. The same genomic DNA samples were also digested with HpaII and hybridized with the IAP probe (upper blot). The appearance of more low molecular weight bands and the disappearance of high molecular weight bands indicate that the genomic DNA in these samples is more extensively demethylated than in 5-azadC–treated cultures.
To study the combined effect of demethylation and Xist deletion on an endogenous X-inactivated gene, we analyzed Hprt reactivation in adenovirus-Cre–infected or –uninfected Xist conditional mutant cells, with or without 5-azadC treatment (Fig. 6 C). In adenovirus-Cre–infected (Xist deleted) cultures without drug treatment, we observed an ∼100-fold enrichment for HAT-resistant cells over uninfected (Xist wild type) cells. 5-azadC treatment of Xist wild-type cells resulted in a less significant 10-fold increase in the number colonies per plate. However, 5-azadC treatment of Xist mutant cells resulted in a 50-fold increase in HAT-resistant cells over untreated Xist mutant cells and an almost 5,000-fold increase over untreated Xist wild-type cells (Table ). The combined effect of Xist deletion and demethylation is more significant than either treatment alone, implying a synergistic interaction of Xist RNA and DNA methylation in keeping Hprt silent. It is interesting to note that although the absolute numbers of GFP- and Hprt-positive cells are different, 5-azadC treatment led to comparable enrichment in reactivants for both genes (19- and 12-fold, respectively). However, Xist RNA seems to play a more major role in Hprt silencing than in GFP silencing (100-fold enrichment compared with two- to threefold enrichment). The GFP transgene may not be subject to Xist RNA–mediated silencing the same way endogenous X-linked genes are regulated.
Rate of Hprt Reactivation in Xist-deficient Fibroblasts
To test whether the observed HAT-resistant colonies are the result of independent Hprt reactivation events or proliferation of a few reactivants, we calculated Hprt reactivation rates using the Luria-Delbrück fluctuation analysis (Luria and Delbrück 1943). A large number of cultures of adenovirus-Cre–infected and –uninfected cells were expanded from a few cells (generally <100). During expansion, half of the cultures were treated with 5-azadC. After the desired culture size was reached, cells were selected using HAT, and the number of cultures that yielded HAT-resistant colonies and the reactivation rates were determined (Table ). The positive cultures ranged from those containing one clone to those containing confluent plates, indicating that the reactivation event took place at different times during cultivation. The spontaneous reactivation rate of Hprt was as low as previously reported mutation rates in wild-type cells (10−9) (Chen et al. 1998), demonstrating the remarkable stability of X chromosome silencing. The reactivation rate increased by ∼160-fold after deletion of Xist and 60-fold after 5-azadC treatment, indicating that both Xist RNA and DNA methylation contribute significantly to silencing. However, combining Xist deletion with 5-azadC treatment resulted in an almost 10,000-fold increase, confirming synergism of DNA methylation and Xist expression in the maintenance of the inactive state.
Table 2.
Hprt Reactivation Rates
| Xist RNA | DNA methylation | No. independent cultures | No. positive cultures | Average no. cells/culture | Reactivation rate |
|---|---|---|---|---|---|
| + | + | 46 | 1 | 3.55 × 106 | 6.2 × 10−9 |
| + | 5-azadC | 46 | 33 | 3.13 × 106 | 4.0 × 10−7 |
| Δ | + | 49 | 36 | 1.32 × 106 | 1.0 × 10−6 |
| Δ | 5-azadC | 90 | 54 | 1.77 × 104 | 5.2 × 10−5 |
Hprt reactivation rates were determined using the P0 method of Luria and Delbrück 1943. A number of independent cultures of Xist conditional mutant fibroblasts infected with adenovirus-Cre (Δ) or left uninfected (+) were expanded to the appropriate size. When appropriate, cultures were treated with 5-azadC to inhibit DNA methylation. After HAT selection, the number of cultures with HAT-resistant clones was determined, and Hprt reactivation rates were calculated.
Discussion
We generated fibroblasts in which reactivation of two genes on the X chromosome, a GFP transgene and the endogenous Hprt gene, can be detected at a low frequency. We studied the effect of loss of Xist RNA, demethylation of genomic DNA and inhibition of histone deacetylation, on the maintenance of X inactivation. We observed that deletion of Xist leads to reactivation of GFP and Hprt in a very small proportion of cells and conclude that Xist, though not essential for the maintenance of X inactivation, contributes to the stability of the inactive state. We further showed that Xist RNA, histone deacetylation, and DNA methylation act synergistically to achieve the extraordinary stability of X chromosome silencing, with reactivation rates comparable to mutation rates.
Synergism of X Inactivation Mechanisms
It has been shown that during the early stages of cellular differentiation, an Xist RNA–mediated silencing mechanism initiates X inactivation (Penny et al. 1996; Marahrens et al. 1997; Wutz and Jaenisch 2000), and that Xist becomes dispensable for the maintenance of X inactivation after subsequent differentiation (Brown and Willard 1994; Rack et al. 1994, Csankovszki et al. 1999). However, we now present direct evidence that an Xist RNA–mediated maintenance mechanism contributes to silencing in somatic cells. Deletion of Xist reduces the histone macroH2A1 content of Xi chromatin (Csankovszki et al. 1999), and it is possible that this change in histone composition leads to compromised efficiency of silencing. However, to directly assess the role of histone macroH2A1 in X chromosome inactivation, analysis of a targeted mutation of the gene will be necessary.
DNA methylation is another significant contributor to silencing. Demethylation by 5-azadC has been used before to reactivate genes on Xi (Mohandas et al. 1981; Graves 1982). In this study, we also introduced a Dnmt1 mutation that proved to be ∼100 times as effective as 5-azadC treatment in achieving reactivation of GFP. Demethylation combined with Xist deletion increased reactivation rates to a greater extent than either did alone, indicating synergism of silencing mechanisms in keeping Xi silent. In the case of the GFP transgene, we accomplished ≥30% reactivation in Xist/Dnmt1 double mutant fibroblasts. However, at least in the case of Hprt, the majority of the cells maintained silencing even after deletion of Xist and demethylation, indicating the presence of additional stabilizing mechanisms, such as late replication.
Partial compensation for reduced methylation on Xi by other silencing mechanisms occurs in ICF patients. Xi in these patients lacks methylation on CpG islands (Hansen et al. 2000) and, therefore, presumably histone deacetylation is also compromised. However, Xist RNA localizes normally to Xi and, together with late replication, is able to maintain X inactivation, although with reduced efficiency (Hansen et al. 2000).
Inhibition of histone deacetylation by TSA led to a small increase in the number of GFP reactivants. One possible explanation for the modest effect of histone deacetylase inhibitors is the slow rate of histone acetate turnover on the Xi of differentiated cells leading to limited enrichment in acetylated histones after TSA treatment (Keohane et al. 1998). Indeed, treatment of cells with another histone deacetylase inhibitor, sodium butyrate, did not increase antiacetylated histone staining on Xi (Jeppesen and Turner 1993). Furthermore, TSA only inhibits GFP silencing in demethylated cells. A similar dependence of reactivation of genes by TSA on demethylation has been observed by others studying genes silenced in cancer (Cameron et al. 1999), implying that in these cases DNA methylation is the primary mechanism that recruits histone deacetylases to the silenced loci, possibly via binding of methyl-DNA binding proteins (Jones et al. 1998; Nan et al. 1998). In other cases, histone deacetylase inhibitors alone were sufficient to achieve reactivation, such as reexpression of the FMR1 gene in cells derived from fragile X syndrome patients, whereas synergism of histone deacetylation and DNA methylation was still observed (Chiurazzi et al. 1999).
The synergism of multiple silencing mechanisms to assure a highly stable repressed state possibly reflects the importance of dosage compensation for the proper functioning of the organism. It has been demonstrated that gene dosage imbalance in the embryo (Takagi and Abe 1990), or in the extraembryonic tissues (Marahrens et al. 1997), causes lethality. However, partial reactivation of the chromosome appears to be tolerated in vitro, and it is possible to isolate and culture cells with reactivated X-linked genes. Yet, our fibroblast clones with reactivated Xi-linked genes grew more poorly than fibroblasts of the same genotype that did not reactivate any gene. These results argue that gene dosage imbalance is detrimental to cell growth even in vitro and that clones that reactivated the entire chromosome may not be viable. Therefore, our calculations of reactivation frequencies might be an underestimation, as cells with reactivated chromosomes may die before they can be detected.
Xist RNA and DNA Methylation Contribute Differently to Silencing of Two X-linked Genes
The two genes analyzed, GFP and Hprt, are both subject to X inactivation, but the reactivation rates are different. In the presence of Xist RNA and wild-type levels of methylation and histone H4 acetylation, Hprt is almost never reactivated, whereas GFP reactivation can be readily observed. The difference can be at least partially attributed to the difference in assays. Although reactivation of GFP can be observed almost instantly on FACS®, detection of Hprt reactivants by HAT selection requires that the cells survive reactivation and maintain their proliferative capacity. The smallest detectable HAT-resistant colony contained ∼30 cells, the result of about five cell divisions.
The transgenic nature of GFP might also influence the way the gene responds to silencing mechanisms. Xist RNA may not be able to exert the same level of control over a transgene as over endogenous genes, therefore deletion of Xist has a smaller effect on GFP reactivation than on Hprt reactivation. On the other hand, DNA methylation contributes significantly to the silencing of both genes. Randomly integrated transgenes are frequently methylated (Hertz et al. 1999), which may contribute to their variable expression. GFP is integrated near the centromere and therefore might also be subject to silencing by centric heterochromatin. Nonspecific silencing of GFP by mechanisms unrelated to X inactivation (Eggan et al. 2000) may explain the rapid disappearance of GFP reactivants from the cultures. The observed influence of different mechanisms on Hprt may more faithfully represent silencing of endogenous X-inactivated genes.
Mechanism of X Chromosome Silencing
In recent years, significant progress has been made in deciphering how DNA and histone modifications regulate transcription. Methylation of DNA is thought to change accessibility of chromatin via binding of methylated DNA binding proteins that in turn can recruit histone deacetylases (Jones et al. 1998; Nan et al. 1998). Acetylated NH2-terminal lysines of histones, aside from affecting compactness of chromatin packaging, can bind the bromodomain module present in a wide range of chromatin remodeling proteins (Dhalluin et al. 1999; Jacobson et al. 2000).
The mechanism of Xist RNA–mediated silencing is not as well understood. Xist RNA is involved in preferential localization of histone macroH2A1 to Xi (Csankovszki et al. 1999). However, we do not know whether histone macroH2A1 and Xist RNA interact directly and to what extent histone macroH2A1 participates in silencing. It has been shown that Xist RNA can silence in the absence of DNA methylation (Panning and Jaenisch 1996), or even before cellular differentiation (Wutz and Jaenisch 2000). Xist RNA can also accomplish transcriptional silencing without the chromosome becoming late replicating or hypoacetylated (Wutz and Jaenisch 2000). This study provides evidence that the Xist RNA–mediated silencing mechanism acts synergistically with other silencing factors. Xist RNA, similarly to DNA methylation, appears to act in a localized manner, not as a master switch regulating the entire chromosome. Our observations on the effects of deletion of Xist and earlier studies on 5-azadC reactivated clones (Mohandas et al. 1981; Graves 1982) indicate that reactivation of genes on Xi is neither coordinate nor independent, as reactivation of one gene correlates with reactivation of other linked genes only to a limited extent. Xist mutant clones with reactivated genes, in which the lack of Xist RNA–mediated silencing has phenotypic consequences, provide a useful reagent for further dissecting out how Xist RNA might accomplish silencing.
Acknowledgments
We thank Glen Paradis for help with FACS®; Nicki Watson for help with microscopy; David Humpherys for the preparation of the IAP probe; and Anton Wutz, Ted Rasmussen, Sandra Luikenhuis, and David Akey for discussions and critical reading of the manuscript. This work was conducted using the W.M. Keck Foundation biological imaging facility at the Whitehead Institute.
The work was supported by a grant to R. Jaenisch from the National Institutes of Health/National Cancer Institute (R35-CA44339).
Footnotes
Abbreviations used in this paper: GFP, green fluorescent protein; HAT, hypoxanthine/aminopterin/thymidine; Hprt, hypoxanthine phosphoribosyl transferase; IAP, intracisternal A particle; ICF, immunodeficiency centromeric instability facial anomalies; TSA, trichostatin A; Xa, active X; Xi, inactive X.
References
- Anton M., Graham F.L. Site-specific recombination mediated by an adenovirus vector expressing the Cre recombinase proteina molecular switch for control of gene expression. J. Virol. 1995;69:4600–4606. doi: 10.1128/jvi.69.8.4600-4606.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr M.L., Carr D.H. Correlation between sex chromatin and chromosomes. Acta Cytol. 1962;6:34–45. [PubMed] [Google Scholar]
- Borsani G., Tonlorenzi R., Simmler M.C., Dandolo L., Arnaud D., Capra V., Grompe M., Pizzuti A., Muzny D., Lawrence C. Characterization of a murine gene expressed from the inactive X chromosome. Nature. 1991;351:325–329. doi: 10.1038/351325a0. [DOI] [PubMed] [Google Scholar]
- Brockdorff N., Ashworth A., Kay G.F., Cooper P., Smith S., McCabe V.M., Norris D.P., Penny G.D., Patel D., Rastan S. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature. 1991;351:329–331. doi: 10.1038/351329a0. [DOI] [PubMed] [Google Scholar]
- Brockdorff N., Ashworth A., Kay G.F., McCabe V.M., Norris D.P., Cooper P.J., Swift S., Rastan S. The product of the mouse Xist gene is a 15-kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell. 1992;71:515–526. doi: 10.1016/0092-8674(92)90519-i. [DOI] [PubMed] [Google Scholar]
- Brown C.J., Willard H.F. The human X inactivation centre is not required for maintenance of X-chromosome inactivation. Nature. 1994;368:154–156. doi: 10.1038/368154a0. [DOI] [PubMed] [Google Scholar]
- Brown C.J., Ballabio A., Rupert J.L., Lafreniere R.G., Grompe M., Tonlorenzi R., Willard H.F. A gene from the region of the human X inactivation center is expressed exclusively from the inactive X chromosome. Nature. 1991;349:38–44. doi: 10.1038/349038a0. [DOI] [PubMed] [Google Scholar]
- Cameron E., Bachman K., Myohanen S., Herman J., Baylin S. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- Cedar H. DNA methylation and gene activity. Cell. 1988;53:3–4. doi: 10.1016/0092-8674(88)90479-5. [DOI] [PubMed] [Google Scholar]
- Chen R.Z., Pettersson U., Beard C., Jackson-Grusby L., Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- Cheung W.L., Briggs S.D., Allis C.D. Acetylation and chromosomal functions. Curr. Opin. Cell Biol. 2000;12:326–333. doi: 10.1016/s0955-0674(00)00096-x. [DOI] [PubMed] [Google Scholar]
- Chiurazzi P., Pomponi M.G., Pietrobono R., Bakker C.E., Neri G., Oostra B.A. Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum. Mol. Genet. 1999;8:2317–2323. doi: 10.1093/hmg/8.12.2317. [DOI] [PubMed] [Google Scholar]
- Clemson C.M., McNeil J.A., Willard H.F., Lawrence J.B. XIST RNA paints the inactive X chromosome at interphaseevidence for a novel RNA involved in nuclear/chromosome structure. J. Cell Biol. 1996;132:259–275. doi: 10.1083/jcb.132.3.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson C.M., Chow J.C., Brown C.J., Lawrence J.B. Stabilization and localization of Xist RNA are controlled by separate mechanisms and are not sufficient for X inactivation. J. Cell Biol. 1998;142:13–23. doi: 10.1083/jcb.142.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi C., Pehrson J.R. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature. 1998;393:599–601. doi: 10.1038/31275. [DOI] [PubMed] [Google Scholar]
- Csankovszki G., Panning B., Bates B., Pehrson J., Jaenisch R. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat. Genet. 1999;22:323–324. doi: 10.1038/11887. [DOI] [PubMed] [Google Scholar]
- Dhalluin C., Carlson J.E., Zeng L., He C., Aggarwal A.K., Zhou M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Eggan K., Akutsu H., Hochedlinger K., Rideout W., III, Yanagimachi R., Jaenisch R. X-chromosome inactivation in cloned mouse embryos. Science. 2000;290:1578–1581. doi: 10.1126/science.290.5496.1578. [DOI] [PubMed] [Google Scholar]
- Graves J. 5-azacytidine-induced re-expression of alleles on the inactive X chromosome in a hybrid mouse cell line. Exp. Cell Res. 1982;141:99–105. doi: 10.1016/0014-4827(82)90072-6. [DOI] [PubMed] [Google Scholar]
- Hadjantonakis A., Gertsenstein M., Ikawa M., Okabe M., Nagy A. Generating green fluorescent mice by germline transmission of green fluorescent ES cells. Mech. Dev. 1998;76:79–90. doi: 10.1016/s0925-4773(98)00093-8. [DOI] [PubMed] [Google Scholar]
- Hansen R.S., Canfield T.K., Stanek A.M., Keitges E.A., Gartler S.M. Reactivation of XIST in normal fibroblasts and a somatic cell hybridabnormal localization of XIST RNA in hybrid cells. Proc. Natl. Acad. Sci. USA. 1998;95:5133–5138. doi: 10.1073/pnas.95.9.5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen R.S., Wijmenga C., Luo P., Stanek A.M., Canfield T.K., Weemaes C.M.R., Gartler S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA. 1999;96:14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen R.S., Stöger R., Wijmenga C., Stanek A.M., Canfield T.K., Luo P., Matrazzo M.R., D'Esposito M., Feil R., Gimelli G. Escape from gene silencing in ICF syndromeevidence for advanced replication time as a major determinant. Hum. Mol. Genet. 2000;9:2575–2587. doi: 10.1093/hmg/9.18.2575. [DOI] [PubMed] [Google Scholar]
- Hertz J., Schell G., Doerfler W. Factors affecting de novo methylation of foreign DNA in mouse embryonic stem cells. J. Biol. Chem. 1999;274:24232–24240. doi: 10.1074/jbc.274.34.24232. [DOI] [PubMed] [Google Scholar]
- Hooper M., Hardy K., Handyside A., Hunter S., Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- Jackson-Grusby L., Beard C., Possemato R., Tudor M., Fambrough D., Csankovszki G., Dausman J., Lee P., Wilson C., Lander E., Jaenisch R. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet. 2001;27:31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- Jacobson R.H., Ladurner A.G., King D.S., Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- Jat P.S., Cepko C.L., Mulligan R.C., Sharp P.A. Recombinant retroviruses encoding simian virus 40 large T antigen and polyomavirus large and middle T antigens. Mol. Cell. Biol. 1986;6:1204–1217. doi: 10.1128/mcb.6.4.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen P., Turner B.M. The inactive X chromosome in female mammals is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression. Cell. 1993;74:281–289. doi: 10.1016/0092-8674(93)90419-q. [DOI] [PubMed] [Google Scholar]
- Jones P., Veenstra G., Wade P., Vermaak D., Kass S., Landsberger N., Strouboulis J., Wolffe A. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Keohane A.M., O'Neill L P., Belyaev N.D., Lavender J.S., Turner B.M. X-Inactivation and histone H4 acetylation in embryonic stem cells. Dev. Biol. 1996;180:618–630. doi: 10.1006/dbio.1996.0333. [DOI] [PubMed] [Google Scholar]
- Keohane A., Lavender J., O'Neill L., Turner B. Histone acetylation and X inactivation. Dev. Genet. 1998;22:65–73. doi: 10.1002/(SICI)1520-6408(1998)22:1<65::AID-DVG7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Lei H., Oh S., Okano M., Juttermann R., Goss K., Jaenisch R., Li E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- Lock L.F., Takagi N., Martin G.R. Methylation of the Hprt gene on the inactive X occurs after chromosome inactivation. Cell. 1987;48:39–46. doi: 10.1016/0092-8674(87)90353-9. [DOI] [PubMed] [Google Scholar]
- Luria S.E., Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marahrens Y., Panning B., Dausman J., Strauss W., Jaenisch R. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 1997;11:156–166. doi: 10.1101/gad.11.2.156. [DOI] [PubMed] [Google Scholar]
- Marahrens Y., Loring J., Jaenisch R. Role of the Xist gene in X chromosome choosing. Cell. 1998;92:657–664. doi: 10.1016/s0092-8674(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Migeon B.R. X-chromosome inactivationmolecular mechanisms and genetic consequences. Trends Genet. 1994;10:230–235. doi: 10.1016/0168-9525(94)90169-4. [DOI] [PubMed] [Google Scholar]
- Mohandas T., Sparkes R., Shapiro L. Reactivation of an inactive human X chromosomeevidence for X inactivation by DNA methylation. Science. 1981;211:393–396. doi: 10.1126/science.6164095. [DOI] [PubMed] [Google Scholar]
- Monk M., Harper M. Sequential X chromosome inactivation coupled with cellular differentiation in early mouse embryos. Nature. 1979;281:311–313. [Google Scholar]
- Nan X., Ng H.H., Johnson C.A., Laherty C.D., Turner B.M., Eisenman R.N., Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Norris D.P., Brockdorff N., Rastan S. Methylation status of CpG-rich islands on active and inactive mouse X chromosomes. Mamm. Genome. 1991;1:78–83. doi: 10.1007/BF02443782. [DOI] [PubMed] [Google Scholar]
- Okano M., Bell D.W., Harber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Panning B., Jaenisch R. DNA hypomethylation can activate Xist expression and silence X-linked genes. Genes Dev. 1996;10:1991–2002. doi: 10.1101/gad.10.16.1991. [DOI] [PubMed] [Google Scholar]
- Penny G.D., Kay G.F., Sheardown S.A., Rastan S., Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996;379:131–137. doi: 10.1038/379131a0. [DOI] [PubMed] [Google Scholar]
- Priest J.H., Heady J.E., Priest R.E. Delayed onset of replication of human X chromosomes. J. Cell Biol. 1967;35:483–487. doi: 10.1083/jcb.35.2.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rack K.A., Chelly J., Gibbons R.J., Rider S., Benjamin D., Lafreniere R.G., Oscier D., Hendriks R.W., Craig I.W., Willard H.F. Absence of the XIST gene from late-replicating isodicentric X chromosomes in leukaemia. Hum. Mol. Genet. 1994;3:1053–1059. doi: 10.1093/hmg/3.7.1053. [DOI] [PubMed] [Google Scholar]
- Sauer B., Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi N., Abe K. Detrimental effects of two active X chromosomes on early mouse development. Development. 1990;109:189–201. doi: 10.1242/dev.109.1.189. [DOI] [PubMed] [Google Scholar]
- Walsh C., Chaillet J., Bestor T. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- Wutz A., Jaenisch R. A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol. Cell. 2000;5:695–705. doi: 10.1016/s1097-2765(00)80248-8. [DOI] [PubMed] [Google Scholar]
- Xu G.L., Bestor T.H., Bourc'his D., Hsieh C.L., Tommerup N., Bugge M., Hulten M., Qu X., Russo J.J., Viegas-Pequignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- Yoshida M., Horinouchi S., Beppu T. Trichostatin A and trapoxinnovel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays. 1995;17:423–430. doi: 10.1002/bies.950170510. [DOI] [PubMed] [Google Scholar]