

Abstract
Gap1p, the general amino acid permease of Saccharomyces cerevisiae, is regulated by intracellular sorting decisions that occur in either Golgi or endosomal compartments. Depending on nitrogen source, Gap1p is transported to the plasma membrane, where it functions for amino acid uptake, or to the vacuole, where it is degraded. We found that overexpression of Bul1p or Bul2p, two nonessential components of the Rsp5p E3–ubiquitin ligase complex, causes Gap1p to be sorted to the vacuole regardless of nitrogen source. The double mutant bul1Δ bul2Δ has the inverse phenotype, causing Gap1p to be delivered to the plasma membrane more efficiently than in wild-type cells. In addition, bul1Δ bul2Δ can reverse the effect of lst4Δ, a mutation that normally prevents Gap1p from reaching the plasma membrane. Evaluation of Gap1p ubiquitination revealed a prominent polyubiquitinated species that was greatly diminished in a bul1Δ bul2Δ mutant. Both a rsp5-1 mutant and a COOH-terminal truncation of Gap1p behave as bul1Δ bul2Δ, causing constitutive delivery of Gap1p to the plasma membrane and decreasing Gap1p polyubiquitination. These results indicate that Bul1p and Bul2p, together with Rsp5p, generate a polyubiquitin signal on Gap1p that specifies its intracellular targeting to the vacuole.
Keywords: ubiquitin, Golgi, BUL1, E4, GAP1
Introduction
Saccharomyces cerevisiae encodes 23 amino acid permeases (Nelissen et al. 1997) which can be categorized according to their regulation in response to nitrogen availability in the growth medium (Sophianopoulou and Diallinas 1995). One class of permeases displays either constitutive expression or partial downregulation under nitrogen starvation conditions (Horak 1986). This class of permeases probably imports amino acids for use in protein synthesis and is exemplified by Can1p, a basic amino acid permease (Hoffmann 1985), and the histidine permease Hip1p (Tanaka and Fink 1985). The second class of permeases includes high capacity permeases that are induced on poor nitrogen sources. Permeases of this class include Gap1p, the general amino acid permease which can transport all naturally occurring amino acids (Grenson et al. 1970; Jauniaux and Grenson 1990), and Put4p, a proline-specific permease (Vandenbol et al. 1989). These permeases are probably used by the yeast cell to import amino acids for use as a nitrogen source (Jauniaux and Grenson 1990).
Plasma membrane proteins, such as permeases, are delivered to the cell surface by the secretory pathway. The trans-Golgi compartment is a major branch point in this pathway, where proteins destined for the cell surface are sorted from Golgi complex–resident proteins and proteins destined for the vacuolar/lysosomal compartments (Glick 2000; Lemmon and Traub 2000). Sorting at this branch point in the pathway often involves recycling of proteins between the trans-Golgi and the prevacuolar compartment (PVC) (also known as the late endosome). For example, Vps10p, a Golgi receptor for a soluble vacuolar protease, maintains its intracellular distribution by cycling from the trans-Golgi compartment to the PVC and then back to the trans-Golgi compartment (Marcusson et al. 1994).
Movement of Gap1p and Put4p through the late secretory pathway is regulated by the quality of the external nitrogen source. In cells grown on a relatively poor nitrogen source such as urea, Gap1p is sorted to the plasma membrane, where it is active for transport. In contrast, in cells grown on a relatively rich nitrogen source such as glutamate, Gap1p travels through the secretory pathway to the Golgi compartment, but is directed to the vacuolar sorting pathway and is degraded without ever having reached the cell surface (Roberg et al. 1997b). Recessive mutations in any of four genes, SEC13, LST4, LST7, or LST8, produces a similar effect as growth on glutamate: Gap1p and Put4p proteins are expressed, but their activity is very low even on poor nitrogen sources (Roberg et al. 1997a).
Some plasma membrane proteins, such as Ste2p, Ste6p, and Fur4p, are regulated by ubiquitination and consequent internalization via the endocytic pathway (Galan et al. 1996; Hicke and Riezman 1996; Hicke 1997; Kölling and Losko 1997; Losko et al. 2001). Gap1p is also regulated in this manner, and Gap1p ubiquitination and endocytosis are triggered by a shift to a rich nitrogen source. Gap1p ubiquitination requires Rsp5p, an E3 ubiquitin ligase which catalyzes the addition of a ubiquitin moiety to lysine residue(s) in target proteins (Huibregtse et al. 1995; Springael and André 1998). After internalization, Gap1p is degraded in the vacuole (Springael and André 1998).
In this paper we show that ubiquitination also plays a role in controlling Gap1p sorting within the Golgi or endosomal compartments. We demonstrate that the Bul1p and Bul2p proteins, which form a complex with Rsp5p (Yashiroda et al. 1996, Yashiroda et al. 1998), specify the polyubiquitination of Gap1p protein and that this polyubiquitination is required as a signal for the sorting of Gap1p from the trans-Golgi compartment to the vacuole.
Materials and Methods
Strains, Plasmids, and Media
The yeast strains used in this study (listed in Table ) are all in the S288C genetic background. A distinguishing feature of the S288C background is expression of high levels of Gap1p and Put4p permeases when ammonia is used as a nitrogen source (Courchesne and Magasanik 1983). Complete gene deletions of LST4, BUL1, and BUL2 were constructed by gene replacement through homologous recombination (Wach et al. 1994). Oligonucleotides used to generate the PCR products for gene replacement were as follows: LST4, OSH43, and OSH44; BUL1, OSH62, and OSH63; and BUL2, OSH64, and OSH65. The sequences of all oligonucleotides are provided below. Correct gene deletions were confirmed by PCR. All plasmids used in this study are listed in Table and the details of plasmid construction are given below. Medium was prepared as described (Roberg et al. 1997b).
Table 1.
Yeast Strains Used in this Study (All Are Isogenic with S288C)
| Name | Former name | Genotype | Origin |
|---|---|---|---|
| CKY4 | — | Mat a ura3 | Kaiser strain collection |
| CKY482 | — | Mat α ura3 leu2 gap1::LEU2 | Kaiser strain collection |
| CKY694 | NRY169 | Mat α ura3 pep12::TRP1 | Kaiser strain collection |
| CKY695 | SHY13-6B | Mat α ura3 lst4::kanMX6 | This study |
| CKY698 | SHY48-2A | Mat a ura3 bul1::kanMX6 bul2::kanMX6 | This study |
| CKY699 | SHY60-44B | Mat α ura3 lst4::kanMX6 bul1:kanMX6 bul2::kanMX6 | This study |
| CKY700 | SHY125-8B | Mat a ura3 vps45Δ | Kaiser strain collection |
| CKY701 | SHY138-11C | Mat α ura3 leu2 gap1::LEU2 bul1::kanMX6 bul2::kanMX6 | This study |
| CKY712 | SHY159-2B | Mat α ura3 rsp5-1 | This study |
| CKY713 | SHY159-3A | Mat α ura3 lst4::kanMX6 rsp5-1 | This study |
| CKY714 | SHY160-3D | Mat α ura3 bul1::kanMX6 bul2::kanMX6 rsp5-1 | This study |
| CKY702 | SHY171-1A | Mat α ura3 leu2 gap1::LEU2 lst4::kanMX6 | This study |
| CKY715 | SHY212-1D | Mat α ura3 leu2 gap1::kanMX6 | This study |
| CKY703 | SLY71 | Mat a ura3 leu2 his3 gap1::LEU2 | This study |
| CKY704 | SLY173 | Mat α ura3 leu2 his3 gap1::LEU2 bul1::kanMX6 bul2::kanMX6 | This study |
Table 2.
Plasmids Used in this Study (See Materials and Methods for Further Details)
| Genotype | Name | Other name; description |
|---|---|---|
| — | pRS206-2μ | URA3 2μ (Sikorski and Hieter 1989) |
| — | pRS316 | URA3 CEN (Sikorski and Hieter 1989) |
| — | pRS305-2μ | LEU2 2μ (Sikorski and Hieter 1989) |
| pBUL1 | pCK249 | pSH43; pRS316 containing BUL1 ORF plus 5′ and 3′ regions |
| pBUL1 | pCK228 | pSH44; pRS306 2μ containing BUL1 ORF plus 5′ and 3′ regions |
| p bul1P 157Q,P158A | pHY37 | pRS316 containing BUL1 with double point mutations P157Q, P158A (Yashiroda et al. 1998) |
| pBUL2 | pCK229 | pSH47; pRS306 2μ containing BUL2 ORF plus 5′ and 3′ regions |
| pBUL2 | pCK250 | pSH54; pRS305 2μ containing BUL2 ORF plus 5′ and 3′ regions |
| pPGAP1-lacZ | pMS29 | pGAP1-lacZ fusion at codon 53 of GAP1; CEN (Stanbrough and Magasanik 1995) |
| pHA-GAP1 | pPL257 | GAP1 with the HA1 epitope inserted at codon 62; CEN (Ljungdahl et al. 1992) |
| pGAP1-GFP | pCK230 | pSH40; GAP1-sGFP fusion under the GAP1 promoter; CEN |
| PADH1-HA-GAP1 | pCK227 | pEC224; encodes the HA-GAP1 ORF and terminator fused behind the ADH1 promoter in pRS316 |
| PCUP1-UBI-c-myc | pCK231 | pSL1; UBI-c-myc expression cassette fused behind the CUP1 promoter based on pRS423 HIS3 2μ |
| PCUP1-UB1 | pCK232 | pSL2; pSL1 without the c-myc tag |
| The four plasmids below are isogenic, differing only in sequence noted | ||
| pGAP1 | pCK233 | pSH50; GAP1 with CYC1 terminator |
| pGAP1Δ2 | pCK234 | pSH51; pSH50 with stop codon causing loss of last 11 codons |
| pHA-GAP1 | pCK235 | pSH49; pSH50 with HA epitope as in pPL257 above |
| pHA-GAP1Δ2 | pCK236 | pSH52; pSH51 with HA epitope as in pPL257 above |
Screen for Resistance to l-Azetidine-2-Carboxylic Acid
Strain CKY4 (Mat a ura3; Table ) was transformed with DNA from a high copy (2μ) plasmid library, grown in synthetic minimal glucose medium for 5 h, and plated on solid SD medium containing 50 mg/liter l-azetidine-2-carboxylic acid (ADCB) (SD-ADCB; Sigma-Aldrich). Colonies that formed after incubation at 24°C for 4–5 d were tested for growth on SD-ADCB plates. Plasmids recovered from the resistant transformants were retransformed into CKY4 to verify their ability to confer ADCB resistance. Plasmids pADR3 and pADR13 carried the complete coding sequences of ZDS1, RCE1, and BUL1. Plasmid pADR10 contained the complete coding sequences of DAT1, CTK3, and BUL2. The BUL1 and BUL2 genes were each subcloned into a high copy vector, as described above, and were then tested for their ability to confer ADCB resistance.
Assays for Amino Acid Uptake and β-Galactosidase
Amino acid uptake assays were performed as described (Roberg et al. 1997b). Strains to be assayed for GAP1 transcription were transformed with plasmid pMS29 carrying the PGAP1-lacZ fusion (Stanbrough and Magasanik 1995), and specific β-galactosidase activity was measured as described previously (Guarente 1983).
Fluorescence Microscopy
Strains expressing Gap1–green fluorescent protein (GFP) were cultured overnight in SD medium to exponential phase and viewed directly using a fluorescence microscope (Eclipse; Nikon) coupled to a CCD Hamamatsu video camera. Image analysis was performed using Openlab software from Improvision, Inc.
Membrane Protein Preparation, Western Blotting, Cell Fractionation, Equilibrium Density Centrifugation, and Antibodies
Protocols are described in Roberg et al. 1997b. Antibodies were used as follows: anti-hemagglutinin (HA) antibody, either 12CA5 (BabCO) at 1:1,000 dilution, or 16B12 (BabCO) at 1:500 dilution; mouse anti-Dpm1p (Molecular Probes) at 1:200; rabbit anti-Pma1p (gift of A. Chang, Albert Einstein College of Medicine, New York, NY) at 1:10,000 dilution; and HRP-coupled sheep anti–rabbit and HRP-coupled sheep anti–mouse (both Amersham Pharmacia Biotech) at 1:10,000 dilution.
Immunoprecipitation and Western Blotting of Ubiquitin Conjugates
Detection of ubiquitin conjugates was performed as described previously (Kölling and Hollenberg 1994), with some modifications. Cells were grown overnight in SD Urea medium to exponential phase and 6 × 108 cells were collected by centrifugation, washed in cold 10 mM NaN3, and suspended in 150 μl lysis buffer (0.3 M sorbitol, 10 mM Hepes, pH 7.5, 10 mM NaN3) with protease inhibitors 1 mM PMSF, 0.5 mg/ml leupeptin, 0.7 mg/ml pepstatin. Cells were lysed by vortexing with glass beads. 1 vol of 2× sample buffer (4% SDS, 125 mM Tris-Cl, pH 6.8, 20% glycerol, 20 mM DTT, 0.02% bromophenol blue) was added, and the lysate was incubated at 37°C for 1 h. 4 vol of IP dilution buffer (1.25% Triton X-100, 190 mM NaCl, 6 mM EDTA, 60 mM Tris-Cl, pH 7.5) was added and insoluble material was removed by centrifugation at 12,000 g. Samples were preadsorbed by 30 min incubation at 25°C with 40 μl of a 20% suspension of protein G–Sepharose 4 fast flow (Amersham Pharmacia Biotech) in IP buffer (1% Triton X-100, 0.2% SDS, 150 mM NaCl, 5 mM EDTA, 50 mM Tris-Cl, pH 7.5) and the beads were removed by centrifugation. Immunoprecipitations were carried out by overnight incubation at 4°C with 8 μl of antiserum (rat anti-HA antibody [3F10]; Boehringer), followed by incubation for 3 h at room temperature with 40 μl of protein G–Sepharose suspension. The beads were washed three times with IP buffer, diluted 1:2 with 2× sample buffer with 100 mM DTT, and incubated for 1 h at 37°C. Half of the immunoprecipitated material was loaded onto a 10% SDS gel. Western blotting was performed as above. Antibodies were used as follows: mouse anti-HA antibody, 16B12 (BAbCO) at 1:500 dilution; mouse anti–c-myc antibody (9E10) at 1:1,000 dilution; and HRP-coupled sheep anti–mouse (both Amersham Pharmacia Biotech) at 1:5,000 dilution.
Oligos Used for Gene Deletion
Oligonucleotides used are as follows: OSH43, GTCTTGTGTGTGGCC-TTGTAGAGAAGGTGAAGAGGGAGAGTTTATCGTACGCTGC-AGGTCGAC, OSH44, TTAGGTAACTGGAATATATTAAACATGTAAAGAAGGAGAAAACAGAATTCGAGCTCGTTTAAAC, OSH62, CGAAAAGAGACTGTTCGTGTGTGTCAACAGGTATATCGTACGCTAACGTACGCTGCAGGTCGAC and OSH63, TATATCTATAAGAAAAGTAACGAGAATTTTTTCTAATGTTTTTTTAGAATTCGAGCTCGTTAAAC, OSH64, GAAGCAGCAGATTTGAGAT-ATATTCTGGGGAACAAAAGAAGTATTACGTACGCTGCAGGTCGAC, and OSH65, CAATTATTTGTAAAACTGCGAGATTACT-GTTAGTGTTGTATGGTCTAGAATTCGAGCTCGTTTAAAC.
Plasmid Construction
The plasmid pCK228 (pBUL1), which carries the BUL1 gene and surrounding genomic sequences, was constructed by PCR amplification of BUL1 sequences from the plasmid template pADR3 (screen isolate; see below) using oligos OSH68 and OSH69. The resulting fragment was digested with BglII and SalI and ligated into an appropriately digested 2μ URA3 ampR shuttle plasmid pRS306-2μ (Sikorski and Hieter 1989). The plasmid pCK229 (pBUL2), which carries the BUL2 gene, was constructed by PCR amplification of BUL2 sequences from the plasmid template pADR10 (screen isolate; see below) using oligos OSH72 and OSH73. The resulting PCR fragment was digested with BglII and SalI and ligated into pRS306-2μ (Sikorski and Hieter 1989). Plasmids pCK228 and pCK229 conferred ADCB resistance in a manner similar to the original plasmid isolates from the screen. pCK249 (see pBUL1 in Fig. 9 D) was constructed as pCK228 using pRS316 instead of pRS306 2μ. pCK250 (see pBUL2 in Fig. 8 C) was constructed as pCK229 using pRS305 2μ instead of pRS306 2μ. pCK230 (pGAP1-GFP) was constructed by PCR amplification of the GAP1 promoter and coding sequence from the plasmid template pPL247 (which contains GAP1 promoter, ORF, and terminator; Ljungdahl et al. 1992) using OSH13 and OSH23. This fragment was digested with XhoI and ligated into XhoI cut plasmid pPS1527, which contains the S65T GFP coding region and the NUF2 terminator, and is URA3 CEN ampR (Ferrigno, P., and P. Silver, Harvard Medical School, Boston, MA). pCK233 (see pGAP1 used in Fig. 7 and Fig. 9) and pCK235 (see pHA-GAP1 used in Fig. 7 and Fig. 9) were constructed by replacing the 2.2-kb XbaI-AgeI fragment from pSL7 (see below) with the XbaI-AgeI fragment from pPL247 and pPL257, respectively (Ljungdahl et al. 1992). pCK234 (pGAP1Δ2) and pCK236 (pHA-GAP1Δ2) were constructed in parallel using pSL8 as the starting plasmid. To construct pSL7 and pSL8 a 2.8-kb EcoRI-HindIII partial digest of pCM252 (Belli et al. 1998) was subcloned into YCplac33 cut with EcoRI and HindIII, resulting in plasmid pSL6. The GAP1 ORF was isolated using oligos SL17 and SL18, which created a 1.8-kb 5′-BamHI GAP1 3′-PstI fragment which was subcloned into pSL6 to create pSL7. A GAP1 ORF with a stop codon after codon 591 (deletion of the last 11 amino acids, GAP1Δ2) was isolated with oligo SL17 (see above) and oligo SL22, cccctgcagTtaCTTTGTGGCCATAATTGCCT, resulting in a 1.8-kb 5′-BamHI GAP1Δ2 3′-PstI fragment which was subcloned into pSL6 to create pSL8. A BamHI-ClaI fragment from YEp96 or YEp105 (provided by A. Varshavsky, California Institute of Technology, Pasadena, CA) (Ellison and Hochstrasser 1991) was ligated into BamHI/ClaI–digested pRS423 (HIS3 2μ) to make pCK231 (PCUP1-UBI-c-myc) and pCK232 (PCUP1-UBI), respectively.
Figure 9.

Bul1p and Bul2p regulate Gap1p activity in conjunction with Rsp5p. (A) Wild-type (CKY4), rsp5-1 (CKY712), lst4Δ (CKY695), rsp5-1 lst4Δ (CKY713), bul1Δ bul2Δ (CKY698), and rsp5-1 bul1Δ bul2Δ (CKY714) strains were cultured in minimal medium (SD Ammonia) at 34°C. (B) Wild-type (CKY4) and rsp5-1 (CKY712) strains were transformed with a multicopy plasmin carrying BUL1 (pCK228) or the corresponding vector. Transformants were grown at 34°C in minimal medium (SD Ammonia). (D) Wild-type (CKY4), bul1Δ bul2Δ (CKY698), and lst4Δ bul1Δ bul2Δ (CKY699) strains were transformed with a low copy vector carrying BUL1 (pCK249), bul1P157Q, P158A (pHY37), or the corresponding vector. Transformants were cultured in minimal medium (SD Ammonia) at 24°C. (A, B, and D) Strains were assayed for [14C]citrulline uptake and values are expressed as a percentage of the relevant wild-type strain. The data represent the mean for three independent experiments (A), or three separate transformants (B and D); error bars represent one standard deviation. (C) Wild-type (CKY4), rsp5-1 (CKY712), bul1Δ bul2Δ (CKY698), and rsp5-1 bul1Δ bul2Δ (CKY714) strains were transformed with pHA-GAP1 (pPL257), and protein extracts made from exponentially growing cultures in minimal medium (SD Ammonia) at 24°C or 34°C were subject to SDS-PAGE and Western blotting with anti-HA antibody. 12.5 times more material was loaded onto this gel compared with the gel in Fig. 3, in order to visualize the ubiquitinated forms of HA-Gap1p, which are denoted with an asterisk.
Figure 8.
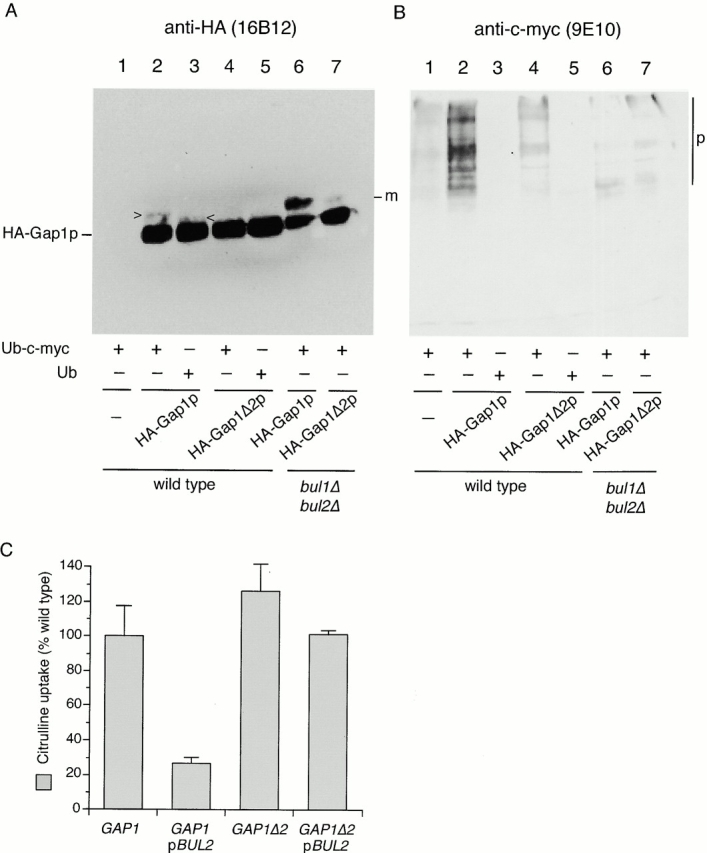
The Gap1Δ2p truncation is a poor substrate for Bulp-mediated polyubiquitination. (A) Wild-type (CKY703; lanes 1–5) and bul1Δ bul2Δ (CKY704; lanes 6 and 7) strains were transformed with combinations of empty vector (pRS316; lane 1), pHA-GAP1 (pCK235; lanes 2, 3, and 6), or pHA-GAP1Δ2 (pCK236; lanes 4, 5, and 7) and either pPCUP1-UBI-c-myc (pCK231; lanes 1, 2, 4, 6, and 7) or pPCUP1-UBI (pCK232; lanes 3 and 5). Strains were cultured overnight in minimal medium (SD Urea) at 24°C and CuSO4 was added to 0.1 μM to induce the CUP1 promoter 3 h before harvesting in exponential phase. Anti-HA (3F10) immunoprecipitates were subject to SDS-PAGE and Western blotting with either (A) anti-HA (16B12) or (B) anti–c-myc (9E10). The following forms of Gap1p are marked: m, monoubiquitinated HA-Gap1(Δ2)p; p, polyubiquitinated HA-Gap1(Δ2)p; >, HA-Gap1p-Ub-c-myc; and <, HA-Gap1p-Ub. (C) A gap1::kanMX6 strain (CKY715) was transformed with combinations of plasmids carrying GAP1 (pCK233) or GAP1Δ2 (pCK234) and either a multicopy vector or one carrying BUL2 (pBUL2; pCK250) or the corresponding vector. Transformants were grown on minimal medium (SD Ammonia) at 24°C, and [14C]citrulline uptake activities were determined. Values are expressed as a percentage of the activity of the gap1Δ [pGAP1] strain. The data represents the mean for three separate transformants; error bars represent one standard deviation.
Figure 7.

The GAP1Δ2 truncation bypasses lst4Δ to restore Gap1p activity in the plasma membrane. (A) Wild-type (CKY482), lst4Δ (CKY702), and a bul1Δ bul2Δ (CKY701) strain were transformed with pGAP1 (pCK233) or pGAP1Δ2 (pCK234). Three transformants of each strain were cultured in minimal medium (SD Ammonia) at 24°C and assayed for [14C]citrulline uptake. Values are the mean and standard deviation for three transformants for each strain and are expressed as a percentage of the activity in wild-type (gap1Δ [pGAP1]).
Results
Overexpression of BUL1 or BUL2 Decreases Gap1p Activity
The toxic proline analogue ADCB enters cells primarily through the Put4p proline permease. To identify new genes that control the sorting of the nitrogen-regulated permeases we conducted a primary screen for genes that when overproduced could confer resistance to ADCB. A wild-type strain (CKY4) was transformed with an S. cerevisiae genomic library in a multicopy (2 μm) vector, and transformants that were resistant to 50 mg/liter ADCB were isolated. Sequencing and subcloning of plasmids from two clones isolated from the screen indicated that overexpression of either BUL1 or BUL2 (pBUL1; pCK228 and pBUL2; pCK229) could confer ADCB resistance (Fig. 1 A; see Materials and Methods). To eliminate possible effects of Gap1p on ADCB uptake we verified that overexpression of either BUL1 or BUL2 also conferred ADCB resistance to a gap1Δ strain (Fig. 1 A).
Figure 1.
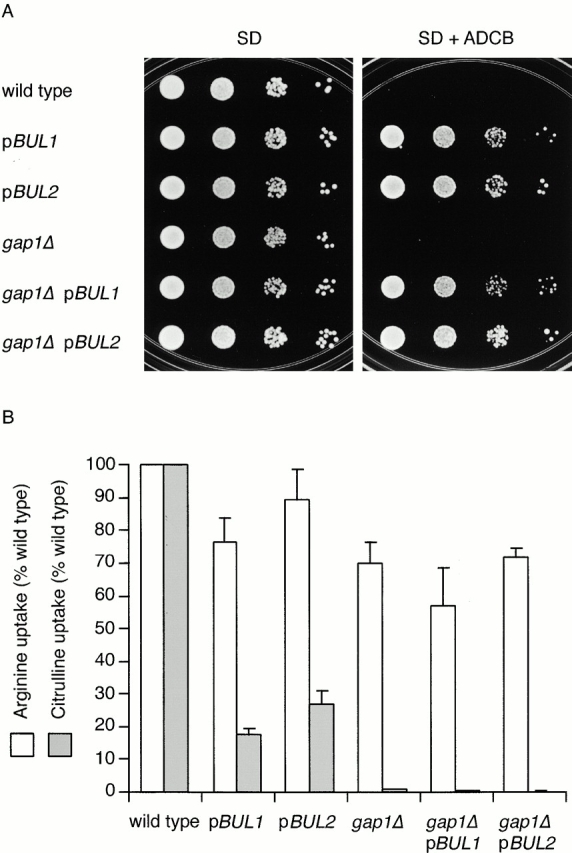
Overexpression of either BUL1 or BUL2 decreases Put4p and Gap1p activity. (A) Wild-type (CKY4) and gap1Δ (CKY482) strains transformed with an empty vector (pRS306-2μ), pBUL1 (pCK228), or pBUL2 (pCK229) were spotted as serial dilutions on minimal medium containing no drug (SD Ammonia) or ADCB at 50 mg/liter (SD Ammonia plus ADCB) and incubated at 24°C. (B) The same strains were grown in minimal medium at 24°C and assayed for import of [14C]arginine or [14C]citrulline. Uptake velocities are expressed as a percentage of the wild-type strain carrying an empty vector. The data represent the mean for at least four separate transformants; error bars represent one standard deviation.
To assess the effect of overexpression of either BUL1 or BUL2 on Gap1p activity, we assayed the rate of [14C]citrulline uptake by yeast cells, a specific measure of Gap1p activity. Introduction of a BUL1 or BUL2 multicopy plasmid into a wild-type strain caused Gap1p activity to be reduced four- to fivefold below that of a strain carrying an empty vector (Fig. 1 B). Arginine is transported into yeast cells by both Gap1p and the dedicated arginine permease Can1p, whose activity is not significantly altered by nitrogen source. A gap1Δ strain (CKY482) grown in minimal medium imported arginine at a rate of ∼70% of an isogenic wild-type strain, indicating that Gap1p was responsible for about one third of the arginine uptake. Overexpression of BUL1 or BUL2 has no effect on Can1p activity because both wild-type and gap1Δ strains containing either pBUL1 or pBUL2 imported arginine at rates similar to a gap1Δ strain (Fig. 1 B). Thus, overexpression of either BUL1 or BUL2 reduced Gap1p and Put4p activity without significantly affecting Can1p.
BUL1 and BUL2 encode related proteins (55% identity), each with a predicted molecular mass of 110 kD. Bul1p was first identified as a protein that binds to the Rsp5p E3 ubiquitin ligase, an enzyme involved in the attachment of ubiquitin moieties to proteins destined for degradation (Yashiroda et al. 1996). BUL2 was shown to be a functional homologue of BUL1 by the growth defects exhibited by a bul1Δ bul2Δ strain under different stress-related growth conditions (Yashiroda et al. 1998).
The Effect of Overexpression of BUL1 or BUL2 on Gap1p Can Be Suppressed by a Block in Golgi Complex to Vacuole Trafficking
Strains overexpressing BUL1 or BUL2 exhibited the same selective defect in Gap1p and Put4p activity that we had observed previously for sec13-1, lst4-1, and lst7-1 mutants (Roberg et al. 1997a). The loss of Gap1p activity in these mutants is the consequence of a failure to traffic Gap1p to the plasma membrane (Roberg et al. 1997a). Trafficking to the plasma membrane can be partially restored by blocking the vacuolar limb of the intracellular sorting pathway by deletion of PEP12, a t-SNARE of the PVC required for vacuolar trafficking (Becherer et al. 1996). We determined whether a pep12Δ mutation could similarly suppress the effect of overexpression of BUL1 or BUL2. Overexpression of BUL1 reduces Gap1p activity to <20% of wild-type (CKY4 with pCK228), but the presence of pep12Δ in the genetic background (CKY694 with pCK228) increased [14C]citrulline uptake more than threefold, bringing Gap1p activity to ∼70% of that of a wild-type strain (Fig. 2). We included a second Golgi to PVC sorting mutant, vps45Δ, in this analysis (Cowles et al. 1994). A vps45Δ strain (CKY700) overexpressing either BUL1 or BUL2 had twofold higher Gap1p activity than an isogenic wild-type strain overexpressing the BUL gene (Fig. 2). Thus, overexpression of either BUL gene caused a reduction in Gap1p activity, but this reduction could be partially suppressed by mutations in the vacuolar protein sorting VPS pathway. These observations raised the possibility that the BUL gene products regulate the entry of Gap1p into the Golgi to vacuole pathway.
Figure 2.

The effects of BUL1 or BUL2 overexpression can be suppressed by mutations in the Golgi to vacuole pathway. Wild-type (wt, CKY4), pep12Δ (CKY694), or vps45Δ (CKY700) strains containing either BUL1 or BUL2 on a multicopy plasmid (pCK228 or pCK229) were grown on minimal medium (SD Ammonia) at 24°C. The activities for [14C]citrulline uptake are expressed as a percentage of the activity of an isogenic wild-type strain. The data represent the mean for four separate transformants; error bars represent one standard deviation.
Deletion of BUL1 and BUL2 Increases Gap1p Activity
We constructed chromosomal deletions of BUL1 and BUL2 and evaluated their effects on Gap1p expression and activity (Fig. 3). Deletion of either BUL1 or BUL2 or both genes together did not produce any observable alteration in growth on rich or minimal media over a wide range of temperatures. However, the bul deletion mutations gave rise to a pronounced increase in Gap1p activity: bul1Δ and bul2Δ strains exhibited a 2.3- and 1.9-fold increase of Gap1p activity, respectively (data not shown), and a bul1Δ bul2Δ double mutant (CKY698) increased Gap1p activity by 2.6-fold (Fig. 3 A). Analysis of GAP1 expression levels showed that the increase in Gap1p activity in the bul1Δ bul2Δ strain was not due to an increase in GAP1 transcription because PGAP1-lacZ reporter expression is decreased modestly in the double mutant (Fig. 3 B). Consistent with the observed increase in Gap1p activity, we also noted that a bul1Δ bul2Δ strain is hypersensitive to ADCB, indicating that Put4p activity may be increased as well. Both the high Gap1p and Put4p activity exhibited by a bul1Δ bul2Δ mutant and the decrease in Gap1p and Put4p activity exhibited by strains overexpressing Bul1p or Bul2p indicate that in wild-type cells the BUL genes act negatively on Gap1p and Put4p activity by a posttranscriptional mechanism.
Figure 3.
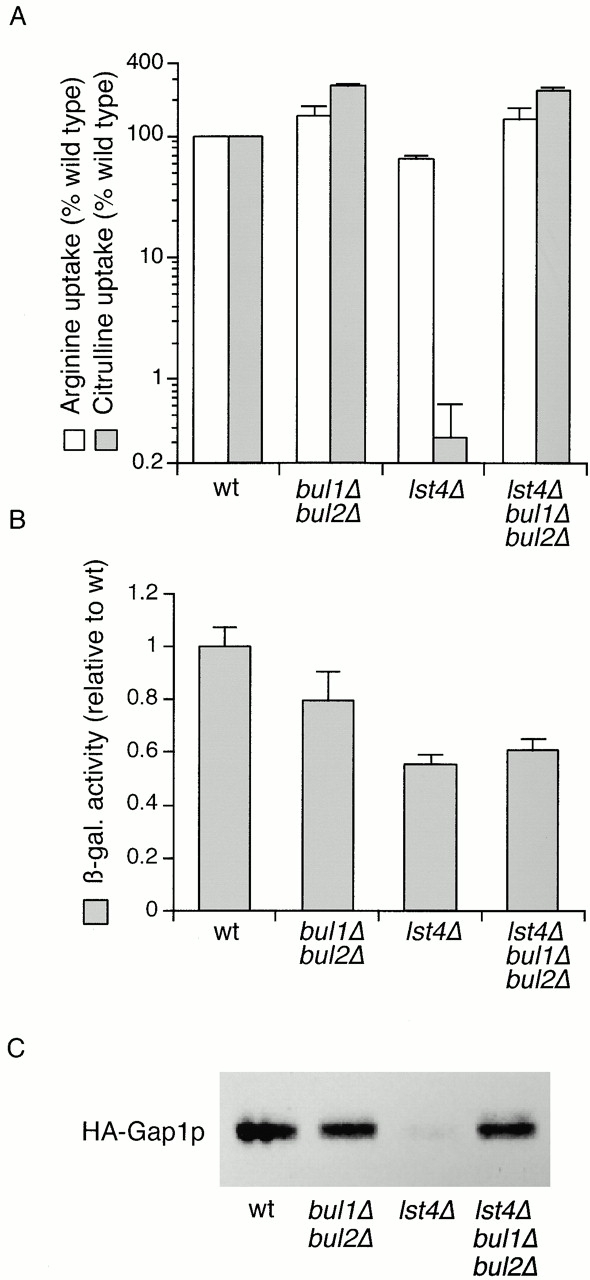
Mutations in BUL1 and BUL2 suppress the effect of lst4Δ on Gap1p activity. (A) Wild-type (wt, CKY4), bul1Δ bul2Δ (CKY698), lst4Δ (CKY695), and lst4Δ bul1Δ bul2Δ (CKY699) strains were cultured in minimal medium (SD Ammonia) at 24°C and assayed for [14C]citrulline and [14C]arginine uptake. Data are expressed as a percentage of the wild-type strain for each amino acid assayed. The mean and one standard deviation for at least three independent experiments are shown. (B) The same strains were transformed with PGAP1-lacZ (pMS29) and assayed for β-galactosidase activity after growth in minimal medium (SD Ammonia) at 24°C. The mean specific activity for three transformants is given relative to the activity in wild-type. (C) The same strains were transformed with pHA-GAP1 (pPL257), and protein extracts made from exponentially growing cultures in minimal medium (SD Ammonia) at 24°C were subject to SDS-PAGE and Western blotting with anti-HA antibody. Each lane contains an extract from the same number of yeast cells.
Deletion of BUL1 and BUL2 Restores Gap1p Activity in lstΔ
We showed previously that sec13-1, lst4-1, and lst7-1 mutations dramatically decrease the export of Gap1p to the plasma membrane, due to a trafficking defect at the level of the Golgi compartment or PVC. We had previously used the sec13-1 allele to represent this class of mutants, but SEC13 also has an essential part in ER to Golgi complex trafficking, causing unwanted complications for analysis of permease traffic in the Golgi compartment. Therefore, we cloned the LST4 gene (ORF YKL176C) and constructed a complete deletion of the coding region (Helliwell, S.B., and C.A. Kaiser, manuscript in preparation; see Materials and Methods). An lst4::kanMX6 (lst4Δ) strain has no apparent growth defect on rich or minimal medium. We established that an lst4Δ strain (CKY695) behaved similarly to the original lst4-1 strain (Roberg et al. 1997a) by assessing Gap1p activity (Fig. 3). We confirmed that HA-Gap1p traveled to the vacuole independently of the plasma membrane and was degraded in the vacuole in an lst4Δ strain by using the same criteria used to demonstrate the effects of sec13-1 (Roberg et al. 1997b; Helliwell, S.B., and C.A. Kaiser, manuscript in preparation). Finally, we performed sucrose gradient fractionation on an lst4Δ strain and HA-Gap1p cofractionated with the ER and Golgi markers, but there was no HA-Gap1p detectable in plasma membrane fractions marked by Pma1p (see below). Thus, lst4Δ and the lst4-1 alleles similarly prevent delivery of Gap1p to the plasma membrane. We also compared Gap1p activity in lst4Δ and a strain in which lst4Δ was combined with end3Δ, a mutation demonstrated to block endocytosis (Benedetti et al. 1994). The lst4Δ end3Δ strain exhibited no significant increase in Gap1p activity over an lst4Δ strain (data not shown), indicating that the Gap1p transport step defined by lst4Δ occurs before the endocytosis step defined by end3Δ. This observation is consistent with those made earlier, in which the reduction of Gap1p activity in sec13-1 is not suppressed by end3Δ (Roberg et al. 1997b).
The effects of overexpression or deletion of the BUL1 and BUL2 genes on Gap1p activity suggested a function antagonistic to that of the SEC13 and LST gene products, so we wished to elucidate the relationship between the LST4 and BUL gene functions. We assayed the effects of bul1Δ bul2Δ, lst4Δ and lst4Δ bul1Δ bul2Δ deletions on Gap1p activity (Fig. 3 A). The bul1Δ bul2Δ strain (CKY698) exhibited significantly more Gap1p activity than a wild-type strain (CKY4). The lst4Δ mutation almost completely abolished [14C]citrulline uptake. However, in the lst4Δ bul1Δ bul2Δ strain (CKY699) [14C]citrulline uptake was similar to that of the bul1Δ bul2Δ strain, showing that lst4Δ does not block Gap1p trafficking to the plasma membrane when combined with bul1Δ bul2Δ mutations. This epistasis relationship suggested that Bul1p and Bul2p acted on Gap1p before the sorting step governed by Lst4p and therefore before Gap1p reaches the plasma membrane. In an lst4Δ background, neither single bulΔ mutant had the same effect as the bul1Δ bul2Δ double mutant. Gap1p activity in bul1Δ lst4Δ or bul2Δ lst4Δ double mutants was much lower than that of wild-type (similar to that of an lst4Δ single mutant) (data not shown). Apparently, there is sufficient functional overlap between BUL1 and BUL2 that both genes must be deleted in order to bypass completely the effect of an lst4Δ mutation. Uptake of arginine was slightly affected for the three mutant strains shown here, but these differences could be attributed to the contribution of Gap1p to arginine uptake.
Since lst4Δ shares all of its known Gap1p-related phenotypes with sec13-1, we also evaluated Gap1p activity in an sec13-1 bul1Δ bul2Δ strain. An sec13-1 strain exhibited almost undetectable Gap1p activity but an sec13-1 bul1Δ bul2Δ strain had more Gap1p activity than wild-type (data not shown). Thus, Bul1p and Bul2p appear to act on Gap1p before the trafficking step(s) controlled by Lst4p and Sec13p.
The effects on Gap1p due to loss of BUL1 and BUL2 occur posttranscriptionally, and we ascertained that the effects of the lst4Δ mutation did not stem from altered transcription (Fig. 3 B). Expression of the PGAP1-lacZ reporter was reduced approximately twofold by an lst4Δ mutation, but this relatively small effect on transcription was not sufficient in itself to account for the >200-fold decrease in Gap1p activity caused by lst4Δ. We also evaluated the effect of lst4Δ, bul1Δ, and bul2Δ mutations on the steady state levels of HA-Gap1p expressed from a centromeric plasmid pPL257 (Ljungdahl et al. 1992). Comparison of HA-Gap1p in wild-type and bul1Δ bul2Δ strains indicated that there is little difference in the steady state protein level (Fig. 3 C). An lst4Δ strain exhibited greatly decreased but still detectable levels of HA-Gap1p, indicating that the protein is less stable than in a wild-type (compare lanes 1 and 3). The level of HA-Gap1p in an lst4Δ bul1Δ bul2Δ mutant was similar to wild-type, showing that the bul1Δ bul2Δ mutation could greatly increase HA-Gap1p stability in an lst4Δ background.
BUL1 and BUL2 Allow Intracellular Retention of Gap1p
Next, we examined the effect of mutations in BUL1 and BUL2 on the intracellular location of Gap1p. We performed fluorescence microscopy using a low copy plasmid expressing GAP1-GFP in wild-type (CKY4) and bul1Δ bul2Δ (CKY698) strains (Fig. 4A and Fig. B). The wild-type strain exhibited GFP fluorescence at the cell surface, a fainter signal visible at the perinuclear/ER membrane in some cells, and additional strong internal punctate foci of Gap1p-GFP that appeared not to correspond to ER or plasma membrane. In contrast, Gap1-GFP fluorescence in the bul1Δ bul2Δ strain was almost exclusively at the cell periphery, indicative of plasma membrane localization.
Figure 4.

Gap1-GFP efficiently localizes to the plasma membrane in bul1Δ bul2Δ and lst4Δ bul1Δ bul2Δ mutants. Wild-type (CKY4), bul1Δ bul2Δ (CKY698), lst4Δ (CKY695), and lst4Δ bul1Δ bul2Δ (CKY699) strains were transformed with pGAP1-GFP (pCK230), cultured in minimal medium (SD Ammonia) at 24°C, and imaged live using a UV fluorescence microscope with a GFP filter (GFP), or with Nomarski optics (DIC). Exposure times for lst4Δ were four times longer than for the other strains. Bar, 2 μm.
Fractionation of cell extracts on sucrose density gradients allows the separation of plasma membrane fractions from those of the Golgi complex and ER, as determined by the presence of marker proteins Pma1p (plasma membrane), GDPase activity (Golgi complex), and Dpm1p (ER). Two significant pools of HA-Gap1p could be detected in a wild-type strain. One pool, accounting for approximately two thirds of the total HA-Gap1p, fractionated with Golgi and ER markers, whereas the other pool, accounting for the remaining one third of the HA-Gap1p, corresponded to the plasma membrane fraction (Fig. 5 A). In contrast, in a bul1Δ bul2Δ strain, a significantly greater proportion of HA-Gap1p fractionated with the plasma membrane, with very little HA-Gap1p fractionating with the ER and Golgi markers, demonstrating a shift in the ratio of internal- and plasma membrane–localized HA-Gap1p (Fig. 5 B). Thus, by three different experimental criteria, loss of BUL function causes constitutive Gap1p secretion: a bul1Δ bul2Δ mutant strain exhibited increased Gap1p activity, Gap1-GFP is localized almost exclusively to the plasma membrane, and HA-Gap1p fractionated mainly with the plasma membrane marker on sucrose density gradients.
Figure 5.
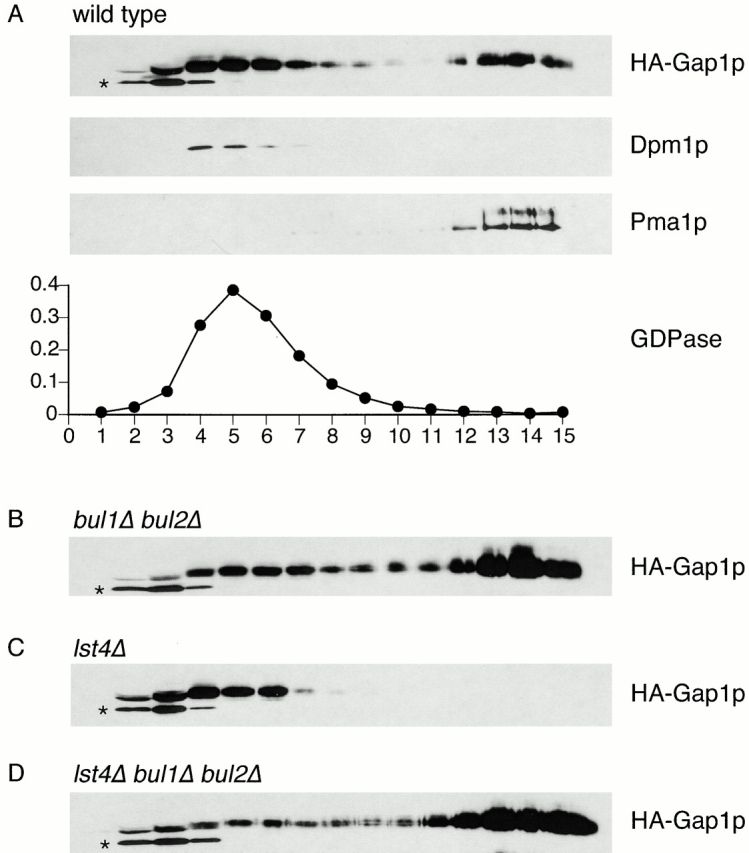
Cell fractionation demonstrates plasma membrane localization of HA-Gap1p in bul1Δ bul2Δ and lst4Δ bul1Δ bul2Δ mutants. (A) Wild-type (CKY4), (B) bul1Δ bul2Δ (CKY698), (C) lst4Δ (CKY695), and (D) lst4Δ bul1Δ bul2Δ (CKY699) strains were transformed with pHA-GAP1 (pPL257), grown on minimal medium (SD Ammonia) at 24°C, and crude cell extracts were subjected to isopycnic fractionation on a 20–60% sucrose density gradient. Fractions were assayed for HA-Gap1p, the Golgi marker GDPase by enzymatic assay, the ER marker Dpm1p, and the plasma membrane marker Pma1p after SDS-PAGE and Western blotting. Analysis of GDPase, Dpm1p, and Pma1p was also performed for strains (B) bul1Δ bul2Δ, (C) lst4Δ, and (D) lst4Δ bul1Δ bul2Δ; the fractionation of these markers was the same as for the wild-type. Sucrose gradient concentration increases from left to right in the figure. The asterisks indicate an endogenous soluble protein recognized by the anti-HA antibody.
The observations made with the bul1Δ bul2Δ mutant caused us to reevaluate our ideas of Gap1p trafficking in wild-type cells. Even when cells were grown on a medium that gave maximum Gap1p activity in wild-type, this activity was not as great as that possible in a bul1Δ bul2Δ mutant strain. In a bul1Δ bul2Δ genetic background the amount of Gap1p at the cell surface was increased at the expense of the internal pool of Gap1p. Thus, Bul1p and Bul2p appear to be necessary for the maintenance of an internal pool of Gap1p.
Bul1p and Bul2p Act before Lst4p in Gap1p Trafficking
We assessed the localization of Gap1-GFP in lst4Δ (CKY695) and lst4Δ bul1Δ bul2Δ (CKY699) strains (Fig. 4C and Fig. D) using fluorescence microscopy. The lst4Δ mutant clearly displayed Gap1p-GFP localizing to internal punctate structures, with no clear plasma membrane or perinuclear ER staining. However, in an lst4Δ bul1Δ bul2Δ strain, Gap1p-GFP was localized almost exclusively to the plasma membrane. We also analyzed the distribution of HA-Gap1p in these strains by cell fractionation (Fig. 5). The lst4Δ strain had no detectable HA-Gap1p cofractionating with the plasma membrane marker Pma1p. In contrast, HA-Gap1p was found predominantly in plasma membrane fractions in an lst4Δ bul1Δ bul2Δ strain, a distribution similar to that of the bul1Δ bul2Δ strain. These observations confirm that Bul1p and Bul2p specify the location of Gap1p by acting to prevent Gap1p trafficking to the cell surface. Moreover, the phenotype of an lst4Δ bul1Δ bul2Δ strain indicates that Bul1p and Bul2p functions are needed in order for Lst4p to have an effect on Gap1p trafficking.
Deletion of BUL1 and BUL2 Decreases Gap1p Polyubiquitination
Partitioning of Gap1p between the secretory pathway and the vacuolar targeting pathway appears to depend on the activity of Bul1p and Bul2p, proteins that bind to the E3 ubiquitin ligase Rsp5p (Yashiroda et al. 1996). The simplest interpretation for these findings is that Bul1p and Bul2p affect internal Gap1p sorting by setting the ubiquitination state of Gap1p itself. Therefore, we compared ubiquitination of Gap1p in wild-type and bul1Δ bul2Δ cells. To detect ubiquitinated proteins, we expressed myc epitope–tagged ubiquitin from a copper-regulated promoter (Ecker et al. 1987; Ellison and Hochstrasser 1991; Hochstrasser et al. 1991). We also used an HA-GAP1 construct that was expressed from the constitutive ADH1 promoter to eliminate possible differences in GAP1 transcription in the different strains under examination (Fig. 3). In cells grown in urea or ammonia as the sole nitrogen source, the absolute level of HA-Gap1p expression from the ADH1 promoter was similar to the level expressed from the endogenous GAP1 promoter. To detect ubiquitinated Gap1p, extracts from cells expressing HA-Gap1p were immunoprecipitated with anti-HA antibody, and these samples were then probed by immunoblotting with anti-myc antibody to detect ubiquitinated forms of HA-Gap1p.
In a wild-type genetic background (CKY703), HA-Gap1p was significantly modified with c-myc–ubiquitin (Fig. 6 B, lane 3), and the low gel mobility of these forms indicates an increase in mass consistent with polyubiquitination (Fig. 6 B, p). In contrast, the bul1Δ bul2Δ mutant (CKY704) exhibited much less polyubiquitinated HA-Gap1p (Fig. 6 B, compare lanes 5 and 3). Moreover, the residual ubiquitinated HA-Gap1p detected in this strain also exhibited a distribution biased to lower molecular weight forms than were apparent in wild-type. Immunoblotting with anti-HA demonstrates that only HA-Gap1p was immunoprecipitated by the anti-HA antibody (Fig. 6 A; compare lane 1 with 2–5); therefore, the proteins detected in Fig. 6 B (lane 3) represent HA-Gap1p conjugated to Ub-myc. The HA-Gap1p identified in the bul1Δ bul2Δ mutant with anti-HA antibody (Fig. 6 A, lanes 4 and 5) differs from HA-Gap1p from wild-type in that a second immunoreactive band is visible just above the main form of HA-Gap1p (Fig. 6 A, m). This lower mobility form of Gap1p probably corresponds to HA-Gap1p carrying a small number of ubiquitin moieties; however, it was not recognized by anti-myc antibodies, presumably because of a relative inefficiency of anti-myc detection of monovalent Ub-myc species.
Figure 6.

Bul1p and Bul2p specify polyubiquitination of Gap1p. Wild-type (CKY703; lanes 1–3) and bul1Δ bul2Δ (CKY704; lanes 4 and 5) strains were transformed with combinations of empty vector (pRS316; lane 1) or vector-carrying HA-Gap1p, (pCK227; lanes 2–5), and either pPCUP1-UBI-c-myc (pCK231; lanes 1, 3, and 5) or pPCUP1-UBI (pCK232; lanes 2 and 4). Strains were cultured overnight in minimal medium (SD Urea) at 24°C, and CuSO4 was added to 0.1 μM to induce the CUP1 promoter 3 h before harvesting in exponential phase. Anti-HA (3F10) immunoprecipitates were subject to SDS-PAGE and Western blotting with either (A) anti-HA (16B12) antibody or (B) anti–c-myc (9E10) antibody. m, monoubiquitinated HA-Gap1p; p, polyubiquitinated HA-Gap1p. (C) Lanes 1 and 2 correspond to lane 5 from A and B, respectively, except that sixfold more yeast extract was used for immunoprecipitation and Western blotting.
To demonstrate that the putative monoubiquitinated form of HA-Gap1p that increased in intensity in the bul1Δ bul2Δ strain represented HA-Gap1p-Ub-myc, we repeated the experiment with the bul1Δ bul2Δ mutant expressing Ub-myc (as in Fig. 6A and Fig. B, lane 5), but used six times more cell extract (Fig. 6 C). Anti-HA immunoblotting from this scaled up experiment revealed several distinct forms of HA-Gap1p migrating slightly more slowly than the main (unubiquitinated) form visualized with the anti-HA antibody (Fig. 6 C, lane 1). The anti-myc antibody immunoblot also revealed several discrete bands (Fig. 6 C, lane 2), the two fastest migrating of which comigrate with two of the slow moving HA-Gap1p forms detected with the anti-HA antibody. This indicates that these forms correspond to Ub-myc covalently attached to HA-Gap1p, and most likely correspond to HA-Gap1p with one, two, or three ubiquitin moieties appended. Thus, in a bul1Δ bul2Δ strain there was a great reduction of polyubiquitinated Gap1p and a corresponding increase in mono-ubiquitinated Gap1p, suggesting that Bul1p and Bul2p are normally involved in the polyubiquitination of Gap1p.
A COOH-terminal Truncation of Gap1p Affects Sorting and Polyubiquitination
A cis-acting allele of Gap1p with a deletion of 11 COOH-terminal amino acid residues, Gap1Δ2p, has already been shown to perturb both ubiquitination and endocytosis of Gap1p (Hein and André 1997; Springael and André 1998). Therefore, we wished to assess whether Gap1Δ2p was also defective in its capacity to be sorted from the Golgi complex to the vacuole. We compared Gap1p activity in wild-type (CKY482), lst4Δ (CKY702), and bul1Δ bul2Δ (CKY701) strains that were expressing either GAP1 (pCK233) or GAP1Δ2 (pCK234) (Fig. 7). Strikingly, Gap1Δ2p suppressed the effects of lst4Δ, because a GAP1Δ2 lst4Δ strain exhibited 100-fold more Gap1p activity than an otherwise isogenic GAP1 lst4Δ strain (Fig. 7).
It seemed possible that Gap1Δ2p was active in lst4Δ because the truncated form of the protein was not a substrate for polyubiquitination, so we compared the ubiquitination state of HA-Gap1Δ2p to that of HA-Gap1p. Polyubiquitinated HA-Gap1p was readily detected in a wild-type strain and the amount of this polyubiquitinated form was greatly decreased in a bul1Δ bul2Δ strain (Fig. 8 B, lanes 2 and 6). Parallel analysis of HA-Gap1Δ2p revealed very little polyubiquitination in both wild-type and bul1Δ bul2Δ strains (Fig. 8 B, lanes 4 and 7). The low level of HA-Gap1Δ2p polyubiquitination in a wild-type strain was similar to that of HA-Gap1p in a bul1Δ bul2Δ mutant. Thus, Gap1Δ2p represents a form of Gap1p that is inefficiently polyubiquitinated, reproducing the effects of bul1Δ bul2Δ on wild-type Gap1p.
If Gap1Δ2p can no longer act as an efficient substrate for Bulp-mediated polyubiquitination, then the intracellular distribution and activity of Gap1Δ2p should not be affected by an increase in Bulp activity. We assessed Gap1p activity in a gap1Δ strain (CKY715) expressing either GAP1 (pCK233) or GAP1Δ2 (pCK234) in combination with an empty multicopy plasmid or one expressing BUL2 (pCK250). Overexpression of BUL2 decreased wild-type Gap1p activity fourfold (Fig. 1 B and 8 B), but Gap1p activity in a GAP1Δ2 strain was largely resistant to the effect of overexpression of BUL2. This finding is consistent with the idea that Bul1p overexpression exerts its effect on wild-type Gap1p by altering the ubiquitination state of Gap1p itself, rather than acting on some other cellular component involved in Gap1p sorting.
Bul1p and Bul2p Regulate Gap1p Activity in Conjunction with E3 Ubiquitin Ligase Rsp5p
Rsp5p is a homology to EG-AP COOH terminus E3 ubiquitin protein ligase that is responsible for ubiquitination of Gap1p before its endocytosis (Huibregtse et al. 1995; Springael and André 1998). Bul1p and Bul2p do not contain sequences related to the catalytic domains of known ubiquitin protein ligases, so we surmised that Rsp5p provides the catalytic activity for Bul1p- and Bul2p-controlled polyubiquitination. Therefore, a mutant lacking Rsp5p activity should have a similar effect on the sorting of Gap1p as a bul1Δ bul2Δ deletion mutant. We compared Gap1p activity in wild-type (CKY4), bul1Δ bul2Δ (CKY698), and a strain carrying the rsp5-1 temperature-sensitive allele (CKY712) growing at 34°C (this strain arrests growth above 35°C) (Fig. 9 A). Both bul1Δ bul2Δ and rsp5-1 strains displayed Gap1p activity greater than threefold more than the wild-type, suggesting that Rsp5p, like Bul1p and Bul2p, allows for the partial retention of Gap1p in an intracellular compartment. To determine whether Rsp5p also acts on Gap1p before the transport step defined by Lst4p, we assayed Gap1p activity in lst4Δ rsp5-1 (CKY713). In the lst4Δ rsp5-1 strain grown at 34°C, Gap1p activity was similar to that of an rsp5-1 strain and was much greater than for an lst4Δ alone (CKY695), showing that in the absence of Rsp5p lst4Δ does not prevent Gap1p from reaching the plasma membrane (Fig. 9 A). Consistent with the hypothesis that Rsp5p and Bulp function at the same step in Gap1p transport, the combination of rsp5-1 with bul1Δ bul2Δ (CKY714) caused no additional increase in Gap1p activity over that of the rsp5-1 or bul1Δ bul2Δ strains.
We also expected that strains lacking the catalytic activity of Rsp5p should not be affected by an increase in BUL gene dosage. To test this, we measured [14C]citrulline uptake in wild-type (CKY4) and rsp5-1 (CKY712) strains carrying either a multicopy plasmid expressing BUL1 (pBUL1; pCK228) or the corresponding empty vector (Fig. 9 B). The rsp5-1 mutant grown at 34°C was refractory to BUL1 overexpression, maintaining high levels of Gap1p activity, indicating that Rsp5p activity is required for BUL1 overexpression to have an effect on Gap1p sorting.
We wished to evaluate Gap1p ubiquitination in the rsp5-1 mutant to see whether a reduction in ubiquitination, and in particular monoubiquitination, correlated with the effect of rsp5-1 on Gap1p intracellular trafficking. In a wild-type (CKY4) strain at 34°C, monoubiquitinated forms of HA-Gap1p were visible migrating slightly more slowly than the principal HA-Gap1p form on immunoblots, but these slower migrating forms were absent in the rsp5-1 (CKY712) strain expressing HA-Gap1p at the same temperature (Fig. 9 C). Comparison of bul1Δ bul2Δ (CKY698) and wild-type strain expressing HA-Gap1p by this method shows an increase in the monoubiquitinated forms of Gap1p in the strain lacking Bul1p and Bul2p, as observed previously (Fig. 6 A, 8 A, and 9 C). These monoubiquitinated forms of HA-Gap1p were not evident in the rsp5-1 bul1Δ bul2Δ mutant (CKY714), confirming that Rsp5p is responsible for the formation of the monoubiquitinated Gap1p species observed in a bul1Δ bul2Δ strain.
Bul1p interacts with Rsp5p via a proline-rich motif in Bul1p known as the PPXY motif, and mutation of this motif within Bul1p (Bul1P157Q, P158A) produces a protein that is stable, but cannot bind to Rsp5p (Yashiroda et al. 1998). We investigated the effects of this bul1P157Q , P158A mutation on bul1Δ bul2Δ–related Gap1p sorting. Wild-type (CKY4), bul1Δ bul2Δ (CKY698), and lst4Δ bul1Δ bul2Δ (CKY699) strains were transformed with a low copy vector carrying no insert (vector), bul1P157Q , P158A (pHY37), or BUL1 (pCK249). In all three genetic backgrounds, the activity of Gap1p in the strain expressing bul1P157Q , P158A was similar to that in the strain expressing the empty vector, indicating that the PPXY motif is crucial for Bul1p function in Gap1p sorting. Wild-type BUL1 expressed from the same vector in bul1Δ bul2Δ and lst4Δ bul1Δ bul2Δ strains complemented the loss of BUL1 and BUL2 from the chromosome, reducing Gap1p activities to values similar to those of a wild-type and an lst4Δ strain, respectively.
The biochemical and genetic evidence for a functional interaction between the Bul proteins and Rsp5p, together with the similar consequences of mutations in these genes for Gap1p sorting, show that the Bul1p and Bul2p proteins probably exert their effect on Gap1p sorting as components of an Rsp5p-based ubiquitin ligase complex. The different components of this complex have different effects on the ubiquitination state of Gap1p, but the polyubiquitination of Gap1p by the complex appears to be the crucial step for targeting Gap1p to the vacuole.
Discussion
We have identified Bul1p and Bul2p, two nonessential components of a protein complex containing the E3 ubiquitin ligase Rsp5p, as proteins that both specify Gap1p polyubiquitination and intracellular transport of the Gap1p permease from the late secretory pathway to the vacuole.
Multiple observations indicate that BUL1 and BUL2 control Gap1p entry into the vacuolar-sorting pathway from the trans-Golgi complex. Overexpression of either BUL1 or BUL2 reduces delivery of Gap1p to the plasma membrane, an effect that is partially suppressed by pep12 or vps45 mutants compromised in vesicular transport from the Golgi complex to PVC. Conversely, deletion of BUL1 and BUL2 causes more efficient delivery of Gap1p to the plasma membrane than in wild-type. Most significantly, an lst4Δ mutant, which alone completely shifts the sorting of Gap1p to the vacuolar pathway regardless of nitrogen source, can be completely suppressed by bul1Δ bul2Δ. This suppression is evident in the high Gap1p activity in an lst4Δ bul1Δ bul2Δ triple mutant and is most easily explained if we postulate that newly synthesized Gap1p encounters the sorting event specified by Bul1p and Bul2p before the step that depends on Lst4p.
The overall scheme for cellular trafficking of Gap1p suggested by these observations is depicted in Fig. 10. Newly synthesized Gap1p is transported to the trans-Golgi, where it can be sorted either to the PVC in a BUL-dependent manner, or to the plasma membrane. One hypothesis to explain the relationship between LST4 and the BUL1 and BUL2 genes is that the overall partitioning of newly synthesized Gap1p between the plasma membrane and the vacuole is controlled both by sorting at the trans-Golgi and by recycling from the PVC to the Golgi complex, which in turn depends on Lst4p. The idea is that in a wild-type cell Gap1p cycles between the PVC and the Golgi complex multiple times, even when cells are growing on a nitrogen source that gives high Gap1p activity in the plasma membrane. Thus, if Lst4p were required for Gap1p retention in or retrieval from the PVC, deletion of LST4 should both decrease the probability of delivery of Gap1p to the plasma membrane and speed delivery to the vacuole. These expectations are in agreement with the finding that Gap1p is absent from the plasma membrane and is degraded relatively rapidly in an lst4Δ strain. Finally, the high levels of active Gap1p in the plasma membrane of both bul1Δ bul2Δ and lst4Δ bul1Δ bul2Δ mutants are consistent with the idea that in the absence of Bul1p and Bul2p function, Gap1p never enters the vacuolar pathway and therefore never encounters the Lst4p-dependent sorting steps. The proposed recycling of Gap1p between the trans-Golgi and the PVC is similar to the mechanism by which the prohormone-processing enzyme Kex2p, and a soluble vacuolar protease receptor Vps10p, are retained in the late secretory pathway (Marcusson et al. 1994; Redding et al. 1996). However, the BUL1, BUL2, and LST4 genes appear to specifically affect Gap1p and Put4p cycling and do not appear to play a role in the proper localization of either Vps10p or Kex2p because CPY sorting and mating are normal in bul1Δ bul2Δ or lst4Δ mutants (data not shown).
Figure 10.
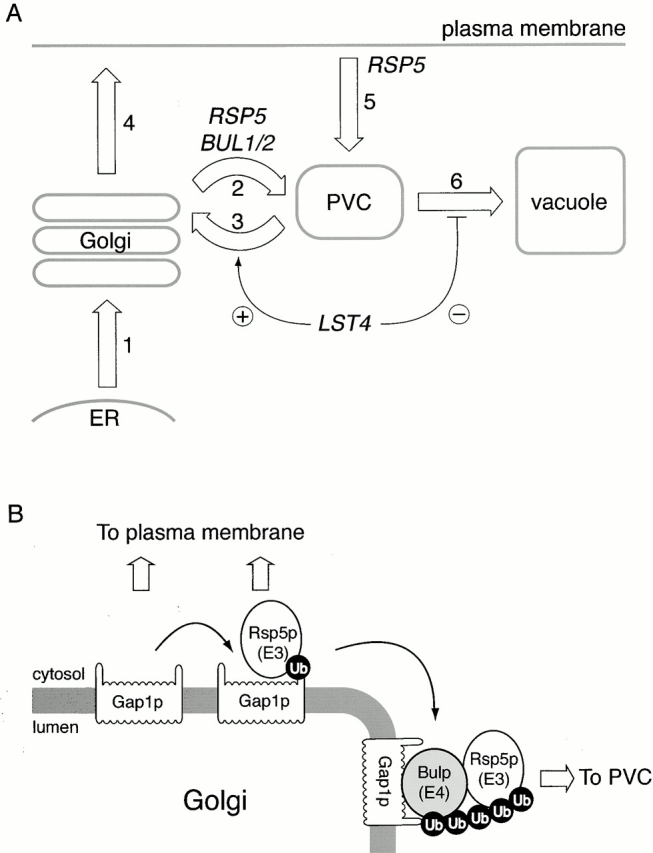
Overview for how Gap1p trafficking is genetically controlled. (A) The epistasis relationship between rsp5-1 or bul1Δ bul2Δ and lst4Δ suggests that Rsp5p, Bul1p, and Bul2p promote Gap1p transport from the Golgi complex to the PVC (step 2), and that Lst4p acts either to retrieve Gap1p from the PVC back to the Golgi (step 3), or to prevent Gap1p moving on the vacuole (step 6). The numbers denote different intracellular transport routes for Gap1p. (B) Intracellular sorting of Gap1p appears to be specified by its ubiquitination state. The Rsp5p–Bul1p–Bul2p complex polyubiquitinates Gap1p, whereas Rsp5p alone monoubiquitinates Gap1p. Only Gap1p that has been polyubiquitinated appears competent for sorting to the PVC.
Physiological Regulation of Gap1p Transport, a Possible Intracellular Storage Compartment
The hypothesis that Gap1p recycles in the late secretory pathway as part of its normal itinerary can explain some of the puzzling aspects of Gap1p physiology. Gap1p is delivered to the cell surface relatively slowly; in the steady state, cells harbor about two thirds of the total Gap1p in an intracellular form, even when grown under conditions that give maximum Gap1p activity (Fig. 4 and Fig. 5; Roberg et al. 1997b). This distribution differs markedly from other well-studied plasma membrane proteins such as Pma1p and Gas1p, which are found almost exclusively at the plasma membrane in steady state (Roberg et al. 1999). We suggest that Gap1p engaged in cycling between the trans-Golgi and the PVC could account for the abundant intracellular pool of Gap1p and could also explain why eventual plasma membrane delivery of Gap1p takes so long. The overall partitioning of Gap1p between the plasma membrane and the vacuole is specified by the nitrogen source. In glutamate-grown cells, Gap1p enters the secretory pathway and is degraded in the vacuole without ever reaching the cell surface, but the time taken for Gap1p to reach the vacuole is slow: the half time for Gap1p turnover is ∼55 min in glutamate-grown cells (Roberg et al. 1997b). Because Gap1p is significantly delayed in the vicinity of the Golgi complex and PVC whether Gap1p is delivered to the plasma membrane or to the vacuole, it seems likely that some recycling takes place regardless of the ultimate outcome of the sorting decision.
Gap1p engaged in cycling between the Golgi complex and PVC can be thought of as an uncommitted, internal storage form of Gap1p, ready to be directed to the cell surface after a change in the quality of nitrogen source. Indeed, we found that cells that have been grown on glutamate, which have an intracellular pool of Gap1p but none at the cell surface, will rapidly redistribute active Gap1p to the cell surface when transferred to urea medium (Roberg et al. 1997b). The physiological significance of the recycling form of Gap1p may be to provide cells with the means to very rapidly adjust Gap1p activity at the cell surface in response to changing needs for amino acids to be used as a nitrogen source.
Polyubiquitination Controls Gap1p Intracellular Trafficking
We have noted a correlation between the intracellular trafficking of Gap1p and its ubiquitination state. Transacting bul1Δ bul2Δ or rsp5-1 mutations or a cis-acting GAP1Δ2 mutation cause a great reduction in the amount of polyubiquitinated Gap1p and a concomitant redirection of Gap1p to the cell surface. Rsp5p, Bul1p, and Bul2p all appear to be components of the same ubiquitin ligase complex, but they have different effects on Gap1p ubiquitination. The bul1Δ bul2Δ mutation blocks the formation of polyubiquitinated Gap1p and increases the amount of monoubiquitinated forms of Gap1p. The rsp5-1 mutation prevents all Gap1p ubiquitination, consistent with the presumptive role of Rsp5p as the catalytic subunit of the complex. Since bul1Δ bul2Δ and rsp5-1 have the same effect on Gap1p sorting, it seems that polyubiquitination is the key determinant for Gap1p trafficking from the Golgi complex to the vacuole. The Gap1Δ2p truncation removes the last 11 amino acids from Gap1p, but does not remove any lysine residues, which are the direct targets for ubiquitin attachment (Hein and André 1997). Therefore, the Gap1Δ2p truncation must prevent Gap1p polyubiquitination in a way that we do not yet understand. Perhaps the COOH terminus of Gap1p is necessary to engage an E3 complex that contains Bul1p or Bul2p.
Rsp5p, the E3 ubiquitin protein ligase to which Bul1p and Bul2p bind, was first identified as the NPI1 gene (nitrogen permease inactivator) required for ubiquitination- and ammonium-induced endocytosis of Gap1p (Grenson 1983; Hein et al. 1995; Springael and André 1998). The effect of npi1 mutants on endocytosis was documented in the Σ1278b genetic background. Because the S288C strain we use does not respond to ammonium by downregulating Gap1p activity (Courchesne and Magasanik 1983), we do not have a ready way to assess whether bul1Δ bul2Δ mutants affect nitrogen-regulated endocytosis of Gap1p. Nevertheless, even in the S288C genetic background both the rate of Gap1p delivery to the plasma membrane and the rate of Gap1p endocytosis should affect the steady state level of Gap1p activity. Therefore, we wondered whether the high Gap1p activity in a bul1Δ bul2Δ lst4Δ triple mutant might be a consequence of a possible defect in Gap1p endocytosis caused by bul1Δ bul2Δ. This possibility seemed unlikely, because when lst4Δ was combined with end3Δ, a mutation known to block the general endocytic pathway (Benedetti et al. 1994), no increase in Gap1p activity could be detected (not shown). This result is consistent with the idea that bul1Δ bul2Δ greatly increases Gap1p activity in an lst4Δ background by increasing the amount of Gap1p delivered to the cell surface, rather than by decreasing the rate of endocytosis.
Another possible explanation for the observation that in a bul1Δ bul2Δ genetic background lst4Δ does not exert an effect on Gap1p sorting is that Lst4p could be a negative regulator of the Rsp5p–Bul1p–Bul2p ubiquitin ligase complex. According to this hypothesis, an lst4Δ mutation should cause an increase in the amount of polyubiquitinated Gap1p. Although we have found that in vivo assays for Gap1p ubiquitination can reliably reveal defects in Gap1p ubiquitination, we have not yet been able to detect increases in the amount of ubiquitinated Gap1 in strains exhibiting increased traffic of Gap1p to the vacuole, such as overproducers of Bul1p or Bul2p. It may be that the size of the in vivo pool of polyubiquitinated Gap1p is self-limiting because polyubiquitination acts as a signal that leads directly to vacuolar degradation of the polyubiquitinated species.
Bul1p and Bul2p, HECT E3 Ubiquitin Ligase Adapters
Rsp5p has been implicated in a range of cellular processes all regulated by ubiquitin conjugation, including endocytosis, mitochondrial inheritance, and transcription factor regulation (Wendland et al. 1998; Fisk and Yaffe 1999; Hoppe et al. 2000). Rsp5p has several defined motifs: the catalytic HECT domain, a C2 calcium–dependent lipid binding domain, and three WW motifs which bind the amino acid motif PPXY in substrate proteins (Huibregtse et al. 1995; Wang et al. 1999; Chang et al. 2000). The WW motifs are thought to determine the substrate specificity of ubiquitin ligase, because an Rsp5p variant comprising only the catalytic HECT domain and the third WW domain provides essential Rsp5p function at normal temperatures, and only this third WW domain interacts with Spt23p, an Rsp5p substrate (Hoppe et al. 2000). Bul1p and Bul2p also contain a PPXY motif that is required for their binding to Rsp5p, but Bul1p and Bul2p do not appear to be targets of the E3 ligase activity of Rsp5p and they have no sequence homology to ubiquitin ligases (Yashiroda et al. 1996). Analysis of a mutant Bul1p in which the PPXY motif is mutated to QAXY indicates that Bul1p requires this motif to have an effect on Gap1p intracellular transport and activity. In addition, Gap1p activity is no longer reduced upon Bul1p overexpression in a strain with reduced Rsp5p activity. Thus, our results suggest that Bul1p and Bul2p modulate the extent of Rsp5p-mediated polyubiquitination of substrate membrane proteins.
Here we show that the Golgi sorting of Gap1p and probably also Put4p is in part controlled by an Rsp5p ubiquitin ligase activity. It is likely that ubiquitination can influence the Golgi sorting of other proteins as well. For example, it has been shown recently that the tryptophan permease, Tat2p, is sorted from the Golgi complex to the vacuole under conditions of nutrient deprivation (Beck et al. 1999). Intracellular trafficking of Tat2p to the vacuole can be blocked by mutation of lysine residues in Tat2p that act as ubiquitin acceptor sites, strongly suggesting a role for ubiquitination as a signal for sorting of Tat2p from the Golgi complex to the vacuole (Beck et al. 1999). We are currently examining whether Rsp5p and Bul1p or Bul2p have a part in regulating ubiquitination of Tat2p.
The biochemical activity required for adding ubiquitin monomers to monoubiquitinated substrates was recently defined as E4, based on the discovery that S. cerevisiae Ufd2p promotes the efficient polyubiquitination of a monoubiquitinated proteasomal substrate only in the presence of the relevant E3 ubiquitin ligase activity (Koegl et al. 1999). Bul1p and Bul2p most likely define a novel E4 activity that utilizes the E3 activity of Rsp5p to increase the number of ubiquitin monomers that are added to Gap1p.
Acknowledgments
We thank Paul Ferrigno, Pam Silver, Alexander Varshavsky, Amy Chang, Yoshiko Kikuchi, John Huibregtse, Esther Chen, and Neil Rowley for strains, plasmids, or antibodies, Carolyn Sevier and Esther Chen for critically reading the manuscript, and members of the Kaiser lab for helpful discussion and encouragement.
S.B. Helliwell acknowledges support from the Swiss National Science Foundation (grant 823A-053450) and the Novartis Foundation. S. Losko acknowledges support from the Deutsche Forschungsgemeinschaft (grant LO794/1-1). This work was supported by National Institutes of Health grant GM56933 to C.A. Kaiser.
Footnotes
Abbreviations used in this paper: ADCB, l-azetidine-2-carboxylic acid; GFP, green fluorescent protein; HA, hemagglutinin; PVC, prevacuolar compartment.
References
- Becherer K.A., Rieder S.E., Emr S.D., Jones E.W. Novel syntaxin homologue, Pep12p, required for the sorting of lumenal hydrolases to the lysosome-like vacuole in yeast. Mol. Biol. Cell. 1996;7:579–594. doi: 10.1091/mbc.7.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T., Schmidt A., Hall M.N. Starvation induces vacuolar targeting and degradation of the tryptophan permease in yeast. J. Cell Biol. 1999;146:1227–1238. doi: 10.1083/jcb.146.6.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belli G., Gari E., Piedrafita L., Aldea M., Herrero E. An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res. 1998;26:942–947. doi: 10.1093/nar/26.4.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti H., Raths S., Crausaz F., Riezman H. The END3 gene encodes a protein that is required for the internalization step of endocytosis and for actin cytoskeleton organization in yeast. Mol. Biol. Cell. 1994;5:1023–1037. doi: 10.1091/mbc.5.9.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A., Cheang S., Espanel X., Sudol M. Rsp5 WW domains interact directly with the carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 2000;275:20562–20571. doi: 10.1074/jbc.M002479200. [DOI] [PubMed] [Google Scholar]
- Courchesne W.E., Magasanik B. Ammonia regulation of amino acid permeases in Saccharomyces cerevisiae . Mol. Cell. Biol. 1983;3:672–683. doi: 10.1128/mcb.3.4.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles C.R., Emr S.D., Horazdovsky B.F. Mutations in the VPS45 gene, a SEC1 homologue, result in vacuolar protein sorting defects and accumulation of membrane vesicles. J. Cell Sci. 1994;107:3449–3459. doi: 10.1242/jcs.107.12.3449. [DOI] [PubMed] [Google Scholar]
- Ecker D.J., Khan M.I., Marsh J., Butt T.R., Crooke S.T. Chemical synthesis and expression of a cassette adapted ubiquitin gene. J. Biol. Chem. 1987;262:3524–3527. [PubMed] [Google Scholar]
- Ellison M.J., Hochstrasser M. Epitope-tagged ubiquitin. A new probe for analyzing ubiquitin function. J. Biol. Chem. 1991;266:21150–21157. [PubMed] [Google Scholar]
- Fisk H.A., Yaffe M.P. A role for ubiquitination in mitochondrial inheritance in Saccharomyces cerevisiae . J. Cell Biol. 1999;145:1199–1208. doi: 10.1083/jcb.145.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan J.M., Moreau V., André B., Volland C., Haguenauer-Tsapis R. Ubiquitination mediated by the Npi1p/Rsp5p ubiquitin-protein ligase is required for endocytosis of the yeast uracil permease. J. Biol. Chem. 1996;271:10946–10952. doi: 10.1074/jbc.271.18.10946. [DOI] [PubMed] [Google Scholar]
- Glick B.S. Organization of the Golgi apparatus. Curr. Opin. Cell Biol. 2000;12:450–456. doi: 10.1016/s0955-0674(00)00116-2. [DOI] [PubMed] [Google Scholar]
- Grenson M. Inactivation-reactivation process and repression of permease formation regulate several ammonia-sensitive permeases in the yeast Saccharomyces cerevisiae . Eur. J. Biochem. 1983;133:135–139. doi: 10.1111/j.1432-1033.1983.tb07438.x. [DOI] [PubMed] [Google Scholar]
- Grenson M., Hou C., Crabeel M. Multiplicity of the amino acid permeases in Saccharomyces cerevisiae. IV. Evidence for a general amino acid permease. J. Bacteriol. 1970;103:770–777. doi: 10.1128/jb.103.3.770-777.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 1983;101:181–191. doi: 10.1016/0076-6879(83)01013-7. [DOI] [PubMed] [Google Scholar]
- Hein C., André B. A C-terminal di-leucine motif and nearby sequences are required for NH4(+)-induced inactivation and degradation of the general amino acid permease, Gap1p, of Saccharomyces cerevisiae . Mol. Microbiol. 1997;24:607–616. doi: 10.1046/j.1365-2958.1997.3771735.x. [DOI] [PubMed] [Google Scholar]
- Hein C., Springael J.Y., Volland C., Haguenauer-Tsapis R., André B. NPl1, an essential yeast gene involved in induced degradation of Gap1 and Fur4 permeases, encodes the Rsp5 ubiquitin-protein ligase. Mol. Microbiol. 1995;18:77–87. doi: 10.1111/j.1365-2958.1995.mmi_18010077.x. [DOI] [PubMed] [Google Scholar]
- Hicke L. Ubiquitin-dependent internalization and down-regulation of plasma membrane proteins. FASEB J. 1997;11:1215–1226. doi: 10.1096/fasebj.11.14.9409540. [DOI] [PubMed] [Google Scholar]
- Hicke L., Riezman H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 1996;84:277–287. doi: 10.1016/s0092-8674(00)80982-4. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M., Ellison M.J., Chau V., Varshavsky A. The short-lived MAT alpha 2 transcriptional regulator is ubiquitinated in vivo. Proc. Natl. Acad. Sci. USA. 1991;88:4606–4610. doi: 10.1073/pnas.88.11.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann W. Molecular characterization of the CAN1 locus in Saccharomyces cerevisiae. A transmembrane protein without N-terminal hydrophobic signal sequence. J. Biol. Chem. 1985;260:11831–11837. [PubMed] [Google Scholar]
- Hoppe T., Matuschewski K., Rape M., Schlenker S., Ulrich H.D., Jentsch S. Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell. 2000;102:577–586. doi: 10.1016/s0092-8674(00)00080-5. [DOI] [PubMed] [Google Scholar]
- Horak J. Amino acid transport in eukaryotic microorganisms. Biochim. Biophys. Acta. 1986;864:223–256. doi: 10.1016/0304-4157(86)90001-8. [DOI] [PubMed] [Google Scholar]
- Huibregtse J.M., Scheffner M., Beaudenon S., Howley P.M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA. 1995;92:2563–2567. doi: 10.1073/pnas.92.7.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauniaux J.C., Grenson M. GAP1, the general amino acid permease gene of Saccharomyces cerevisiae. Nucleotide sequence, protein similarity with the other bakers yeast amino acid permeases, and nitrogen catabolite repression. Eur. J. Biochem. 1990;190:39–44. doi: 10.1111/j.1432-1033.1990.tb15542.x. [DOI] [PubMed] [Google Scholar]
- Koegl M., Hoppe T., Schlenker S., Ulrich H.D., Mayer T.U., Jentsch S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell. 1999;96:635–644. doi: 10.1016/s0092-8674(00)80574-7. [DOI] [PubMed] [Google Scholar]
- Kölling R., Hollenberg C.P. The ABC-transporter Ste6 accumulates in the plasma membrane in a ubiquitinated form in endocytosis mutants. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:3261–3271. doi: 10.1002/j.1460-2075.1994.tb06627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kölling R., Losko S. The linker region of the ABC-transporter Ste6 mediates ubiquitination and fast turnover of the protein. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2251–2261. doi: 10.1093/emboj/16.9.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon S.K., Traub L.M. Sorting in the endosomal system in yeast and animal cells. Curr. Opin. Cell Biol. 2000;12:457–466. doi: 10.1016/s0955-0674(00)00117-4. [DOI] [PubMed] [Google Scholar]
- Ljungdahl P.O., Gimeno C.J., Styles C.A., Fink G.R. SHR3a novel component of the secretory pathway specifically required for localization of amino acid permeases in yeast. Cell. 1992;71:463–478. doi: 10.1016/0092-8674(92)90515-e. [DOI] [PubMed] [Google Scholar]
- Losko S., Kopp F., Kranz A., Kölling R. Uptake of the ATP binding cassette (ABC)–transporter Ste6 into the yeast vacuole is blocked in the doa4 mutant. Mol. Biol. Cell. 2001;12:1047–1059. doi: 10.1091/mbc.12.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcusson E.G., Horazdovsky B.F., Cereghino J.L., Gharakhanian E., Emr S.D. The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell. 1994;77:579–586. doi: 10.1016/0092-8674(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Nelissen B., De Wachter R., Goffeau A. Classification of all putative permeases and other membrane plurispanners of the major facilitator superfamily encoded by the complete genome of Saccharomyces cerevisiae . FEMS Microbiol. Rev. 1997;21:113–134. doi: 10.1111/j.1574-6976.1997.tb00347.x. [DOI] [PubMed] [Google Scholar]
- Redding K., Brickner J.H., Marschall L.G., Nichols J.W., Fuller R.S. Allele-specific suppression of a defective trans-Golgi network (TGN) localization signal in Kex2p identifies three genes involved in localization of TGN transmembrane proteins. Mol. Cell. Biol. 1996;16:6208–6217. doi: 10.1128/mcb.16.11.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberg K.J., Bickel S., Rowley N., Kaiser C.A. Control of amino acid permease sorting in the late secretory pathway of Saccharomyces cerevisiae by SEC13, LST4, LST7 and LST8 Genetics 147 1997. 1569 1584a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberg K.J., Rowley N., Kaiser C.A. Physiological regulation of membrane protein sorting late in the secretory pathway of Saccharomyces cerevisiae J. Cell Biol 137 1997. 1469 1482b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberg K.J., Crotwell M., Espenshade P., Gimeno R., Kaiser C.A. LST1 is a SEC24 homologue used for selective export of the plasma membrane ATPase from the endoplasmic reticulum. J Cell Biol. 1999;145:659–672. doi: 10.1083/jcb.145.4.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R.S., Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sophianopoulou V., Diallinas G. Amino acid transporters of lower eukaryotesregulation, structure and topogenesis. FEMS Microbiol. Rev. 1995;16:53–75. doi: 10.1111/j.1574-6976.1995.tb00155.x. [DOI] [PubMed] [Google Scholar]
- Springael J.Y., André B. Nitrogen-regulated ubiquitination of the Gap1 permease of Saccharomyces cerevisiae . Mol. Biol. Cell. 1998;9:1253–1263. doi: 10.1091/mbc.9.6.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanbrough M., Magasanik B. Transcriptional and posttranslational regulation of the general amino acid permease of Saccharomyces cerevisiae . J. Bacteriol. 1995;177:94–102. doi: 10.1128/jb.177.1.94-102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J., Fink G.R. The histidine permease gene (HIP1) of Saccharomyces cerevisiae . Gene. 1985;38:205–214. doi: 10.1016/0378-1119(85)90219-7. [DOI] [PubMed] [Google Scholar]
- Vandenbol M., Jauniaux J.C., Grenson M. Nucleotide sequence of the Saccharomyces cerevisiae PUT4 proline-permease-encoding genesimilarities between CAN1, HIP1 and PUT4 permeases. Gene. 1989;83:153–159. doi: 10.1016/0378-1119(89)90413-7. [DOI] [PubMed] [Google Scholar]
- Wach A., Brachat A., Pohlmann R., Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae . Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Wang G., Yang J., Huibregtse J.M. Functional domains of the Rsp5 ubiquitin-protein ligase. Mol. Cell. Biol. 1999;19:342–352. doi: 10.1128/mcb.19.1.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland B., Emr S.D., Riezman H. Protein traffic in the yeast endocytic and vacuolar protein sorting pathways. Curr. Opin. Cell Biol. 1998;10:513–522. doi: 10.1016/s0955-0674(98)80067-7. [DOI] [PubMed] [Google Scholar]
- Yashiroda H., Oguchi T., Yasuda Y., Toh E.A., Kikuchi Y. Bul1, a new protein that binds to the Rsp5 ubiquitin ligase in Saccharomyces cerevisiae . Mol. Cell. Biol. 1996;16:3255–3263. doi: 10.1128/mcb.16.7.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiroda H., Kaida D., Toh-e A., Kikuchi Y. The PY-motif of Bul1 protein is essential for growth of Saccharomyces cerevisiae under various stress conditions. Gene. 1998;225:39–46. doi: 10.1016/s0378-1119(98)00535-6. [DOI] [PubMed] [Google Scholar]