

Abstract
In this study, we investigated cardiomyocyte cytoarchitecture in a mouse model for dilated cardiomyopathy (DCM), the muscle LIM protein (MLP) knockout mouse and substantiated several observations in a second DCM model, the tropomodulin-overexpressing transgenic (TOT) mouse. Freshly isolated cardiomyocytes from both strains are characterized by a more irregular shape compared with wild-type cells. Alterations are observed at the intercalated disks, the specialized areas of mechanical coupling between cardiomyocytes, whereas the subcellular organization of contractile proteins in the sarcomeres of MLP knockout mice appears unchanged. Distinct parts of the intercalated disks are affected differently. Components from the adherens junctions are upregulated, desmosomal proteins are unchanged, and gap junction proteins are downregulated. In addition, the expression of N-RAP, a LIM domain– containing protein located at the intercalated disks, is upregulated in MLP knockout as well as in TOT mice. Detailed analysis of intercalated disk composition during postnatal development reveals that an upregulation of N-RAP expression might serve as an early marker for the development of DCM. Altered expression levels of cytoskeletal proteins (either the lack of MLP or an increased expression of tropomodulin) apparently lead to impaired function of the myofibrillar apparatus and to physiological stress that ultimately results in DCM and is accompanied by an altered appearance and composition of the intercalated disks.
Keywords: dilated cardiomyopathy, N-RAP, tropomodulin, adherens junction, gap junction
Introduction
The contractile tissue of the heart is composed of individual cardiomyocytes that are connected to each other at specific intercellular junctions, the intercalated disks. Cardiomyocytes are highly differentiated bipolar cells that are almost completely filled with contractile elements, the myofibrils. The basic unit of a myofibril is the sarcomere, which is defined as the region in between two neighboring Z-disks, where the thin (actin) filaments are anchored. Contraction is the result of an interaction of the actin filaments with the myosin heads from the thick filaments located in the central region of the sarcomere, where they are held in place by a specific structure, the M-band. A similar high degree of organization can also be seen at the intercalated disks, situated at the ends of the cardiomyocytes, where three different types of junctions can be distinguished: adherens junctions, where the myofibrils are anchored; desmosomes, which also serve for mechanical stabilization by integrating intermediate filaments; and gap junctions, which mediate ion transfer for electrical coupling in order to guarantee synchronous contraction.
In recent years, many cardiomyopathies have been linked to mutations either in sarcomeric or in cytoskeletal proteins, resulting in familial hypertrophic cardiomyopathy and in dilated cardiomyopathy (DCM), respectively, and several mouse models have been generated to study the structural and functional changes that accompany cardiomyopathies (for reviews see Vikstrom and Leinwand 1996; Leiden 1997; Bonne et al. 1998; Seidman and Seidman 1998; Towbin 1998; Chien 1999). Both hypertrophic cardiomyopathy as well as DCM can lead to alterations in the histologic appearance of cardiac tissue, which include myocyte disarray as well as myocyte loss and fibrosis. It has been proposed that mutations in components of the force-generating apparatus tend to lead to cardiac hypertrophy, whereas changes in the force transmitting and cytoskeletal structures are characteristic of DCM (Chien 1999). So far, relatively little is known about how these mutations affect cardiomyocyte structure or the ways the cells attach to each other. Therefore, we decided to investigate cardiomyocyte and intercalated disk architecture in a mouse model for DCM, the muscle LIM protein (MLP) knockout mouse (Arber et al. 1997) and, to verify our observations on the intercalated disk structure in a second model for this disease, the tropomodulin-overexpressing transgenic (TOT) mouse (Sussman et al. 1998a).
MLP is a member of the LIM-only class of the LIM domain protein family that possesses two LIM domains. Depending on the developmental status of the cell, it can be localized in the nucleus as well as in the cytoplasm, where it is preferentially associated with actin filaments and with the Z-disk region of muscle cells (Arber et al. 1994). The two LIM domains are responsible for the differential targeting: the first LIM domain seems to be involved in nuclear targeting and the interaction with α-actinin, a component of the Z-disk, whereas the second domain mediates association with actin filaments as well as binding to spectrin, a protein of the membrane cytoskeleton (Arber and Caroni 1996; Flick and Konieczny 2000). MLP is highly expressed during differentiation of all kinds of striated muscle, but its expression in the adult is restricted to cardiac and slow twitch fibers of skeletal muscle (Arber et al. 1994; Schneider et al. 1999). Mice that are homozygous null for MLP display symptoms of DCM with a dramatic increase in heart size (Arber et al. 1997). Two different phenotypes were described: an early postnatal phenotype that results in death by the end of the second week after birth and an adult phenotype, where the animals reach a normal age but show all the features of DCM, such as thinning of the left ventricular wall with concomitant chamber enlargement and reduced contractile function (Arber et al. 1997).
Tropomodulin is a pointed end actin-capping protein and is thought to be essential for the maintenance of thin filament length in the sarcomere (Gregorio and Fowler 1995; Sussman et al. 1998b). Alterations of its expression levels are deleterious in cultured cardiomyocytes (Gregorio and Fowler 1995; Sussman et al. 1998b) as well as in transgenic mice, where an excess of tropomodulin expression leads to the development of juvenile DCM (Sussman et al. 1998a).
Our results from the analysis of the cardiomyocyte phenotype in these mice reveal that, although sarcomere structure in MLP knockout mice appears normal in the light microscope, the intercalated disk exhibits major alterations in both mouse models for DCM: adherens junction proteins that are associated with myofibril attachment are upregulated, whereas connexin-43, the major constituent of ventricular gap junctions, is downregulated. These structural changes might provide a working hypothesis for the cause of the functional alterations that are seen in the failing heart.
Materials and Methods
Mouse Strains
MLP knockout mice (Arber et al. 1997), TOT mice (Sussman et al. 1998a), and their nontransgenic control strains (OBF-E52 and FVB-N, respectively) were maintained at the animal facility of the Institute of Cell Biology at the Swiss Federal Institute of Technology, Zurich, Switzerland. Age- and sex-matched controls were used together with the transgenic mice for all experiments.
Antibodies
The monoclonal antibodies against myomesin (clone B4; Grove et al. 1984) and the polyclonal antibodies against myosin binding protein C (MyBP-C; Bähler et al. 1985), MLP (Arber et al. 1997), and N-RAP (Luo et al. 1997) were characterized in our laboratories. The monoclonal antibodies against sarcomeric α-actinin (clone EA-53), vinculin (clone hVin), and the polyclonal antibodies against pan-cadherin, α-catenin, and β-catenin were purchased from Sigma-Aldrich. The monoclonal antibody against plakoglobin (γ-catenin) and the monoclonal antibody against desmoglein were from Transduction Laboratories, and the polyclonal antibody against connexin-43 was from Chemicon. The polyclonal antibody against desmoplakin (North et al. 1999) was a gift from Dr. Alison North (University of Manchester, Manchester, UK).
The secondary antibodies for immunofluorescence were bought from Jackson ImmunoResearch Laboratories (Cy3-conjugated anti–mouse and anti–rabbit Igs) and Cappel (FITC-conjugated anti–mouse and anti–rabbit Igs). Rhodamine–phalloidin was obtained from Molecular Probes, and Cy5-conjugated phalloidin was a gift from Prof. H. Faulstich (Max-Plank Institute for Medical Research, Heidelberg, Germany). Incubations with the secondary antibodies on their own gave no significant signal in either freshly isolated cardiomyocytes or semithin cryosections.
HRP-conjugated anti–mouse Igs (Dako) and anti–rabbit Igs (Calbiochem) were used for immunoblotting.
Freshly Isolated Cardiomyocytes
Muscle strips (aproximately 1 × 3 mm) from equivalent regions of the left ventricle were tied onto plastic plates at extended length and digested in 1 mg/ml collagenase (type 2; Worthington Biochemical Corp.) and 1 mg/ml verapamil (Knoll AG) in digestion solution (137 mM NaCl, 5 mM KCl, 4 mM NaHCO3, 5.5 mM glucose, 2 mM MgCl2, 2.5 mM CaCl2, 10 mM Pipes, pH 6.5; Draeger et al. 1989) at 37°C for 90 min. After washing three times on ice with washing solution (137 mM NaCl, 5 mM KCl, 4 mM NaHCO3, 5.5 mM glucose, 1.1 mM Na2HPO4×2H2O, 0.4 mM KH2PO4, 2 mM EGTA, 5 mM MES, 0.5 mM DTT, 100 mg/l streptomycin, pH 6.1), the pieces were cut off from the plates and placed in 500 μl cold washing solution, depending on the size of the muscle pieces. Individual cardiomyocytes were isolated by gently pipetting the digested tissue pieces up and down using a wide-mouthed Pasteur pipette. The cardiomyocytes were spun onto gelatine-coated slides using a Cytospin centrifuge (Shandon Southern), fixed for 15 min in 4% paraformaldehyde in PBS, and then permeabilized for 10 min in 0.2% Triton X-100 in PBS and incubated in a mixture of the primary antibodies diluted in 1% BSA/TBS (155 mM NaCl, 2 mM MgCl2, 2 mM EGTA, 20 mM Tris-base, pH 7.6) at 4°C overnight in a humid chamber. After washing with PBS containing 0.002% Triton X-100, secondary antibody incubations were carried out for a minimum of 3 h at room temperature (RT) to ensure complete penetration of the antibodies into the cells. After thorough washing in PBS containing 0.002% Triton X-100, the specimens were mounted in 0.1 M Tris-HCl (pH 9.5) glycerol (3:7) containing 50 mg/ml n-propyl gallate as anti-fading reagent (Messerli et al. 1993a) and sealed with nail polish.
Semithin Frozen Sections
Muscle strips were dissected from equivalent regions of the left ventricle, tied onto plastic plates, and fixed in 4% paraformaldehyde in PBS for 90 min at RT. After 10-min washes in cytoskeleton buffer (137 mM NaCl, 5 mM KCl, 1.1 mM Na2PO4, 0.4 mM KH2PO4, 4 mM NaHCO3, 5.5 mM glucose, 2 mM EGTA, 5 mM MES, 10 mg/ml streptomycin, pH 6.1) (North et al. 1994) three times, they were cut into smaller pieces and left to infiltrate with a polyvinyl pyrrolidone–sucrose mixture for 3 h on a rotating table at RT (Tokuyasu 1989). The pieces were then mounted on aluminium pins and plunge frozen in liquid nitrogen. Semithin sections (0.25 μm thick) were cut with glass knives in a Reichert Ultracryomicrotome at −70°C (Leica). The sections were retrieved on gelatine-coated coverslips, blocked first for 10 min in 0.02 M glycine/TBS, and then for 30 min in 5% preimmune goat serum in 1% BSA/TBS, and incubated in a mixture of the primary antibodies diluted with 1% BSA/TBS at 4°C overnight. After washing in 0.1% BSA in TBS five times, the secondary antibodies were applied together with Cy5-conjugated phalloidin for 1h. The coverslips were washed five times for 5 min in TBS and mounted on slides in gelvatol (Airvol 203; Air Products) containing 5 mg/ml n-propyl gallate (North et al. 1994).
Confocal Microscopy
Confocal images were recorded on an inverted microscope DM IRB/E equipped with a true confocal scanner TCS NT and a PL APO 63×/1.32 oil immersion objective (Leica) using an argon–krypton mixed gas laser. Image processing was done on a Silicon Graphics workstation using Imaris (Bitplane AG), three-dimensional multichannel image processing software specialized for confocal microscopy images (Messerli et al. 1993b).
Immunoblotting
Left ventricles were dissected, and small tissue pieces were homogenized by freeze slamming and solubilized in a modified version of SDS sample buffer (Ehler et al. 1999). Equivalent amounts of protein were run on 8–22%-gradient polyacrylamide minigels (Bio-Rad Laboratories) using the buffer system of Laemmli 1970. The proteins were blotted overnight onto nitrocellulose (Hybond-C extra; Amersham-Pharmacia Biotech; Towbin et al. 1979). After correct transfer had been established by Ponceau red staining (Serva), nonspecific binding sites were blocked by incubation in 5% nonfat dry milk in washing buffer (0.9% NaCl, 9 mM Tris, pH 7.4, 0.1% Tween-20) for 1 h at RT. Incubations with the primary and secondary HRP-conjugated antibodies were carried out as described previously (Ehler et al. 1999). Results from the chemiluminescence reaction were visualized on Fuji medical x-ray films. Only bands of the expected molecular weight as judged by Kaleidoscope prestained standards (Bio-Rad Laboratories) were present with all the antibodies used. The figures show representative blots performed with extracts from one individual animal; the experiments were repeated at least two times with extracts from at least three different animals.
Electron Microscopy
Tissue pieces from equivalent regions of the left ventricle were tied onto plastic plates, washed for 5 min in 0.1 M sodium cacodylate buffer (SCB), pH 7.2, and fixed in 2% glutaraldehyde (Fluka; Buchs) in 0.1 M SCB overnight at 4°C. After washing three times in SCB, the samples were postfixed for 1 h in 1% osmium tetroxide in SCB, dehydrated stepwise to 70% ethanol, and stained en bloc for 1 h in 2% uranyl acetate in 70% ethanol. After dehydration was completed, the cells were infiltrated with an epoxy resin based on Epon812 (Fluka) in a series of ascending mixtures of epoxy resin/propylene oxide. The specimens were then imbedded in rubber moulds filled with 100% resin and heat polymerized for 48 h at 60°C. Ultrathin sections were cut on a Reichert Diatom ultramicrotome (Leica), retrieved on carbon and Formvar-coated copper grids (Provac AG) and stained with 2% uranyl acetate in 95% methanol and Reynolds lead citrate. Preparations were examined in a JEM100C transmission electron microscope at 80 kV.
Solid Phase Binding Assays
N-RAP fragments were expressed in Escherichia coli and purified as described previously (Luo et al. 1999). Rat MLP was expressed as a histidine-tagged protein (Arber and Caroni 1996) and purified by the same methods using a plasmid containing the entire MLP sequence cloned into the BamH1 and HindIII sites of the pQE-9 vector (QIAGEN). Wells from Nunc MaxiSorp microtiter plates (Nalge Nunc) were coated with 100 μl of purified recombinant N-RAP fragments at a concentration of 0.1 μM in 6 M urea, 50 mM Tris-HCl (pH 8.0), 5 mM EGTA, and 10 mM DTT overnight at 4°C. After blocking for 1–3 h at 37°C with 0.5% BSA in PBS-T (PBS + 0.2% Tween-20), the wells were incubated overnight at 4°C with varying concentrations of purified MLP in overlay buffer (100 mM KCl, 50 mM Tris-HCl, pH 7.4, 1 mM EGTA, 2 mM MgCl2, 2 mM ATP, 0.3 mM DTT, 0.2% Tween-20). After four washes with PBS-T, the wells were incubated with the polyclonal anti-MLP antibody diluted 1:2,000 in PBS-T + 0.5% BSA for 1 h at RT followed by another four washes. As secondary antibodies, HRP-conjugated anti–rabbit Igs (Amersham Pharmacia Biotech) were applied for 1 h at a dilution of 1:2,000 in PBS-T + 0.5% BSA, and after four washes, the color reaction was performed by incubating the wells for 30 min with 100 μl of substrate solution (0.1 mg/ml 3,3′,5,5′-tetramethylbenzidine dihydrochloride, 0.01% H2O2, and 0.1 M sodium acetate, pH 5.2). The reaction was stopped by addition of an equal volume of 1 M H2SO4, and the color reaction was analyzed in an ELISA plate reader (Dynatech) by measuring the absorbency at 450 nm. Each data point represents the measurement of triplicate or quadruplicate wells. The data were analyzed as previously described (Luo et al. 1999).
Image Analysis
Evaluation of the protein expression levels as analyzed by immunoblotting as well as the evaluation of the degree of complexity of the intercalated disk in the electron micrographs were carried out using NIH Image, followed by statistical analysis in KaleidaGraph (Synergy Software).
Results
Mice that are deficient for MLP display a phenotype that is very similar to the human disease DCM, including enlargement of all four chambers of the heart, wall thinning, and reduced left ventricle function, as shown by echocardiography (Arber et al. 1997). To characterize the alterations in cardiomyocyte cytoarchitecture during the development of DCM, we analyzed the structure of freshly isolated cardiomyocytes obtained from adult MLP−/− mice. As shown previously, cardiomyocytes from MLP−/− are characterized by a more irregular overall shape than their wild-type counterparts (Arber et al. 1997). To investigate whether these changes of morphology are also reflected by changes in myofibril structure and composition, we looked at the distribution of several components of the sarcomere in freshly isolated cardiomyocytes. Although the myofibrils themselves run in a much more irregular way in MLP−/− compared with wild-type cardiomyocytes, no gross alterations of sarcomere structure were observed. There was no effect on either thick filament structure, as shown by staining for myosin-binding protein C, which is localized in double bands in the A-band (Fig. 1A and Fig. B) or thin filament structure, as shown by phalloidin, which stains F-actin in the I-bands (Fig. 1C and Fig. D). The localization pattern of α-actinin (Fig. 1E and Fig. F), an integral component of the Z-disk, and of myomesin (Fig. 1G and Fig. H), an M-band–associated protein, were indistinguishable in wild-type and MLP−/− cardiomyocytes (Fig. 1E and Fig. G, and Fig. F and Fig. H, respectively). Therefore, sarcomere structure appeared to be normal in MLP−/− cardiomyocytes, as judged by light microscopy.
Figure 1.

Sarcomeric organization is normal in freshly isolated cardiomyocytes from MLP−/− mice. Confocal microscope images of freshly isolated cardiomyocytes from wild-type (A, C, E, and G) and MLP−/− mice (B, D, F, and H) stained for components of the A-band (MyBP-C, A and B), the I-band (F-actin visualized by rhodamine–phalloidin in C and D), the Z-disk (sarcomeric α-actinin; E and F), and the M-band (myomesin; G and H). No alterations in the cross-striations were observed; however, the individual myofibrils do not run as strictly parallel in the MLP−/− cardiomyocytes as they do in wild type. Bar, 5 μm.
However, alterations can be observed when the distribution pattern of vinculin is analyzed by confocal microscopy in freshly isolated cardiomyocytes. Vinculin is a component of both intercalated disks and costameres, where myofibrils are anchored laterally to the membrane and via integrins to the extracellular matrix (Pardo et al. 1983). Fig. 2 displays single confocal sections through an individual freshly isolated cardiomyocyte from a wild-type (A, C, and E) or a MLP−/− mouse (B, D, and F) taken at three different levels: the substrate (A and B), through the middle of the cell (C and D), and from the top (E and F). Although wild-type cardiomyocytes displayed perfectly aligned costameres, a disturbed pattern was detected in the top (B) and bottom (F) optical sections in cardiomyocytes from MLP−/− mice. In wild-type cardiomyocytes, vinculin molecules were organized in a somewhat parallel pattern along the intracellular side of the plasma membrane (Fig. 2A and Fig. E). A similar parallel pattern was seen in optical sections taken from the middle of cardiomyocytes from wild-type and MLP−/− mice, indicating normal Z-disk association of vinculin (Terracio et al. 1990). However, in regions underneath the plasma membrane in cardiomyocytes from MLP−/− mice, vinculin seemed to be localized in dots that were distributed irregularly, and the parallel striations were partially lost (Fig. 2B and Fig. F). In addition, the freshly isolated MLP−/− cardiomyocytes were more irregularly shaped than the rod-shaped wild-type cardiomyocytes. These irregularities were particularly prominent at the remnants of the intercalated disks of the MLP−/− cardiomyocytes, which exhibited vinculin staining that was more conspicuous compared with wild-type cardiomyocytes. Our observations suggest that both cell–matrix as well as cell–cell contact structures are affected in cardiomyocytes from MLP−/− mice.
Figure 2.

The organization of the costameric protein vinculin is disturbed in MLP−/− cardiomyocytes. Single confocal sections of freshly isolated cardiomyocytes from wild-type (A, C, and E) and MLP−/− mice (B, D, and F) stained with antibodies to vinculin. Optical sections were taken from the bottom (A and B), through the middle of the cell (C and D), and from the top (E and F). MLP−/− cardiomyocytes display a more irregular overall shape and, instead of a striated arrangement, they show a patchy distribution of vinculin at their surface.
To analyze whether similar alterations could be observed for other components of the intercalated disk, we compared the expression levels of several intercalated disk–associated proteins on immunoblots of whole muscle samples from the ventricles of wild-type and MLP−/− mice. The expression levels of all investigated adherens junction–associated proteins were elevated, as shown in Fig. 3 (A, cadherin, +82%; B, β-catenin, +34%; C, plakoglobin; +29%; D, α-catenin, +97%; E, vinculin, +51%; and H, N-RAP, +152%), whereas connexin-43, the major gap junctional protein in the ventricle, appeared downregulated in MLP−/− hearts (Fig. 3 F, −34%). The expression of desmosomal proteins like desmoplakin and desmoglein was not significantly affected (Fig. 3G and Fig. H, +4.5% and +3%, respectively). Therefore, the absence of MLP seems to result in an altered protein composition of the intercalated disks with an upregulation of adherens junction–associated proteins and a downregulation of gap junction proteins.
Figure 3.

Expression levels of intercalated disk proteins are changed in adult MLP−/− hearts. Immunoblots on whole heart muscle samples of wild-type (WT) or MLP−/− mice with antibodies against cadherin (A), β-catenin (B), plakoglobin (C), α-catenin (D), vinculin (E), connexin-43 (F), desmoplakin (G), desmoglein (H), and N-RAP (I). J shows a part of a Ponceau-stained filter. Adherens junction–associated proteins appear upregulated (A–E, and H); the gap junction component connexin-43 seems downregulated (F), and the expression levels of the desmosomal markers desmoplakin and desmoglein are unaffected (G). Representative blots with extracts from one animal per lane are shown.
Immunofluorescence analysis of semithin cryosections of left ventricular tissue of wild-type (Fig. 4A, A′, B, B′, G, G′, H, and H′) and MLP−/− mice (Fig. 4C, Fig. C′, D, D′, I, I′, J, and J′) that were stained for different components of the intercalated disk together with Cy5-phalloidin to visualize F-actin (Fig. 4A′–L′) revealed differences in the organization of the intercalated disk at the level of the light microscope. When the localization of β-catenin, for example, was compared between wild-type and MLP−/− hearts, it was apparent that not only more intercalated disks were stained, reflecting the irregular morphology with the increased number of cell–cell contacts, but that also the signal at the intercalated disk itself was broader in MLP−/− than in wild type (Fig. 4, compare C with A). In sections stained for the gap junction component connexin-43, a marked reduction was apparent in MLP−/− hearts (Fig. 4 D). The signal for desmosomes, as identified with a desmoplakin antibody, seemed indistinguishable between wild type and MLP−/− (Fig. 4G and Fig. I). Interestingly, when the localization of N-RAP, an intercalated disk–associated LIM domain protein was analyzed, a duplication of the N-RAP signal was invariably found in the MLP−/− hearts. Although in the wild type only a single band was stained at the intercalated disk (Fig. 4 H), comparable to the signal obtained for the β-catenin antibody or for vinculin (data not shown), in the case of MLP−/−, a doublet for N-RAP was always observed, associated with more pronounced actin staining (Fig. 4 J).
Figure 4.

Changes of intercalated disk morphology as seen by immunofluorescence. Semithin cryosections of wild type (A, A′, B, B′, G, G′, H, and H′); MLP−/− (C, C′, D, D′, I, I′, J, and J′) and TOT mice (E, E′, F, F′, K, K′, L, and L′) were stained with antibodies against β-catenin (A, C, and E), connexin-43 (B, D, and F), desmoplakin (G, I, and K), and N-RAP (H, J, and L), together with Cy5-phalloidin, to visualize F-actin (A′–L′). Although an increased signal can be observed in the case of adherens junction proteins like β-catenin in DCM hearts, the signal for the gap junction protein connexin-43 appears reduced, and that for desmoplakin as a desmosomal marker is unchanged. In wild-type hearts, N-RAP stains only a single band at the intercalated disk, whereas in DCM hearts, a duplication of this band can be observed. Bar, 10 μm.
To investigate whether this intercalated disk phenotype might be a general hallmark of DCM, we analyzed intercalated disk composition in a second mouse model for this disease, the TOT mouse. TOT mice show cardiomyopathic alterations within one month after birth and develop DCM with compromised contractile function and impaired myofibril organization (Sussman et al. 1998a). In addition, the overall shape of freshly isolated cardiomyocytes from TOT mice is less regular than that from their wild-type counterparts, comparable to the morphology of MLP−/− cardiomyocytes (data not shown). Interestingly, similar observations on intercalated disk organization as for MLP−/− mice could also be made on semithin cryosections from TOT hearts (Fig. 4E, Fig. E′, F, F′, K, K′, L, and L′). Adherens junction–associated proteins such as β-catenin showed an increased signal (Fig. 4 E), whereas the expression levels of connexin-43 seemed to be reduced drastically (Fig. 4 F), and the expression of the desmosomal protein desmoplakin was unchanged (Fig. 4 K). Additionally, in TOT mice the duplication of the N-RAP signal could be observed as well (Fig. 4 L), suggesting that this change in N-RAP localization might be a general feature of DCM. Thus, two mouse models of DCM with different etiologies show marked alterations of intercalated disk protein composition. The expression of adherens junction–associated proteins is upregulated, there is a reduced number of gap junctions, and a broader distribution of the intercalated disk–associated LIM domain protein N-RAP is observed.
Electron microscopy revealed a severe alteration of intercalated disk ultrastructure in MLP−/− and TOT hearts. The increase in width for staining of adherens junction–associated proteins as seen by immunofluorescence microscopy is not due to an increased thickness of the protein coat itself but rather to a higher degree of convolution of the membrane at the intercalated disk in MLP−/− hearts (Fig. 5 B) compared with wild type (Fig. 5 A), thus giving the impression of a broader stained region at the level of the light microscope. Random measurements of ID membrane length revealed that MLP−/− IDs showed an increase of 21.0 ± 0.2% (SD) in membrane length per standardized distance measured normal to the long axis of the cell compared with wild type. Analysis of the number of membrane loops per micrometer revealed an increase as well with 2.3 ± 0.2 (SD) loops for the MLP−/− compared with 1.5 ± 0.2 (SD) loops for the wild type, again pointing out a higher degree of convolution. Gap junctions (Fig. 5 B, arrowheads) were only rarely detected in randomly chosen fields of the sections of MLP−/− hearts, consistent with the drastically reduced staining for connexin-43 in the immunofluorescence experiments. In addition to the more irregular appearance of the IDs, detachment of neighboring cardiomyocytes was also occasionally observed (data not shown). Therefore, the increase in stained ID area as for example in the vinculin staining in Fig. 2 or in the β-catenin staining in Fig. 4 C might be explained by the broader area that is covered by the intercalated disk due to the higher degree of convolution. The same alteration, namely a more convoluted plasma membrane, can be observed when intercalated disk structure is analyzed in hearts from TOT mice (Fig. 5 C).
Figure 5.
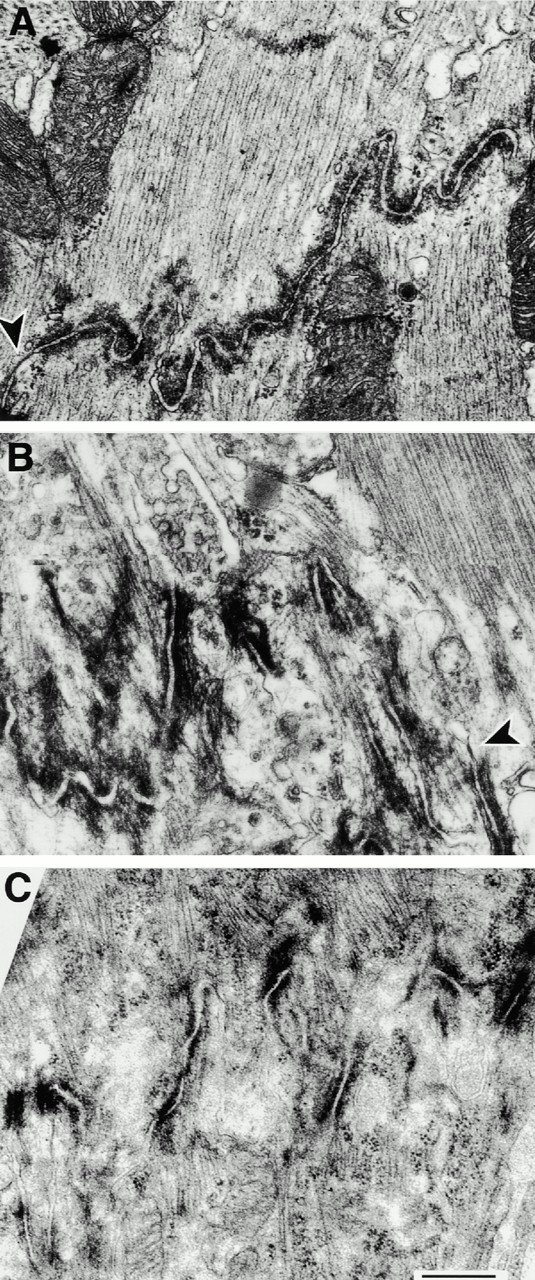
Electron microscopy of the altered intercalated disk morphology in MLP−/− hearts. Negatively stained resin sections of wild-type (A), MLP−/− (B), and TOT hearts (C). Compared with wild-type, the intercalated disks of MLP−/−, as well as of TOT mice, are much more convoluted and only rarely show gap junctions (arrowheads). Bar, 500 nm.
Since all the investigations described above were carried out in adult animals that had already developed DCM, we wanted to know whether the altered composition of the intercalated disk can be observed earlier in development, when heart function is still more or less normal. Immunoblots on SDS samples of left ventricles taken at different developmental stages are shown in Fig. 6; we compared samples from newborn mice (Fig. 6, lanes 1 and 2), from juveniles (3 wk, lanes 3 and 4) and from adults (3–5 mo, lanes 5 and 6). The results showed that changes in expression levels of most ID-associated proteins were only apparent at the adult stage, when the phenotype of DCM is already well developed (Fig. 6, B–D and F). Therefore, the upregulation of adherens junction–associated proteins and the downregulation of connexin-43 seems to be a secondary effect that follows impaired function of the cardiomyocytes. In contrast, N-RAP exhibited increased expression at earlier times. In the case of MLP−/− mice, an upregulation of N-RAP expression could already be detected at birth (Fig. 6 E); in the case of the TOT mice, this became only obvious by the juvenile stage (Fig. 6 G). This suggests that elevated expression of N-RAP might serve as an early marker for DCM, even before the alterations in the expression levels of other intercalated disk–associated proteins become apparent.
Figure 6.
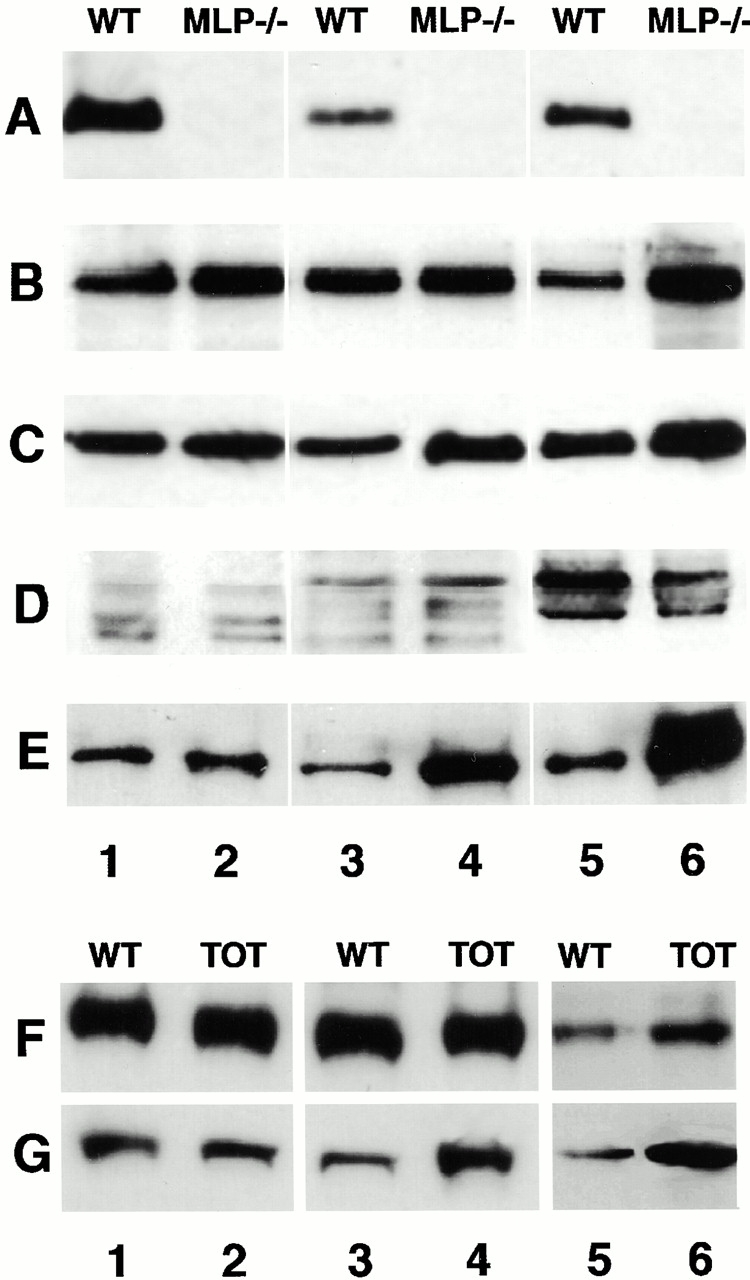
Changes in expression levels of intercalated disk proteins during postnatal development. Immunoblots of whole heart muscle samples of wild-type (WT) and MLP−/− mice as well as of TOT of different developmental stages (newborn, lanes 1 and 2; 21-d-old, lanes 3 and 4; adult, lanes 5 and 6) with antibodies against MLP (A), cadherin (B and F), plakoglobin (C), connexin-43 (D), and N-RAP (E and G). Although the differences in the expression levels of most intercalated disk proteins only become apparent by the adult stage (cadherin, plakoglobin, and connexin-43), N-RAP expression is already elevated in samples from newborn MLP−/− mice as well as from juvenile TOT mice compared with wild type. Representative blots with extracts from one animal per lane are shown.
N-RAP is a recently characterized nebulin-related protein that possesses a LIM domain and is concentrated at the intercalated disks in heart and at myotendinous junctions in skeletal muscle (Luo et al. 1997, Luo et al. 1999; Herrera et al. 2000). Since LIM domains can act as protein–protein interaction interfaces (Schmeichel and Beckerle 1994) and both MLP (Arber et al. 1997) and N-RAP (Luo et al. 1997) are concentrated at intercalated disks, we investigated whether MLP and N-RAP can directly bind to each other. Different fragments of N-RAP containing either most of the nebulin-related super repeats (N-RAP-SR), the NH2-terminal half of the protein including the LIM domain (N-RAP-NH) or the NH2-terminal part without the LIM domain (N-RAP-IB) were recombinantly expressed in E. coli, and their interaction with full-length MLP was monitored in a solid phase binding assay. Strong interaction with MLP was detected for both N-RAP-IB and N-RAP-NH (Fig. 7). However, since MLP binding to the fragment containing the LIM domain was no greater than to N-RAP-IB, the interaction between these two proteins is probably not mediated by the N-RAP LIM domain. The absence of MLP in MLP−/− mice might lead to increased stress at the site of the intercalated disk, leading to an upregulation of N-RAP expression. Further evidence for the importance of a feedback between MLP and N-RAP expression comes from the observation that in adult TOT mice, which show an upregulation of N-RAP, expression of MLP was markedly downregulated (Fig. 8).
Figure 7.

Interaction between MLP and N-RAP in a solid phase binding assay. Significant binding of recombinantly expressed full-length MLP can be observed to N-RAP-IB (the region between the LIM domain and the nebulin-like repeats) as well as to N-RAP-NH (N-RAP-IB plus the NH2-terminal LIM domain). The apparent dissociation constants are 17 ± 3 and 8 ± 2 nM, respectively. No significant binding is observed to the N-RAP superrepeats (N-RAP-SR).
Figure 8.

MLP expression is downregulated in hearts from adult TOT mice. Immunoblots on ventricular samples from adult TOT mice (right) and their respective wild-type strain (left) with antibodies specific for MLP reveal a decrease in MLP expression at this developmental stage. Representative blots with extracts from one animal per lane are shown.
Discussion
In this study, we report alterations of cardiac cytoarchitecture that are associated with two mouse models of DCM, the MLP knockout mouse and the TOT mouse. The organization of the sarcomere in the MLP knockout mouse was not significantly affected, as judged by the appearance in light microscopy, in contrast to the TOT mouse where severe effects on myofibril structure were reported previously (Sussman et al. 1998a). However, cardiomyocytes from both mouse models presented considerable alterations of their cell–matrix as well as their cell–cell contacts. These changes can be partially explained by the more irregular shape of these cardiomyocytes compared with their wild-type counterparts, thus resulting in an increased number of contacts. However, intrinsic alterations of the expression levels of junctional proteins and changes in their organization were also observed. For example, the costameric protein vinculin showed patchy staining near the plasma membranes in MLP−/− cardiomyocytes compared with the regular well-ordered striations that are observed at the membrane of wild-type cardiomyocytes. In the heart, an additional splice variant of vinculin, metavinculin, can be detected, the expression of which was found to be downregulated in human patients with DCM (Maeda et al. 1997). Metavinculin seems to be implicated in the periodic arrangement of actin attachment sites at the membrane of smooth muscle cells (Ehler et al. 1996) and might also be involved in the regular organization of costameres in cardiomyocytes. In the case of MLP−/− mice, no downregulation of metavinculin expression could be detected by reverse transcriptase–PCR (Ehler, E., and J.-C. Perriard, unpublished results), but since no isoform-specific antibodies are available that cross-react with murine metavinculin, a differential localization resulting in a lack of metavinculin at the costameres cannot be excluded. Increased expression of extracellular matrix components and a general upregulation of vinculin was described in patients with DCM (Schaper et al. 1991, Schaper et al. 1995; Wilke et al. 1995), suggesting that this phenomenon might not be restricted to mouse models for this disease.
In addition to the changes in costamere organization, major alterations were observed at the specific cell–cell contacts of cardiomyocytes, the intercalated disks. The ultrastructural alterations observed in the electron microscope with a higher degree of convolution of the membrane and even membrane detachment are quite reminiscent of intercalated disks observed in animals with muscular dystrophy, which can also show symptoms of DCM, in desmin knockout mice that develop cardiomyopathy or in samples from old animals (Forbes and Sperelakis 1972, Forbes and Sperelakis 1985; Thornell et al. 1997).
The importance of an interplay in the stoichiometry of intercalated disk–associated proteins has been demonstrated previously, e.g., in the plakoglobin knockout mouse (Ruiz et al. 1996). Mice that are homozygous null for plakoglobin, which can be found both in desmosomes and adherens junctions, die around embryonic day 12 due to a heart defect. So far, little attention has been paid to the expression levels of adherens junction–associated proteins in cardiac disease. The organization of these proteins changes during development, since at the time of birth, most intercalated disk–associated proteins are still distributed over the entire membrane of the cardiomyocytes and restriction to the bipolar end is only achieved postnatally (Peters et al. 1994; Angst et al. 1997). In the case of the inbred hamster strain Bio14.6, which shows histological and functional symptoms similar to hypertrophic cardiomyopathy (Sakamoto et al. 1997), disruption of cell–cell adhesion with reduced expression levels of A-CAM was reported (Fujio et al. 1995). Our studies indicate that the intercalated disk phenotype that we observe might be specific for DCM since we were unable to detect an upregulation of adherens junction–associated molecules in rodent models that develop hypertrophic cardiomyopathy, such as spontaneously hypertensive rats (Ehler, E., and J.-C. Perriard, unpublished observation).
It is still unclear whether a reduced expression of connexin-43 might be a general feature of cardiac disease. Although this phenomenon has been described in hearts of patients after infarction (Smith et al. 1991; Severs 1995), in hypertrophied human hearts (Peters et al. 1993) and in animal models for cardiomyopathies (e.g., the congestive heart failure stage in a chronic aortic stenosis model in guinea pig; Wang and Gerdes 1999), there are examples of a dispersion of gap junctions from the intercalated disks rather than a downregulation of expression levels. This was found in cases of hypertrophic cardiomyopathy in humans (Sepp et al. 1996) and in the Syrian hamster strain UM-X7.1, which develops a progressive cardiomyopathy with dilation and also shows reduced levels of connexin-43 at the intercalated disks, although connexin-43 expression levels seemed to be unaffected (Luque et al. 1994). The etiology of the hypertrophy might be important as well, since during the early hypertrophic response to renovascular hypertension, a marked upregulation of connexin-43 was observed (Peters 1996). Our results with decreased levels of connexin, as shown by immunoblotting, suggest that in this case reduced amounts of protein are available that might account for the scarce occurrence of gap junctions, as seen in the electron microscope. This downregulation is not compensated by the upregulation of other connexin isoforms like connexin-40, since similar expression levels were found in wild-type and MLP−/− ventricles for this protein (data not shown). Reduced electrical communication between cardiomyocytes might result in an imbalance of free Ca2+ levels. The importance of Ca2+ cycling in cardiac disease was recently demonstrated by Minamisawa et al. 1999. A double knockout of phospholamban, the inhibitor of the muscle-specific sarcoplasmatic reticulum Ca2+ ATPase, and of MLP in mice was not only able to restore normal Ca2+ handling and thus contractility, but showed also a rescued phenotype of the cardiac cytoarchitecture (Minamisawa et al. 1999). Preliminary experiments in our group have indicated that this improvement of function goes hand in hand with the disappearance of the intercalated disk alterations as seen in the single MLP knockout mice (Ehler, E., M. Hoshijima, M. Minamisawa, K. Chien, and J.-C. Perriard, unpublished observations).
Our observations on the MLP knockout mice suggest that the biological role of MLP is to act as a stabilizing factor for myofibril attachment both at the lateral membranes and at the intercalated disk. Recently, it was reported that MLP can serve as a bridging molecule between α-actinin and spectrin, thus providing a link between the myofibril and the membrane cytoskeleton (Flick and Konieczny 2000). In this respect, it is interesting to note that in mdx mice, a mouse model for muscular dystrophy, the membrane cytoskeleton of slow twitch fibers that continue to express MLP in the adult (Schneider et al. 1999) is not as severely affected as that of fast twitch fibers (Williams and Bloch 1999). A slight protective effect of MLP can also be deduced from the fact that TOT mice show an upregulation of N-RAP expression later than do MLP−/− because they still express MLP, although at reduced amounts in the adult. A decrease in MLP expression might be a general feature of late stage cardiomyopathy since it was also demonstrated in human patients with chronic heart failure and in rats with right ventricular hypertrophy induced by chronic pressure overload (Zolk et al. 2000; Ecarnot-Laubriet et al. 2000). How the feedback mechanism between expression levels of MLP and N-RAP works is completely unclear at the moment. Nevertheless, our results suggest that an upregulation of N-RAP expression can be regarded as the earliest marker for a developing DCM.
N-RAP binds actin, vinculin, and talin (Luo et al. 1999), as well as MLP (this report), and has been hypothesized to be an essential link between the terminal actin filaments of myofibrils and the complex of proteins linking these structures to the cell membrane (Luo et al. 1997, Luo et al. 1999). Our results show that a disturbance of the molecular stoichiometry of cytoskeletal proteins, such as loss of MLP or upregulation of tropomodulin, leads to an early increase in N-RAP expression. At the same time, the distribution of N-RAP becomes broader at the light microscope level and the intercalated disks become more convoluted. These changes may be an adaptive response aimed at strengthening the link between the myofibrils and the membrane. It remains to be established whether this altered intercalated disk phenotype is a general feature of DCM by investigating other animal models for this disease as well as human patients. In addition, it will be necessary to establish whether an upregulation of N-RAP expression can be used as an early marker for DCM in a clinical setting.
Acknowledgments
We are grateful for the many helpful discussions with all members of the Perriard and the Eppenberger group. Additionally, we thank Dr. Alison North for the desmoplakin antibody and Prof. Heinz Faulstich for Cy5-conjugated phalloidin. We also thank Amy H. Herrera and Jeffrey Davis for excellent technical assistance and gratefully thank Sara Welch for her technical assistance in maintenance of the TOT mouse colony.
This work was supported by the Swiss National Science Foundation grants 31.52417/97 to J.-C. Perriard and 31.40485.94 to H.M. Eppenberger, and by the research grant Cellular and Molecular Cardiology from the Union Bank of Switzerland, Zurich, Switzerland to H.M. Eppenberger. Research on the TOT mice is supported by a National Institutes of Health award (HL58224) as well as American Heart Association Awards Established Investigator and Grant-in-Aid awards to M. Sussman. Research at the Friedrich Miescher Institute (to P. Caroni) is supported by the Novartis Research Foundation.
Footnotes
Abbreviations used in this paper: DCM, dilated cardiomyopathy; MLP, muscle LIM protein; RT, room temperature; SCB, sodium cacodylate buffer; TOT, tropomodulin-overexpressing transgenic.
References
- Angst B.D., Khan L.U., Severs N.J., Whitely K., Rothery S., Thompson R.P., Magee A.I., Gourdie R.G. Dissociated spatial patterning of gap junctions and cell adhesion junctions during postnatal differentiation of ventricular myocardium. Circ. Res. 1997;80:88–94. doi: 10.1161/01.res.80.1.88. [DOI] [PubMed] [Google Scholar]
- Arber S., Caroni P. Specificity of single LIM motifs in targeting and LIM/LIM interactions in situ. Genes Dev. 1996;10:289–300. doi: 10.1101/gad.10.3.289. [DOI] [PubMed] [Google Scholar]
- Arber S., Halder G., Caroni P. Muscle LIM protein, a novel essential regulator of myogenesis, promotes myogenic differentiation. Cell. 1994;79:221–231. doi: 10.1016/0092-8674(94)90192-9. [DOI] [PubMed] [Google Scholar]
- Arber S., Hunter J.J., Ross J.J., Hongo M., Sansig G., Borg J., Perriard J.-C., Chien K.R., Caroni P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88:393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- Bähler M., Moser H., Eppenberger H.M., Wallimann T. Heart c-protein is transiently expressed during skeletal muscle development in the embryo, but persists in cultured myogenic cells. Dev. Biol. 1985;112:345–352. doi: 10.1016/0012-1606(85)90405-1. [DOI] [PubMed] [Google Scholar]
- Bonne G., Carrier L., Richard P., Hainque B., Schwartz K. Familial hypertrophic cardiomyopathyfrom mutations to functional defects. Circ. Res. 1998;83:580–593. doi: 10.1161/01.res.83.6.580. [DOI] [PubMed] [Google Scholar]
- Chien K. Stress pathways and heart failure. Cell. 1999;98:555–558. doi: 10.1016/s0092-8674(00)80043-4. [DOI] [PubMed] [Google Scholar]
- Draeger A., Stelzer E.H., Herzog M., Small J.V. Unique geometry of actin-membrane anchorage sites in avian gizzard smooth muscle cells. J. Cell Sci. 1989;94:703–711. doi: 10.1242/jcs.94.4.703. [DOI] [PubMed] [Google Scholar]
- Ecarnot-Laubriet A., De Luca K., Vandroux D., Moisant M., Bernard C., Assem M., Rochette L., Teyssier R. Downregulation and nuclear relocation of MLP during the progression of right ventricular hypertrophy induced by chronic pressure overload. J. Mol. Cell. Cardiol. 2000;32:2385–2395. doi: 10.1006/jmcc.2000.1269. [DOI] [PubMed] [Google Scholar]
- Ehler E., Babiychuk E., Draeger A. Human fetal lung (IMR-90) cellsmyofibroblasts with smooth muscle-like contractile properties. Cell Motil. Cytoskeleton. 1996;34:288–298. doi: 10.1002/(SICI)1097-0169(1996)34:4<288::AID-CM4>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Ehler E., Rothen B.M., Hämmerle S.P., Komiyama M., Perriard J.-C. Myofibrillogenesis in the developing chicken heartassembly of Z-disk, M-line and the thick filaments. J. Cell Sci. 1999;112:1529–1539. doi: 10.1242/jcs.112.10.1529. [DOI] [PubMed] [Google Scholar]
- Flick M., Konieczny S. The muscle regulatory and structural protein MLP is a cytoskeletal binding partner of βI-spectrin. J. Cell Sci. 2000;113:1553–1564. doi: 10.1242/jcs.113.9.1553. [DOI] [PubMed] [Google Scholar]
- Forbes M.S., Sperelakis N. Ultrastructure of cardiac muscle from dystrophic mice. Am. J. Anat. 1972;134:271–290. doi: 10.1002/aja.1001340302. [DOI] [PubMed] [Google Scholar]
- Forbes M.S., Sperelakis N. Intercalated discs of mammalian hearta review of structure and function. Tissue Cell. 1985;17:605–648. doi: 10.1016/0040-8166(85)90001-1. [DOI] [PubMed] [Google Scholar]
- Fujio Y., Yamada Honda F., Sato N., Funai H., Wada A., Awata N., Shibata N. Disruption of cell-cell adhesion in an inbred strain of hereditary cardiomyopathic hamster (Bio 14.6) Cardiovasc. Res. 1995;30:899–904. [PubMed] [Google Scholar]
- Gregorio C.C., Fowler V.M. Mechanisms of thin filament assembly in embryonic chick cardiac myocytestropomodulin requires tropomyosin for assembly. J. Cell Biol. 1995;129:683–695. doi: 10.1083/jcb.129.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove B.K., Kurer V., Lehner C., Doetschman T.C., Perriard J.C., Eppenberger H.M. Monoclonal antibodies detect new 185,000 dalton muscle m-line protein. J. Cell Biol. 1984;98:518–524. doi: 10.1083/jcb.98.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera A., Elzey B., Law D., Horowits R. Terminal regions of mouse nebulinsequence analysis and complementary localization with N-RAP. Cell Motil. Cytoskeleton. 2000;45:211–222. doi: 10.1002/(SICI)1097-0169(200003)45:3<211::AID-CM4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Leiden J.M. The genetics of dilated cardiomyopathy—emerging cues to the puzzle. N. Engl. J. Med. 1997;337:1080–1081. doi: 10.1056/NEJM199710093371511. [DOI] [PubMed] [Google Scholar]
- Luo G., Zhang J., Nguyen T.-P., Herrera A., Paterson B., Horowits R. Complete cDNA sequence and tissue localization of N-RAP, a novel nebulin-related protein of striated muscle. Cell Motil. Cytoskeleton. 1997;38:75–90. doi: 10.1002/(SICI)1097-0169(1997)38:1<75::AID-CM7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Luo G., Herrera A.H., Horowits R. Molecular interactions of N-RAP, a nebulin-related protein of striated muscle myotendon junctions and intercalated disks. Biochemistry. 1999;38:6135–6143. doi: 10.1021/bi982395t. [DOI] [PubMed] [Google Scholar]
- Luque E.A., Veenstra R.D., Beyer E.C., Lemmanski L.F. Localization and distribution of gap junctions in normal and cardiomyopathic hamster heart. J. Morph. 1994;222:203–213. doi: 10.1002/jmor.1052220207. [DOI] [PubMed] [Google Scholar]
- Maeda M., Holder E., Lowes B., Valent S., Bies R.D. Dilated cardiomyopathy associated with deficiency of the cytoskeletal protein metavinculin. Circulation. 1997;95:17–20. doi: 10.1161/01.cir.95.1.17. [DOI] [PubMed] [Google Scholar]
- Messerli J.M., Eppenberger Eberhardt M.E., Rutishauser B.M., Schwarb P., von Arx P., Koch Schneidemann S., Eppenberger H.M., Perriard J.-C. Remodelling of cardiomyocyte cytoarchitecture visualized by three-dimensional (3D) confocal microscopy Histochemistry 100 1993. 193 202a [DOI] [PubMed] [Google Scholar]
- Messerli J.M., van der Voort H.T.M., Rungger-Brändle E., Perriard J.-C. Three-dimensional visualization of multi-channel volume datathe amSFP algorithm Cytometry 14 1993. 725 735b [DOI] [PubMed] [Google Scholar]
- Minamisawa S., Hoshijima M., Chu G., Ward C., Frank K., Gu Y., Martone M., Wang Y., Ross J.J., Kranias E., Giles W., Chien K. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99:313–322. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- North A.J., Gimona M., Lando Z., Small J.V. Actin isoform compartments in chicken gizzard smooth muscle cells. J. Cell Sci. 1994;107:445–455. doi: 10.1242/jcs.107.3.445. [DOI] [PubMed] [Google Scholar]
- North A., Bardsley W., Hyam J., Bornslaeger E., Cordingley H., Trinnaman B., Hatzfeld M., Green K., Magee A., Garrod D. Molecular map of the desmosomal plaque. J. Cell Sci. 1999;112:4325–4336. doi: 10.1242/jcs.112.23.4325. [DOI] [PubMed] [Google Scholar]
- Pardo J.V., D'Angelo Siliciano J., Craig S.W. Vinculin is a component of an extensive network of myofibril–sarcolemma attachment regions in cardiac muscle fibers. J. Cell Biol. 1983;97:1081–1088. doi: 10.1083/jcb.97.4.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters N.S. New insights into myocardial arrhythmogenesisdistribution of gap-junctional coupling in normal, ischaemic and hypertrophied human hearts. Clin. Sci. 1996;90:447–452. doi: 10.1042/cs0900447. [DOI] [PubMed] [Google Scholar]
- Peters N.S., Green C.R., Poole-Wilson P.A., Severs N.J. Reduced content of connexin-43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864–875. doi: 10.1161/01.cir.88.3.864. [DOI] [PubMed] [Google Scholar]
- Peters N.S., Severs N.J., Rothery S.M., Lincoln C., Yacoub M.H., Green C.R. Spatiotemporal relation between gap junctions and fascia adherens junctions during postnatal development of human ventricular myocardium. Circulation. 1994;90:713–725. doi: 10.1161/01.cir.90.2.713. [DOI] [PubMed] [Google Scholar]
- Ruiz P., Brinkmann V., Ledermann B., Behrend M., Grund C., Thalhammer C., Vogel F., Birchmeier C., Gunthert U., Franke W.W., Birchmeier W. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J. Cell Biol. 1996;135:215–225. doi: 10.1083/jcb.135.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto A., Ono K., Abe M., Jasmin G., Eki T., Murakami Y., Masaki T., Toyo-oka T., Hanaoka F. Both hypertrophic and dilated cardiomyopathies are caused by mutation of the same gene, δ-sarcoglycan, in hamsteran animal model of disrupted dystrophin-associated glycoprotein complex. Proc. Natl. Acad. Sci. USA. 1997;94:13873–13878. doi: 10.1073/pnas.94.25.13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper J., Froede R., Hein S., Buck A., Hashizume H., Speiser B., Friedl A., Bleese N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation. 1991;83:504–514. doi: 10.1161/01.cir.83.2.504. [DOI] [PubMed] [Google Scholar]
- Schaper J., Mollnau H., Hein S., Scholz D., Munkel B., Devaux B. Interactions between cardiomyocytes and extracellular matrix in the failing human heart. Z. Kardiol. 1995;84:33–38. [PubMed] [Google Scholar]
- Schmeichel K.L., Beckerle M.C. The LIM domain is a modular protein-binding interface. Cell. 1994;79:211–219. doi: 10.1016/0092-8674(94)90191-0. [DOI] [PubMed] [Google Scholar]
- Schneider A., Sultan K., Pette D. Muscle LIM proteinexpressed in slow muscle and induced in fast muscle by enhanced contractile activity. Am. J. Physiol. 1999;276:C900–C906. doi: 10.1152/ajpcell.1999.276.4.C900. [DOI] [PubMed] [Google Scholar]
- Seidman C., Seidman J. Molecular genetic studies of familial hypertrophic cardiomyopathy. Basic Res. Cardiol. 1998;93:13–16. doi: 10.1007/s003950050196. [DOI] [PubMed] [Google Scholar]
- Sepp R., Severs N.J., Gourdie R.G. Altered patterns of cardiac intercellular junction distribution in hypertrophic cardiomyopathy. Heart. 1996;76:412–417. doi: 10.1136/hrt.76.5.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severs N.J. Cardiac muscle cell interactionfrom microanatomy to the molecular make-up of the gap junction. Histol. Histopathol. 1995;10:481–501. [PubMed] [Google Scholar]
- Smith J.H., Green C.R., Petters N.S., Rothery S., Severs N.J. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am. J. Pathol. 1991;139:801–821. [PMC free article] [PubMed] [Google Scholar]
- Sussman M.A., Welch S., Cambon N., Klevitsky R., Hewett T.E., Price R., Witt S.A., Kimball T.R. Myofibril degeneration caused by tropomodulin overexpression leads to dilated cardiomyopathy in juvenile mice J. Clin. Invest 101 1998. 51 61a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman M.A., Baque S., Uhm C.S., Daniels M.P., Price R.L., Simpson D., Terracio L., Kedes L. Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils Circ. Res 82 1998. 94 105b [DOI] [PubMed] [Google Scholar]
- Terracio L., Simpson D.G., Hilenski L., Carver W., Decker R.S., Vinson N., Borg T.K. Distribution of vinculin in the Z-disk of striated muscleanalysis by laser scanning confocal microscopy. J. Cell. Physiol. 1990;145:78–87. doi: 10.1002/jcp.1041450112. [DOI] [PubMed] [Google Scholar]
- Thornell L., Carlsson L., Li Z., Mericskay M., Paulin D. Null mutation in the desmin gene gives rise to a cardiomyopathy. J. Mol. Cell. Cardiol. 1997;29:2107–2124. doi: 10.1006/jmcc.1997.0446. [DOI] [PubMed] [Google Scholar]
- Tokuyasu K.T. Use of poly(vinylpyrrolidone) and poly(vinyl alcohol) for cryoultramicrotomy. Histochem. J. 1989;21:163–171. doi: 10.1007/BF01007491. [DOI] [PubMed] [Google Scholar]
- Towbin J. The role of cytoskeletal proteins in cardiomyopathies. Curr. Opin. Cell Biol. 1998;10:131–139. doi: 10.1016/s0955-0674(98)80096-3. [DOI] [PubMed] [Google Scholar]
- Towbin H., Staehelin T., Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheetsprocedure and applications. Proc. Natl. Acad. Sci. USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikstrom K., Leinwand L. Contractile protein mutations and heart disease. Curr. Opin. Cell Biol. 1996;8:97–105. doi: 10.1016/s0955-0674(96)80053-6. [DOI] [PubMed] [Google Scholar]
- Wang X., Gerdes A.M. Chronic pressure overload cardiac hypertrophy and failure in guinea pigsIII. Intercalated disc remodeling. J. Mol. Cell. Cardiol. 1999;31:333–343. doi: 10.1006/jmcc.1998.0886. [DOI] [PubMed] [Google Scholar]
- Wilke A., Schonian U., Herzum M., Hengstenberg C., Hufnagel G., Brilla C.G., Maisch B. The extracellular matrix and cytoskeleton of the myocardium in cardiac inflammatory reaction. Herz. 1995;20:95–108. [PubMed] [Google Scholar]
- Williams M., Bloch R. Extensive but coordinated reorganization of the membrane skeleton in myofibers of dystrophic (mdx) mice. J. Cell Biol. 1999;144:1259–1270. doi: 10.1083/jcb.144.6.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolk O., Caroni P., Böhm M. Decreased expresson of the cardiac LIM domain protein MLP in chronic human heart failure. Circulation. 2000;101:2674–2677. doi: 10.1161/01.cir.101.23.2674. [DOI] [PubMed] [Google Scholar]