

Abstract
Cell migration represents an important cellular response that utilizes cytoskeletal reorganization as its driving force. Here, we describe a new signaling cascade linking PDGF receptor stimulation to actin rearrangements and cell migration. We demonstrate that PDGF activates Cdc42 and its downstream effector N-WASP to mediate filopodia formation, actin stress fiber disassembly, and a reduction in focal adhesion complexes. Induction of the Cdc42 pathway is independent of phosphoinositide 3-kinase (PI3K) enzymatic activity, but it is dependent on the p85α regulatory subunit of PI3K. Finally, data are provided showing that activation of this pathway is required for PDGF-induced cell migration on collagen. These observations show the essential role of the PI3K regulatory subunit p85α in controlling PDGF receptor–induced cytoskeletal changes and cell migration, illustrating a novel signaling pathway that links receptor stimulation at the cell membrane with actin dynamics.
Keywords: N-WASP, Cdc42, PDGF, phosphatidylinositol 3-kinase, actin cytoskeleton
Introduction
Directed cell migration is a critical feature of several physiological and pathological processes, including development, wound healing, atherosclerosis, immunity, angiogenesis, and metastasis. The migratory response involves actin cytoskeleton reorganization, polarization, cell adhesion, and detachment. Thus, migration requires cell communication with adjacent cells and with extracellular matrix components (ECM), and is triggered by a gradient of chemotactic factors, such as platelet-derived growth factor (PDGF; Hynes 1992; Kundra et al. 1994; Huttenlocher et al. 1995). Among the pathways triggered by PDGF receptor (PDGF-R) stimulation, previous studies point to phosphoinositide 3-kinase (PI3K) and the small G proteins of the Rho family, Rac, as essential components mediating PDGF-induced actin cytoskeletal reorganization and cell migration (Fruman et al. 1998; Hall 1998; Heldin et al. 1998).
Class IA PI3Ks are heterodimers composed of a regulatory (p85) and a catalytic (p110) subunit that phosphorylate phosphoinositides at position 3 of the inositol ring. These products, which are present at a low concentration in resting cells, increase after stimulation with various growth factors including PDGF (Panayotou and Waterfield 1992; Fruman et al. 1998). Activation of PI3K mediates cell survival and division, but also participates in regulating cytoskeletal changes, such as PDGF-induced lamellipodium formation (Wennström et al. 1994a).
The small G proteins of the Rho family also play an essential role in regulating actin cytoskeleton dynamics. Small G proteins are activated by the exchange of bound GDP for GTP, stimulated by guanine nucleotide exchange factors (GEF), and inactivated by GTPase activating proteins and GDP dissociation inhibitors (for review see Lim et al. 1996). The study of Rho family member function has been facilitated by the use of genetically engineered constitutive active mutants (such as V12-Cdc42), exhibiting decreased intrinsic GTPase activity, and the use of dominant negative mutants (such as N17-Cdc42) that exhibit greater affinity for GDP than for GTP (Lim et al. 1996). Rho small G proteins control the formation of filopodia (Cdc42), lamellipodia (Rac), and actin stress fibers (Rho A) (Ridley and Hall 1992; Ridley et al. 1992; Nobes and Hall 1995; Kozma et al. 1995), and also control cell polarization (Nobes and Hall 1999) as well as cell adhesion. In fact, Rho A induces formation of focal adhesions, and Rac/Cdc42 induce formation of peripheral focal contacts (Ridley and Hall 1992; Nobes and Hall 1995; Burridge et al. 1988). Several effectors of activated Rho family proteins have been described previously (for reviews see Lim et al. 1996; Aspenstrom 1999); of these, a number of studies have recently focused on N-WASP, a Cdc42 effector that belongs to the family of Wiskott-Aldrich syndrome proteins (WASP) that, in association with the Arp 2/3 complex, can induce actin polymerization (Symons et al. 1996; Rohatgi et al. 1999; Mullins et al. 1998; Zigmond 1998; Machesky and Insall 1999).
PDGF is a physiological chemotactic factor for fibroblasts that triggers several cytoskeletal changes including lamellipodium formation, a decrease in actin stress fibers, and a reduction in adhesion complexes (Bockus and Stiles 1984; Ridley and Hall 1994; Wennström et al. 1994a). Whereas the mechanism involved in PDGF induction of lamellipodia is well-established and known to involve the activation of PI3K and Rac (Hawkins et al. 1995), the mechanisms by which PDGF alters stress fibers and adhesion complexes is unclear. Here, we analyzed the PDGF-R–induced pathways that might be involved in promoting these cytoskeletal changes. These studies revealed that PDGF activates Cdc42, which in turn mediates filopodia formation, a decrease in actin stress fibers, and a reduction in focal adhesion complexes. We find that activation of the Cdc42 pathway is independent of PI3K activity, but dependent on the p85α regulatory subunit of PI3K. We show that the downstream effector of Cdc42, N-WASP, is essential not only for PDGF-induced filopodia formation (Miki et al. 1998), but also contributes to the PDGF-induced disassembly of actin stress fibers and adhesion complexes. Finally, we show that induction of this pathway contributes to PDGF-induced cell migration on collagen. Together, these results reveal a novel signaling function for the p85 PI3K regulatory subunit. The activation of this cascade, involving PDGF-R, the regulatory subunit of PI3K, Cdc42, and N-WASP, represents a new signaling pathway that links cell-surface receptors to actin cytoskeletal dynamics and cell migration.
Materials and Methods
Antibodies and Reagents
The following antibodies (Abs) were used: antihemagglutinin (HA) mAb (12CA5) (BAbCO), anti-myc mAb (9E10) (Evan et al. 1985), anti-p85 polyclonal Ab (catalogue No. 06-195; Upstate Biotechnology), antipaxillin mAb (Transduction Laboratories), anti–N-WASP polyclonal Ab (donated by Drs. H. Miki and T. Takenawa, Institute of Medical Science, Tokyo, Japan; Miki et al. 1998), and Cy2- and Cy3-conjugated anti-mouse and anti-rabbit Abs (Jackson ImmunoResearch Laboratories). Anti-p110β Ab was a gift of Dr. D. Waterfield (Ludwig Institute for Cancer Research, London, UK; Vanhaesebroek et al. 1999). FITC-phalloidin was from Sigma Chemical Co.; PDGF-BB was from Upstate Biotechnology; collagen type VI was from Sigma Chemical Co.; G418 was from GIBCO BRL; and glutathione-Sepharose 4B was from Amersham Pharmacia Biotech.
Cell Culture and Stable Cell Line Preparation
NIH-3T3 cells and derivatives were maintained in DME (BioWhittaker) containing 10% calf serum (GIBCO BRL). Cultures were maintained in a humidified atmosphere at 37°C, 10% CO2. To study cytoskeletal changes after cell stimulation, cells were preincubated for 16 h in medium containing 0.1% serum; this decreased serum-mediated signals but maintained viability. p65PI3K and p85α NIH-3T3 stable cell lines were previously described (Jiménez et al. 1998). NIH-3T3 p110-CAAX stable lines were obtained by transfection (2 × 105 NIH-3T3 cells) with 3 μg of pSG5-N-myc-p110-CAAX (Jiménez et al. 1998) plus 1 μg of pSV-Neo using the Tfx-50 transfection reagent (Promega) as indicated by the manufacturer. Transfected clones were selected in medium containing G418 (2 mg/ml). N-myc-p110-CAAX was poorly recognized in Western blot by anti-myc or anti-p110α Abs. Therefore, p110-CAAX expression was examined in the selected clones by immunoprecipitation of cell lysates using anti-myc mAb, followed by in vitro PI3K assays using phosphatidylinositol-(4,5)-biphosphate (Sigma Chemical Co.) as a substrate as previously described (Jiménez et al. 1998). For studies on collagen, plates were incubated overnight at 4°C with a 20-μg/ml collagen VI solution in water. PAE cells expressing wt-PDGFβ-R and the F740/F751-PDGFβ-R were provided by Drs. P. Hawkins (Barbraham Instititute, Cambridge, UK) and L. Claesson Welsh (Ludwig Institute for Cancer Research, Uppsala, Sweden; Wennström et al. 1994b), and were maintained in Ham's F12 nutrient mixture (GIBCO BRL) containing 10% FCS.
Constructs, Transient Transfections, and Microinjection
The following constructs were used: pSG5-HA-tagged-p65PI3K, pSG5-HA-p85α, pSG5-N-myc-p110, pSG5-N-myc-p110CAAX, LXV-myc-N17-Rac (DN-Rac), were previously described (Jiménez et al. 1998). LXV-myc-N17-Cdc42 (DN-Cdc42), PcEXV-myc-Cdc42 (wtCdc42), and LXV-myc-V12-Cdc42 (v-Cdc42) were donated by Dr. A. Hall (MRC Laboratory for Molecular Cell Biology, London, UK. pEF-Bos-myc-V14-Rho (v-Rho) was donated by Dr. J. Downward (Imperial Cancer Research Fund, London, UK; Rodríguez-Viciana et al. 1997). PcDL-SRa-N-Wasp and PcDL-SRa-Δcof-N-Wasp (lacking amino acids 473–476: KRSK) were donated by Drs. H. Miki and T. Takenawa (Miki et al. 1998), and the Gex-2T-CRIB domain of Pak-1 was previously described (Sander et al. 1998). The SH3Bcr mutant was obtained by PCR and encompassed the region between base pair 30 and 1,115 of p85α; this fragment (containing BamHI sites at both ends) was subcloned into the BamHI site of the pcDNA 3-HA vector. The p50α alternative splice form of p85α was generously donated by Dr. L. Cantley (Harvard Medical School, Boston, MA; Fruman et al. 1996). For immunofluorescence studies, transient transfection was performed using calcium phosphate (2 × 105 NIH-3T3 cells, 5 μg cDNA). For high efficiency transfections (for Cdc42 activity and cell migration assays), cells were transfected using Lipofectamine plus (GIBCO BRL) following the manufacturer's instructions (1.7 × 106 NIH-3T3 cells, 10 μg cDNA), which yielded ∼60% transfection efficiency. Transfected samples were incubated for 16 h in medium containing 10% serum, to allow exogenous protein expression, before starvation in medium containing 0.1% serum for an additional 16 h. For microinjection, cells were seeded sparingly on coverslips and starved for 16 h before microinjection in DME containing 0.1% serum. cDNA (0.2 mg/ml in 10 mM sodium phosphate, pH 7.2, 80 mM sodium chloride) or Abs (0.2 mg/ml anti-p110β Ab or a 1:5 dilution of anti-p85 Ab in PBS) were microinjected using an automated injection system (AIS; Carl Zeiss) equipped with an Eppendorf microinjector 5242. After microinjection, cells were incubated in DME-0.1% serum at 37°C for 3 h (DNA injections) or 45 min (Ab injections) before analysis.
Biochemistry, Immunofluorescence, Videomicroscopy, and Cdc42 Activity Assay
Cells collected by trypsinization were lysed as previously described (Jiménez et al. 1998). For immunoprecipitation, lysates (300 μg) were incubated for 3 h with 1 μg of purified Ab or 2 μl of polyclonal Ab, followed by a 1-h incubation with protein A-Sepharose. Immunoprecipitation, SDS-PAGE, and Western blotting were performed as previously described (Jiménez et al. 1998). For immunofluorescence, cell samples were cultured on coverslips in medium containing 0.1% serum for 16 h, fixed in 4% paraformaldehyde in PBS, permeabilized in 0.3% Triton X-100, and blocked with 0.5% BSA and 1% FCS in PBS. After incubation with primary antibodies, cells were stained with Cy3- or Cy2-conjugated anti-mouse or anti-rabbit Ab. Filamentous actin was stained with FITC-conjugated phalloidin. Confocal microscopy was performed using a Leica laser scanning confocal microscope. For videomicroscopy studies, immunofluorescent videomicroscopy yielded better results than phase-contrast videos, and, thus, cells were transfected either with the GFP control vector or with the GFP control vector plus the vector encoding DN-Rac or DN-Cdc42. After 16 h, cells were incubated in 0.1% serum for an additional 16 h and filmed before and after the addition of PDGF (50 ng/ml); images were taken every 15 s using a Leica laser confocal microscope and TCS-NT software. Preparation of GST or GST-Pak-1 columns and detection of activated Cdc42 was previously described (Sander et al. 1998). In brief, NIH-3T3 cells transfected with cDNA encoding wtCdc42 (or v-Cdc42) were incubated, starved, and stimulated (see Fig. 3 and Fig. 5 B). Cells were lysed, lysates were normalized for protein content, and 400 μg of the samples were incubated on a GST-glutathione-Sepharose column or a GST-Pak-1-glutathione-Sepharose column for 30 min at 4°C. Columns were washed with lysis buffer and column-bound myc-wt-Cdc42 was examined after resolving the retained material in SDS-PAGE by Western blot using anti-myc mAb.
Figure 3.
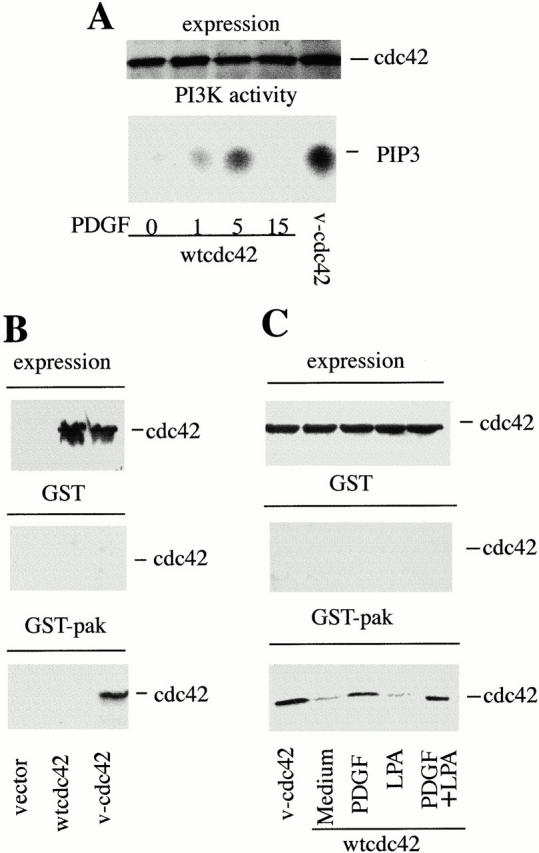
Stimulation of fibroblasts with PDGF activates Cdc42. 1.7 × 106 NIH-3T3 cells were transfected with 10 μg of control cDNA, cDNA encoding myc-wt-Cdc42, or myc-V12-Cdc42 (myc-v-Cdc42). After transfection, cells were incubated as in Fig. 2. In A, PDGF-R stimulation (50 ng/ml) was performed for the indicated periods of time (in minutes). (A) Lysates were resolved in 12% SDS-PAGE (60 μg, lane), transferred to nitrocellulose, and membranes were blotted with anti-myc Ab (top). Alternatively, lysates were immunoprecipitated (300 μg/sample) using anti-myc Ab, and the PI3K activity associated with myc-Cdc42 was measured in vitro (bottom). (B) Cdc42 expression in the transfected samples (indicated) was analyzed as in A, or, alternatively, lysates (300 μg/sample) were passed through a GST-glutathione-Sepharose column (control) or a Pak-1-GST-glutathione-Sepharose column. The Myc-Cdc42 bound to the columns was examined by Western blot using anti-myc Ab. (C) Cells were transfected, starved, and stimulated for 5 min with PDGF and LPA (doses as in Fig. 2 C). Cell lysates were processed as in B. Specific wt-Cdc42 binding to Pak-1 columns is seen in PDGF-stimulated cells. The figure illustrates one representative experiment of three performed with similar results.
Figure 5.

Expression of the p85α regulatory subunit of PI3K inhibits LPA and v-Rho–induced stress fibers. (A) 2 × 105 NIH-3T3 cells were transfected as in Fig. 2 with control cDNA, cDNA encoding HA-p85α, myc-v-Rho, or HA-p85α plus myc-v-Rho. Other cells (serum-starved) were microinjected with HA-p85α plus myc-DN-Cdc42 or with HA-p65PI3K plus N-myc-wt-p110 plus myc-DN-Cdc42. After transfection, cells were incubated, serum-starved, and activated as in Fig. 2. After microinjection, cells were incubated for 3 h before activation as in Fig. 2. Cells were fixed and stained simultaneously with FITC-phalloidin (depicted) and with anti-myc or anti-HA Ab (indirect immunofluorescence, not shown) to detect positive transfected cells (indicated by an arrow). The figure represents one experiment of five performed with similar results. (B) Samples were transfected with wt-Cdc42 combined with control vector, vector encoding p85α, or vector encoding p65PI3K (indicated). After transfection, cells were incubated as in Fig. 2. Lysates were resolved in 10% SDS-PAGE (60 μg per lane), transferred to nitrocellulose, and membranes were blotted with either anti-p85 Ab (top) or anti-myc Ab (middle). Alternatively, lysates were immunoprecipitated (300 μg/sample) using anti-myc Ab, and the associated PI3K activity was examined as in Fig. 3 A. The figure illustrates one representative experiment of three performed with similar results. Bar, 100 μm.
Cell Migration
For migration assays, NIH-3T3 cells were transiently transfected with the indicated constructs. Transfection efficiency (∼60%) was estimated by cotransfecting a plasmid encoding green fluorescent protein (pE-GFP-N; CLONTECH Laboratories, Inc.). Migration was studied in 96-well microchambers using polyvinylpyrrolidone-free filters (10-μm pores; Neuro Probe Inc.) precoated (for 16 h at 4°C) with 20 μg/ml type VI collagen (Sigma Chemical Co.). Assays were performed with different numbers of cells in the top wells and different PDGF concentrations in the serum-free medium in the bottom wells. Linearity was observed (in 5-h assays) using 1–2 × 105 cells/well and 1–100 ng/ml of PDGF. Chambers were incubated for 5 h at 37°C in a humidified atmosphere with 10% CO2. After incubation, filters were removed and the nonmigrating cells in the top part were wiped off. The migrating cells remaining on the bottom part of the filters were fixed and stained with a crystal violet solution (0.5% crystal violet and 20% methanol). Migration was quantitated by densitometry of the corresponding violet spots (National Institutes of Health Image software). The migration index was calculated as the ratio of migration observed in the presence of PDGF and divided by the migration observed with the medium alone. The percent cell migration was calculated by comparing the migration index of control cells (considered 100%) with that of the transfected samples. The proportion of the input NIH-3T3 cells that migrated in response to PDGF (100 ng/ml) was calculated by cell counting and confirmed by measuring the OD at 595 nm of the migrating cells stained with crystal violet. This value was interpolated on a standard curve representing known numbers of cells versus their corresponding OD values.
Results
PDGF Induces Filopodia Formation, a Decrease in Actin Stress Fibers, and a Reduction of Adhesion Complexes via Cdc42
Here, we examine the mechanisms mediating PDGF-induced cytoskeletal changes in NIH-3T3 cells. PDGF treatment not only induces membrane ruffling and lamellipodial extensions, but also a decrease in LPA-stimulated actin stress fibers and focal adhesions (Fig. 1; Bockus and Stiles 1984; Ridley and Hall 1994; Wennström et al. 1994b).
Figure 1.

PDGF receptor stimulation induces actin stress fiber disassembly and a decrease in focal adhesion complexes. NIH-3T3 cells were incubated for 16 h in 0.1% serum. After starvation, cells were incubated for 10 min with medium, or with PDGF (50 ng/ml), or with LPA (10 ng/ml), or with a combination of LPA (10 ng/ml) and PDGF (50 ng/ml). Cells were stimulated, fixed, and stained for filamentous actin using FITC-conjugated phalloidin and for paxillin distribution by indirect immunofluorescence. Images were collected by confocal microscopy. Paxillin images in PDGF-treated samples were overexposed to detect the low intensity peripheral focal adhesion complexes. The figure illustrates one representative experiment of five performed with similar results. Bar, 100 μm.
Previous studies have shown that constitutive active forms of Rac or Cdc42 inhibited Rho A activation and LPA-induced (Rho A–dependent) actin stress fiber formation (Dutartre et al. 1996; Manser et al. 1997; Sander et al. 1999). We subsequently investigated whether Cdc42- or Rac-dependent signaling pathways were responsible for the inhibition of stress fibers and the adhesion complexes observed in PDGF-treated cells using dominant negative (DN) interfering mutants. Expression of DN-Rac in PDGF-treated cells partially restored actin stress fiber formation in ∼10% of the cells, although it efficiently inhibited Rac-dependent lamellipodium formation (in >95% of the cells; Fig. 2 A). In addition, expression of DN-Rac, which increases filopodia stability (Nobes and Hall 1995), revealed the formation of filopodium-like structures in the majority of the transfected cells (Fig. 2 A). In contrast, expression of DN-Cdc42 efficiently restored actin stress fiber formation in PDGF-treated cells (in 60% of the cells), and also diminished Rac-dependent membrane ruffling (in 50% of the cells; Fig. 2 A). Transfection of DN-Rac and DN-Cdc42 in these experiments yielded similar expression levels (not shown). The greater ability of DN-Cdc42 compared with DN-Rac to restore stress fiber formation in PDGF-treated cells was confirmed in microinjection experiments (data not shown).
Figure 2.
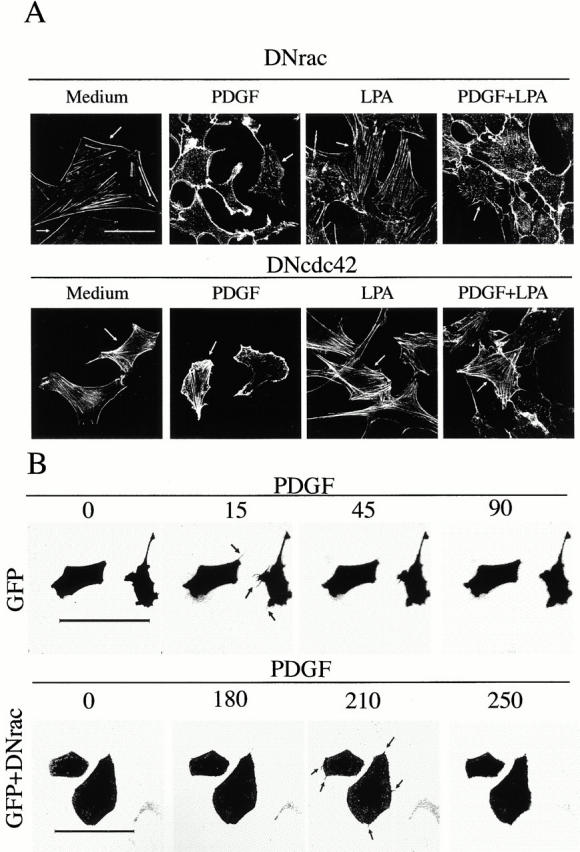
The PDGF-induced decrease in stress fibers is blocked by dominant negative mutants of Cdc42. (A) NIH-3T3 cells (2 × 105) were transfected with cDNA encoding myc-N17-Rac (DN-Rac, 5 μg) or myc-N17-Cdc42 (N17-Cdc42, 5 μg; indicated) using calcium phosphate. Transfected cells were incubated for 16 h in medium containing 10% serum to allow exogenous protein expression, and then incubated for an additional 16-h period in medium containing 0.1% serum. Cells were stimulated as in Fig. 1, fixed, and stained with FITC-phalloidin. Transfected cells (indicated by an arrow) were detected by simultaneous indirect immunofluorescence using anti-myc primary Ab. The figure illustrates that DN-Cdc42 blocks PDGF-induced stress fiber disassembly, whereas DN-Rac inhibits PDGF-induced lamellipodial extensions, enhancing detection of PDGF-induced filopodium-like structures. (B) 2 × 105 NIH-3T3 cells were transfected with cDNA encoding GFP (2.5 μg) or with cDNA encoding GFP plus cDNA encoding DN-Rac (2.5 μg each). Cells were incubated as in A, and videos were filmed before and upon addition of PDGF (50 ng/ml) with images taken every 15 s using a Leica laser confocal microscope and TCS-NT software. The figure shows selected time points after PDGF treatment. One representative experiment of five performed with similar results is shown. Bars, 100 μm.
Neither DN-Rac nor DN-Cdc42 had a significant effect on actin stress fiber content in LPA-treated cells, but DN-Cdc42 expression allowed efficient stress fiber formation in ∼60% of PDGF- plus LPA-treated cells (Fig. 2 A). DN-Cdc42 expression also allowed efficient formation of focal adhesion complexes both in PDGF and in PDGF-plus LPA-treated cells (not shown). These results confirm that PDGF-induced lamellipodium formation is Rac-dependent, but also reveal that the decrease in actin stress fibers and adhesion complexes is mediated mainly via a Cdc42-dependent pathway.
To check that PDGF induces filopodia formation, control cells or cells transfected with DN-Rac or DN-Cdc42 were PDGF-stimulated and examined by time-lapse video microscopy. Evolution of filopodial structures was observed in PDGF-treated cells (Fig. 2 B) and in DN-Rac–expressing cells (Fig. 2 B, filopodia observed for prolonged periods of time). However, filopodia formation was not observed in cells expressing DN-Cdc42 (not shown). We conclude that PDGF induces filopodia formation and inhibits actin stress fibers as well as adhesion complexes via a Cdc42-dependent pathway.
PDGF Activates Cdc42
To determine whether PDGF activates Cdc42, we considered that activated Cdc42 binds to the p85α regulatory subunit of PI3K (Tolias et al. 1995). Thus, we examined the ability of PDGF to induce p85α association to Cdc42 by measuring p85/p110 lipid kinase activity in Cdc42 immune complexes (Fig. 3 A). Whereas recombinant v-Cdc42 stably associated p85/p110 in starved cells, wt-Cdc42 only bound p85/p110 after PDGF-R stimulation; this association was transient, with maximum levels detected at ∼5 min (Fig. 3 A). Formation of this complex was confirmed by Western blotting (not shown).
Activation of Cdc42 by PDGF was validated by an alternative method, using the previously described pull-down assay, based on the ability of activated Cdc42 and Rac to associate to the CRIB region of their effector Pak-1 (Sander et al. 1998). To optimize the method, we first confirmed that the activation of Rac by PDGF results in increased Rac association to the GST-Pak columns (not shown) in agreement with previous reports (Wennström et al. 1994a,Wennström et al. 1994b; Sander et al. 1998). In addition, we confirmed that v-Cdc42, but not wt-Cdc42, bind to GST-Pak-1 columns (Fig. 3 B; Sander et al. 1999). Finally, we find that PDGF treatment, alone or in combination with LPA, induces the association of wt-Cdc42 to GST-Pak-1 columns (Fig. 3 C). Therefore, PDGF-R stimulation induces Cdc42 activation and its transient association to the regulatory subunit of PI3K.
The PI3K Regulatory Subunit Induces Filopodia Formation, a Decrease in Actin Stress Fibers, and a Reduction in Adhesion Complexes
PDGF treatment induces activation and translocation of p85/p110 PI3K to its receptor, an essential event for Rac-mediated lamellipodium formation (Klippel et al. 1992; Wennström et al. 1994a,Wennström et al. 1994b). The molecular basis for Cdc42 activation after PDGF-R stimulation nonetheless remains unclear. We previously noticed that cells expressing p85α or its truncation product p65PI3K (Jiménez, et al. 1998) exhibited filopodium-like extensions and contained decreased numbers of actin stress fibers (our unpublished observations). Thus, we decided to study the ability of PI3K regulatory subunit to mediate actin cytoskeleton changes in greater detail. We have compared the phenotypes of cells expressing p110-CAAX (a constitutive active mutant of the p110 catalytic subunit; Jiménez et al. 1998), the p85α regulatory subunit, or p65PI3K (a p85 mutant that enhances p110 activation; Jiménez et al. 1998). p85α and p65PI3K stable cell lines have been described previously and express transfected proteins at levels similar to that of endogenous p85α (Jiménez et al. 1998); p110-CAAX cell lines, described here, were prepared similarly (Fig. 4 A).
Figure 4.

Increased p85α-PI3K regulatory subunit expression induces a decrease in actin stress fibers and adhesion complexes. (A) N-myc-p110-CAAX stable NIH-3T3 cells were prepared as detailed in Materials and Methods. Lysates of selected clones (200 μg/sample) were immunoprecipitated with anti-myc Ab, and the associated PI3K activity was tested in vitro as in Fig. 3 A. Two representative N-myc-p110-CAAX–positive clones (lanes 2 and 3) are shown. (B) NIH-3T3 stable transfectants expressing p110-CAAX, HA-p85α, or HA-p65PI3K (indicated) were incubated in 0.1% serum for 16 h. Cells were subsequently fixed and stained for filamentous actin using FITC-conjugated phalloidin as in Fig. 1. (C) Cells were treated as in B and stained for paxillin distribution by indirect immunofluorescence as in Fig. 1. (D) NIH-3T3 cells preincubated in medium containing 0.1% serum were microinjected with different cDNAs (0.2 mg/ml) encoding HA-p65PI3K, HA-p85α, or HA-p65PI3K plus cDNA encoding N-myc-wt-p110. Cells were incubated for 3 h and subsequently fixed and stained simultaneously with FITC-phalloidin and anti-HA Ab. Phalloidin staining is illustrated; microinjected HA-positive cells are indicated by an arrow. In the bottom right panel, HA staining is shown for the p85α sample. (E) NIH-3T3 cells were transfected with cDNA encoding either SH3Bcr mutant or p50α, and cells were incubated and stained as in Fig. 2. Quantitation of fluorescence intensity of F-actin (in arbitrary units) in the indicated cross-sections is shown beneath the SH3Bcr and p50 panels. The figure illustrates one representative experiment of four performed with similar results. Bars, 100 μm.
Whereas most p110-CAAX cells exhibited a flattened morphology, extended lamellipodia (60%), and contained stress fiber numbers similar to those of control cells (>90%; Fig. 4 B), cells expressing p85α (90%) were elongated, did not extend lamellipodia, and contained fewer stress fibers than mock-transfected cells (Fig. 4 B). p65PI3K cells showed lamellipodial extensions, as did p110-CAAX cells, but similar to p85α cells, most p65PI3K cells (75%) were elongated and contained fewer actin stress fibers than the controls (Fig. 4 B). In addition, p110-CAAX cells (90%) showed abundant paxillin-containing focal adhesion complexes, whereas p85α cells (90%) and p65PI3K cells (75%; Fig. 4 C) had a lower number of focal adhesion complexes and contained smaller focal contacts located at the peripheral extensions. In conclusion, expression of constitutively activated mutants of PI3K induced lamellipodium formation, whereas expression of p85α and p65PI3K regulatory molecules induced a decrease in actin stress fibers and adhesion complexes.
Similar phenotypes were observed in cells microinjected with plasmids encoding the different PI3K forms and analyzed 3 h later. Microinjection of p110-CAAX (not shown) induced a phenotype similar to that observed in p110-CAAX stable cell lines (Fig. 4 B). In a majority of cells, expression of p65PI3K or p85α (75 and 90%, respectively) induced a decrease in stress fibers compared with the surrounding noninjected cells (Fig. 4 D), although at these short times only 50% of the cells exhibited an elongated morphology. Thus, it appears that whereas the decrease in fibers occurs shortly after p85 expression, the change in cell morphology is more efficient in stable transfectants and, therefore, it may depend on gene expression associated to the prolonged expression of the PI3K regulatory subunit. In addition, the short-term analysis upon microinjection revealed that ∼30% of p65PI3K or p85α-microinjected cells showed formation of filopodium-like structures (Fig. 4 D). The appearance of filopodia in p85α-expressing cells was confirmed by videomicroscopy (not shown). Cells microinjected with p85α or p65PI3K regulatory subunits in combination with wt-p110 also showed a decrease in actin stress fibers and, in the case of p65PI3K, exhibited lamellipodial extensions (Fig. 4 D). Microinjected cells were detected by indirect immunofluorescence using anti-HA Ab, which also revealed the cytosolic–perinuclear localization of the majority of p85α (Fig. 4 D, lower left), similar to that of endogenous p85α and most p65PI3K and p110-CAAX proteins (not shown).
As v-Cdc42 induces filopodia formation and decreases actin stress fibers (Nobes and Hall 1995; Manser et al. 1997), our results showing that p85 and p65PI3K expression also induce these changes suggest that the regulatory subunit of PI3K may trigger a Cdc42-dependent pathway. Considering that p85 associates with Cdc42, we next examined whether p85 association to Cdc42 was required for the regulatory subunit of PI3K to induce actin cytoskeleton changes. Association of Cdc42 to p85 occurs through the Bcr homologous domain of p85 (Zheng et al. 1994). Thus, we examined the phenotype of cells expressing a mutant encompassing the SH3-Bcr regions of p85α and the phenotype of cells expressing p50α, which is an alternative splice form of p85α that lacks the SH3 and the Bcr regions (Fruman et al. 1996). Whereas expression of the SH3-Bcr mutant induced stress fiber loss (in >95% of the cells; Fig. 4 E) and triggered formation of filopodium-like extensions (not shown), p50α expression did not apparently affect actin cytoskeleton (Fig. 4 E). In addition, expression of the SH3-Bcr mutant, but not of p50α, was able to inhibit LPA-induced stress fibers and adhesion complexes (not shown). Quantitative examination of filamentous actin (F-actin) fluorescence intensity along a cross-section of these cells is shown beneath the SH3-Bcr and p50 panels (Fig. 4 E). This examination confirms that in SH3-Bcr–expressing cells, F-actin is located in the cell cortex, and that the pattern with multiple peaks corresponding to actin stress fibers is lost. However, in cells expressing p50α, a similar pattern to that of control cells is obtained. These observations suggest that only the p85 forms containing the Bcr homologous region (p85, p65PI3K, and the SH3-Bcr mutant) are capable of inducing a decrease in actin stress fibers and adhesion complexes.
In conclusion, these studies reveal a novel function for the PI3K regulatory subunit, which induces filopodia formation, a decrease in actin stress fibers, and induces a reduction in focal adhesion complexes, resembling the Cdc42-dependent cytoskeletal changes induced by PDGF. Induction of these changes by p85 appears to depend on the ability of p85α to bind Cdc42, since p50α, which does not bind Cdc42, failed to induce these changes.
The Decrease in Stress Fibers Induced by the PI3K Regulatory Subunit Is Mediated by Cdc42
The increased expression of the PI3K regulatory subunit reduces the basal stress fiber content of cells cultured in low serum conditions (Fig. 4). In addition, p85α expression also inhibited the fibers induced by LPA treatment (in >95% of the cells) and by v-Rho expression (in >75% of the cells) (Fig. 5 A), whereas expression of wt or constitutively active p110 did not significantly affect stress fiber content (not shown).
We previously have shown that the decrease in actin stress fiber formation induced by PDGF was mediated mainly via Cdc42 (Fig. 2). We next examined whether p85α expression also induced inhibition of actin stress fibers via Cdc42. Microinjection of cDNA encoding DN-Cdc42 in combination with p85 cDNA restored actin stress fiber formation (in >95% of the cells; Fig. 5 A), whereas microinjection of DN-Rac showed only a minor effect (restoring stress fibers formation in ∼10% of the cells; not shown). Microinjection of DN-Cdc42 also blocked the decrease in stress fibers in cells expressing p65PI3K plus p110 (in >95% of the cells; Fig. 5 A). Therefore, p85α (and p65PI3K) expression induced a decrease in actin fibers via Cdc42.
We next analyzed whether Cdc42 was activated in cells overexpressing p85α or p65PI3K as described above (Fig. 3A and Fig. B). Examination of Cdc42 bound to PI3K (Fig. 5 B) and examination of Cdc42 bound to GST-Pak-1 columns (not shown) showed that, in cells overexpressing p85α or p65PI3K, a small Cdc42 fraction was activated. In conclusion, overexpression of p65PI3K or p85α mediates a decrease in actin stress fibers via Cdc42.
The PI3K Regulatory Subunit Mediates PDGF-induced Cdc42-dependent Cytoskeletal Changes
Examination of cells expressing PI3K mutants suggested that activation of the Cdc42 pathway is independent of PI3K activity, but could be engaged via the p85 regulatory subunit. To examine whether PI3K activity is required for PDGF-induced Cdc42-dependent cytoskeletal changes, we analyzed the phenotype of cells treated with PDGF in the presence of PI3K activity inhibitors. PI3K inhibitors were effective in blocking PI3K activity–dependent lamellipodium formation (in >95% of the cells; Fig. 6 A, wortmannin; Ly-294002, not shown). However, these inhibitors only moderately affected the ability of PDGF to decrease actin stress fibers (Fig. 6 A), with a partial formation of actin stress fibers observed in ∼15% of the cells. This shows that the PDGF-induced reduction in stress fibers is, to a large extent, independent of PI3K activity.
Figure 6.

The PI3K regulatory subunit controls PDGF-induced cytoskeletal changes. (A) NIH-3T3 cells were cultured 16 h in 0.1% serum and incubated for 1 h with 1 mM wortmannin before stimulation as in Fig. 1. Cells were fixed and stained for filamentous actin using FITC-conjugated phalloidin. (B) NIH-3T3 cells were incubated for 16 h in 0.1% serum and microinjected with anti-p85-PI3K Abs or anti-p110β Abs (see Materials and Methods). Upon microinjection, cells were incubated for 45 min in serum-free medium before stimulation with PDGF and processing as in A. Microinjected cells, identified by immunofluorescence using Cy3 anti-rabbit Ig, are indicated by an arrow. (C) PAE cells expressing either wild-type PDGFβ-R or Phe740/Phe751 PDGFβ-R (indicated) were treated and examined as in Fig. 1. The figure illustrates one representative experiment of three performed with similar results. Bar, 100 μm.
We next examined whether binding of the PI3K regulatory subunit to the PDGF-R (Klippel et al. 1992) was essential for PDGF-induced Cdc42-dependent cytoskeletal changes. To this end, we analyzed the consequences of blocking p85 translocation by sequestering this molecule with microinjected anti-p85 Ab. The antibodies were highly specific, as demonstrated by their specificity in detecting p65PI3K and p85 in Western blot (Fig. 5 B), and their selective ability to immunopurify p85/p110 complexes from [35S]methionine-labeled cells (not shown). Microinjection of anti-p85 Ab reduced PI3K activity–dependent lamellipodium formation in ∼80% of injected cells since it blocked the p85/p110 complex translocation to the cell membrane receptor, indicating that the treatment was effective (Fig. 6 B). In addition, injected cells (80%) showed higher levels of actin stress fibers than the surrounding noninjected cells, suggesting that impairing p85 localization to the receptor inhibits the PDGF-induced Cdc42-dependent decrease in actin stress fibers (Fig. 6 B). As a control, microinjection of p110β-specific Ab had no significant effect on PDGF-induced cytoskeletal changes (Fig. 6 B), although they inhibit p110β kinase activity (not shown) as described previously (Vanhaesebroek et al. 1999; Hooshmand-Rad et al. 2000).
As an alternative approach to confirm that p85 translocation to the PDGF-R was required for PDGF to induce the Cdc42-dependent decrease in actin bundles and adhesion complexes, we examined the phenotypes of cells expressing a PDGFβ-R mutant containing two point mutations at the p85 interaction sites (Tyr740 and Tyr751; Wennström et al. 1994b). For these experiments, wild-type and Phe740/Phe751 PDGFβ-R were expressed in PAE cells, which do not express endogenous PDGFβ-R. In these cells, wt-PDGFβ-R mediated the inhibition of stress fibers and induction of lamellipodia (in ∼80% of the cells), whereas the Phe740/Phe751 PDGFβ-R mutant not only failed to induce lamellipodium formation (Wennström et al. 1994b), but also failed to decrease the levels of actin stress fibers (in >90% of the cells; Fig. 6 C) and adhesion complexes (not shown). In conclusion, activation of the Cdc42 pathway by PDGF is largely independent of PI3K activity, but requires translocation of the PI3K regulatory subunit to its receptor.
N-WASP Participates in Mediating the Cdc42-dependent Decrease in Actin Stress Fibers
It has been shown that α-Pak contributes to mediating the decrease in stress fibers observed in cells expressing v-Rac or v-Cdc42 (Manser et al. 1997; Zhao et al. 1998). However, as the PDGF-induced decrease in stress fibers was, to a large extent, Cdc42-dependent, we searched for a Cdc42-specific effector that might mediate this action and we considered N-WASP. N-WASP is an effector of Cdc42 that mediates filopodia formation, a function for which the cofilin homologous region of N-WASP seems to be essential (Miki et al. 1996, Miki et al. 1998). Thus, we examined the consequences on actin stress fiber formation of transfecting wt-N-WASP or Δcof-N-WASP, which were expressed to a similar extent (Fig. 7 A). Whereas in cells expressing N-WASP, p85α mediated a pronounced decrease in actin stress fibers, Δcof-N-WASP expression restored, to a large extent, actin stress fiber formation in p85α-expressing cells (in >95%; Fig. 7 B). Similar phenotypic changes were observed when p85α and N-WASP plasmids were cotransfected with wt-Cdc42 (not shown). In contrast, expression of wt-Cdc42 alone, N-WASP alone, or Δcof-N-WASP in serum-starved cells had no significant effects on the actin cytoskeleton (our data not shown; Miki et al. 1998). Thus, N-WASP appears to be an essential mediator of the p85α-induced Cdc42-dependent decrease in actin stress fibers.
Figure 7.
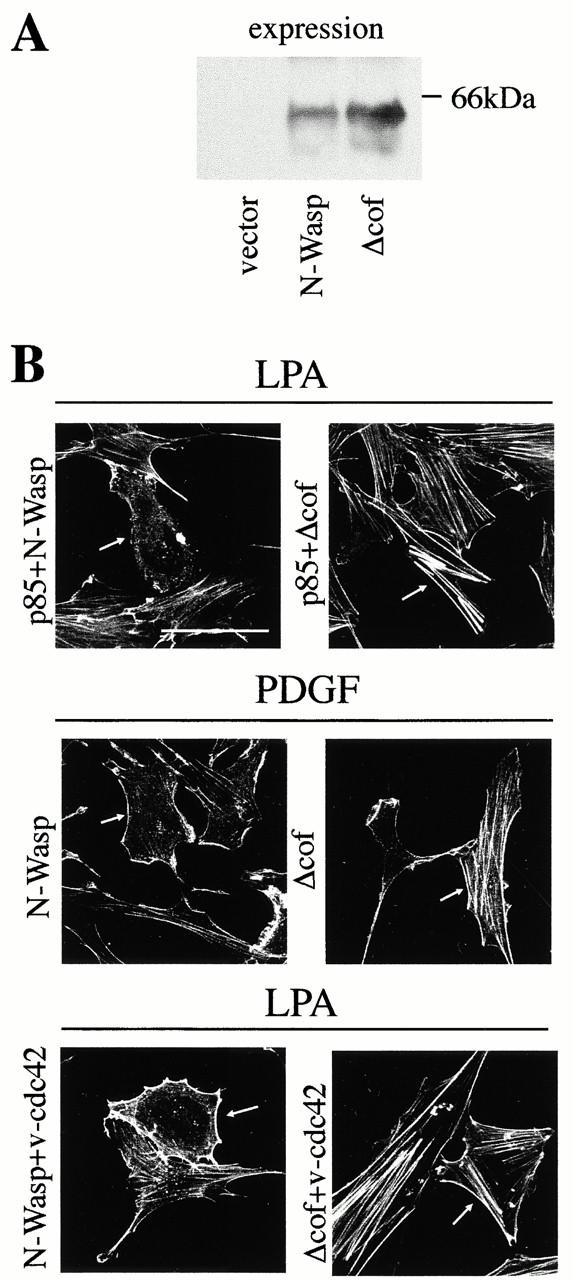
N-WASP mediates the inhibition of actin stress fibers induced by PDGF and by p85α. 2 × 105 NIH-3T3 cells were transfected as in Fig. 2 with a total of 5 μg cDNA. Different combinations of vectors encoding HA-p85α, N-WASP, Δ-cof-N-WASP, and myc-v-Cdc42 were used (indicated). (A) 50 μg of lysates from cells transfected with cDNAs encoding control vector, or N-WASP, or Δ-cof-N-WASP were resolved by SDS-PAGE, transferred onto nitrocellulose, and examined by Western blot using anti-N-WASP Ab. (B) After transfection with the indicated plasmids, cells were cultured and activated as in Fig. 2. Cells were subsequently fixed and stained simultaneously with FITC-phalloidin (depicted) and with anti-myc, anti-HA, or anti–N-WASP Ab (indirect immunofluorescence, not shown) to detect positive transfected cells (indicated by an arrow). Throughout the experiment, Δ-cof-N-WASP expression blocked stress fiber disassembly, which was induced by p85α, PDGF, or v-Cdc42. The figure illustrates one representative experiment of five performed with similar results. Bar, 100 μm.
Δcof-N-WASP expression also interfered with the PDGF-triggered Cdc42-dependent decrease in stress fibers (in 75% of the transfected cells; Fig. 7 B), with similar phenotypic changes observed when the Δcof-N-WASP plasmid was cotransfected with wt-Cdc42. Δcof-N-WASP also increased focal adhesion complexes in PDGF- or PDGF-plus LPA-treated cells (not shown), but did not affect LPA-induced cytoskeletal changes (not shown).
The contribution of N-WASP to the Cdc42-induced decrease in stress fibers was validated by examining the consequences of expressing Δcof-N-WASP on v-Cdc42–expressing cells. Δcof-N-WASP expression not only interfered with v-Cdc42–induced filopodia formation (Miki et al. 1998; our data not shown), but also restored, to a large extent, stress fiber formation in v-Cdc42–expressing cells (in >95%; Fig. 7 B). These experiments using interfering mutants of the Cdc42/N-WASP pathway were also performed using normal murine embryonic fibroblasts, which yielded comparable results (not shown). In conclusion, N-WASP not only mediates Cdc42-induced filopodia formation (Miki et al. 1998), but also contributes to mediate the Cdc42-dependent inhibition of actin stress fibers induced by PDGF/p85α.
The p85/Cdc42/N-WASP Pathway Regulates Cell Migration
We subsequently evaluated the contribution of the Cdc42/N-WASP pathway to PDGF-induced migration using collagen as a substrate. Cells cultured on collagen exhibited cytoskeletal changes (Fig. 8) similar to those observed in the absence of exogenous ECM components. In fact, p85α expression triggered an N-WASP–dependent decrease in actin stress fibers when cells were cultured on collagen (Fig. 8 A), and PDGF treatment also induced cortical actin polymerization and a decrease in LPA-induced stress fibers when cells were cultured on collagen (Fig. 8 A). Moreover, the PDGF-induced decrease in stress fibers was blocked by DN-Cdc42 or Δcof-N-WASP (Fig. 8 A), and PDGF-R stimulation also induced a Cdc42-dependent decrease in focal adhesions (Fig. 8 B). Therefore, activation of the p85/Cdc42/N-WASP pathway by PDGF induced similar cytoskeletal changes when cells were cultured on collagen or in the absence of exogenously added ECM.
Figure 8.
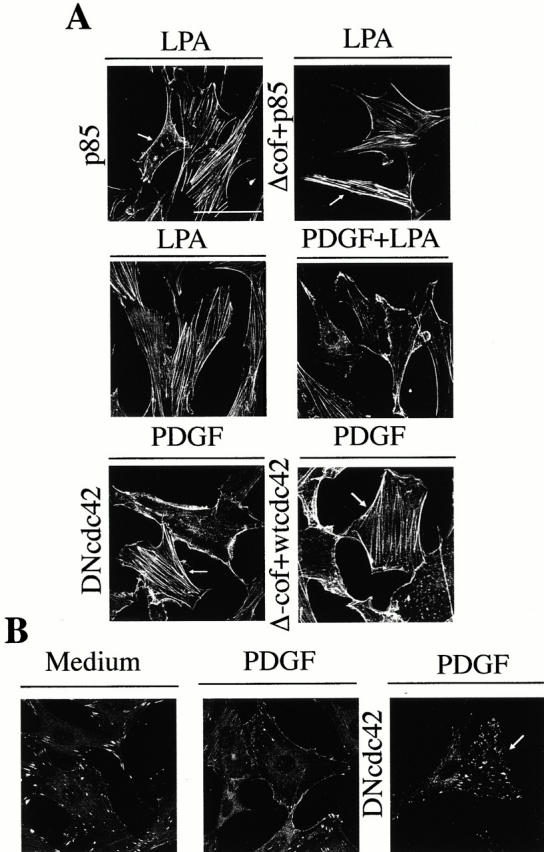
Actin cytoskeleton changes induced by PDGF stimulation or p85α expression in NIH-3T3 cells cultured on collagen. NIH-3T3 cells were cultured on collagen VI–coated plates. The samples (2 × 105 NIH-3T3 cells) were transfected with different combinations of vectors encoding HA-p85α, Δ-cof-N-WASP, myc-wt-Cdc42, and myc-DN-Cdc42 (indicated). After transfection, cells were incubated in complete medium, starved, and subsequently activated as in Fig. 2. (A) Cells were fixed and stained with FITC-phalloidin or, in the case of transfected cells stained simultaneously with FITC-phalloidin (depicted) and anti-myc, anti-HA, or anti-N-WASP Ab to detect transfected cells (indicated by an arrow). (B) Paxillin staining of NIH-3T3 cells cultured on collagen and transfected, starved, and treated as in A (indicated). The figure shows a representative experiment of four performed with similar results. Bar, 100 μm.
We subsequently performed migration assays using collagen-coated microchambers. Under optimized conditions, migration was proportional to the cell numbers and to the dose of PDGF used in the assay, inducing mobilization of ∼20–25% of the 105 input cells (at 5 h with 100 ng/ml PDGF; see Materials and Methods). Cell migration was estimated in quadruplicate by densitometry of migrating cells, and the migration index, representing the x-fold increase in cell migration observed in the presence versus the absence of chemoattractant, was calculated for each condition. Fig. 9 A shows the migration index of control NIH-3T3 cells in response to increasing PDGF doses. We subsequently compared the migration indexes of the transfected samples with those of the control cells. This comparison showed that the migration efficiency of p85α stable cell lines (expressing a two-fold increase in p85α expression, Jiménez et al. 1998) was higher than the migration efficiency of control cells, particularly at suboptimal PDGF doses (10 ng/ml; Fig. 9 B). p110-CAAX cell migration was slightly lower than that of control cells at suboptimal doses of PDGF (10 ng/ml; Fig. 9 B), but similar at 100 ng/ml PDGF (not shown).
Figure 9.

PDGF-induced fibroblast migration is blocked by expression of mutants interfering with the Cdc42/N-WASP pathway. (A) PDGF-induced migration of GFP transiently transfected NIH-3T3 cells was assayed in collagen VI–coated 96-well microchambers and quantitated by densitometry of the spots. Cell migration is expressed as a migration index and calculated as the x-fold increase over the negative control (unstimulated cells). Figure shows the mean of five experiments; SD is indicated. (B) Percent migration (induced by 10 ng/ml PDGF) of p110CAAX- or p85α stable transfectants compared with the migration of control stable transfectants (considered 100%). Figure shows the mean of three experiments; SD is indicated. (C) Percent migration (induced by 100 ng/ml PDGF) of cells transfected with a vector encoding GFP plus the different cDNAs (indicated), compared with the migration of control cells transfected only with cDNA encoding GFP (considered 100%). Figure shows the mean of four experiments; SD is indicated.
To evaluate the contribution of the Cdc42/N-WASP pathway to PDGF-induced migration, we examined the migration efficiency of cells transfected with cDNA encoding interfering mutants of this pathway. The transfection efficiency was monitored after GFP vector expression by flow cytometry and was ∼60% in the different samples. Since Rac previously has been shown to be required for PDGF-induced migration (Ridley et al. 1992), DN-Rac expression was included as a control. When compared with cells transfected with the GFP control vector, DN-Rac– and DN-Cdc42–transfected cells consistently showed a lower migration efficiency than control cells both at 100 ng/ml of PDGF (Fig. 9 C) and at lower doses (not shown), indicating that both Rac and Cdc42 pathways regulate PDGF-induced cell migration. Moreover, whereas transfection of cells with wt-Cdc42 plus N-WASP increased PDGF-induced cell migration, expression of wt-Cdc42 plus the interfering mutant Δcof-N-WASP inhibited PDGF-induced migration on collagen matrix (Fig. 9 C). These results reveal that activation of the PDGF-R/p85/Cdc42/N-WASP pathway plays an essential role in cell migration.
Discussion
Actin cytoskeleton reorganization is an essential process required for cell migration. Here, we describe that stimulation of PDGF-R on fibroblasts activates Cdc42 and its effector N-WASP inducing filopodia formation, a decrease in actin stress fibers and a reduction in focal adhesion complexes. The data presented also support that p85α, the regulatory subunit of PI3K, is capable of activating the Cdc42/N-WASP pathway linking PDGF-R stimulation to Cdc42 activation. These data, integrated into the model depicted in Fig. 10, include the novel description on PDGF's ability to activate Cdc42 and a new role for the PI3K regulatory subunit in controlling actin dynamics and cell migration.
Figure 10.

PDGF-PI3K–derived pathways that affect cytoskeletal organization and cell migration. Model illustrating the signaling pathways initiated by the PDGF-R that affect actin cytoskeleton and cell migration, including the previously described p110/PI3K activity–dependent Rac pathway, which promotes lamellipodial extensions and the novel p85-PI3K regulatory subunit-triggered Cdc42 pathway. Activation of the Rac pathway triggers a number of effectors, of which Por 1, inducing lamellipodium formation, and Pak, mediating inhibition of actin stress fibers in v-Rac and v-Cdc42–expressing cells, are indicated (Lim et al. 1996; Van Aelst et al. 1996). Activation of the Cdc42 pathway also triggers a number of effectors of which, N-WASP promotes filopodia formation, and contributes to mediate the decrease in actin stress fibers and adhesion complexes induced by PDGF. Induction of both pathways control PDGF-induced cell migration on collagen.
We describe that PDGF activates Cdc42 and propose that the regulatory subunit of PI3K is one of the molecules controlling Cdc42 activation by PDGF. We base this conclusion on the observation that microinjection of anti-p85 Abs, which inhibit p85 binding to the PDGF-R, blocked the Cdc42-dependent decrease in actin stress fibers and the appearance of filopodium-like extensions. A similar conclusion was reached after examination of PAE cells expressing a mutant PDGF-R form that does not bind p85 (Wennström et al. 1994b). Stimulation of this mutant receptor failed to induce not only PI3K activity–dependent lamellipodia formation (Wennström et al. 1994b), but also the PDGF-induced decrease in actin stress fibers and adhesion complexes. The similarity between the p85α- and PDGF-induced pathways as well as the ability of the PDGF-R to induce transient Cdc42 and p85α association (Fig. 3) both support the involvement of the PI3K regulatory subunit in PDGF-induced Cdc42 activation. Therefore, we conclude that the change in the subcellular localization of the regulatory subunit of PI3K, induced after PDGF-R stimulation (Klippel et al. 1992; Gillham et al. 1999; Watton and Downward 1999), is required for PDGF-mediated activation of the Cdc42 pathway.
The mechanism by which PDGF induces Cdc42 activation probably involves an additional protein that directly controls the activation of this GTPase such as a GEF (Lim et al. 1996). It is possible that p85α, which contains a number of adaptor protein domains in its sequence (Fruman et al. 1998), brings a GTPase regulator into the complex that mediates Cdc42 activation. In addition, considering that p85 overexpression is sufficient to activate the Cdc42 pathway, p85 may protect Cdc42 from deactivation. Supporting this view, the experiments performed with different forms of the p85 regulatory subunit suggested that activation of the Cdc42 pathway by p85 required direct association of p85 and Cdc42. Alternatively, considering that a fraction of p85 localizes at the cell membrane (data not shown; Gillham et al. 1999; Watton and Downward 1999), p85 may mediate Cdc42 localization at the cell membrane, where many GEFs are found (for review see Hall 1998). The ability of p85 to enhance Cdc42 activation appeared specific, since expression of other molecules such as Pak-1 and N-WASP, which also bind active-Cdc42, did not trigger Cdc42-dependent cytoskeletal changes (data not shown; Manser et al. 1997; Miki et al. 1998). Therefore, p85 overexpression could enhance activation of Cdc42 by protecting it from deactivation or by stabilizing its membrane localization.
The ability of p85 to trigger Cdc42-dependent cytoskeletal changes represents a novel specific signaling capability of the PI3K regulatory subunit in the control of actin dynamics and cell migration, thereby opening a new perspective on the mechanism of action of class IA PI3Ks. Our data also confirm that constitutive activation of PI3K is sufficient to induce lamellipodium formation (Reif et al. 1996; Rodríguez-Viciana et al. 1997). Our view is that PI3K exerts two independent actions on the actin cytoskeleton. The first, described previously, is dependent on the PI3K activity that mediates Rac-dependent lamellipodium formation (Wennström et al. 1994a), and the other, described here, is dependent on the p85 regulatory subunit and induces activation of the Cdc42 pathway.
The greater ability of DN-Cdc42 to restore stress fiber and adhesion complex formation in PDGF-treated cells, compared with DN-Rac, suggests that a Cdc42-specific effector, rather than a Rac effector, is involved in mediating these actions. Nonetheless, we found that DN-Rac expression induced partial recovery of stress fibers in a small proportion of cells, suggesting that Rac effectors such as Pak-1 (effector of Rac and Cdc42; Zhao et al. 1998) could contribute moderately to mediating this effect. In addition, the atypical PKCs λ and ζ also mediate stress fiber loss induced by v-Cdc42 (Coghlan et al. 2000), and could potentially contribute to the PDGF-induced decrease in stress fibers. We considered N-WASP as a potential Cdc42 effector contributing to the decrease in actin fibers since it binds to the PDGF-R via Nck and grb2 adaptor molecules and is an essential mediator of filopodia formation (Miki et al. 1998; for review see Machesky and Insall 1999). We found that expression of an N-WASP mutant lacking the cofilin homologous region inhibited not only filopodia formation (not shown), as previously described (Miki et al. 1998), but also blocked the decrease in stress fibers induced by PDGF-R and p85α (Fig. 7).
WASP family members promote nucleation of new actin filaments through association with the Arp 2/3 complex, thereby triggering Cdc42-induced filopodia formation (Miki et al. 1998; Mullins et al. 1998; Zigmond 1998; Rohatgi et al. 1999). However, the mechanism through which N-WASP mediates the decrease in actin bundles and adhesion complexes remains unclear. N-WASP could mediate this effect directly, since N-WASP is proposed to sever actin polymers (Miki et al. 1996, Miki et al. 1998), however, this function of N-WASP remains controversial (Loisel et al. 1999; Machesky and Insall 1999). Alternatively, N-WASP's participation in decreasing stress fibers may be indirect and mediated via another protein associated to N-WASP (for review see Machesky and Insall 1999). The best candidate to mediate the decrease in actin stress fibers would be a protein directly associated to the cofilin region of N-WASP since deletion of this region blocks N-WASP's ability to decrease stress fibers. Alternatively, mutation of the cofilin region could alter N-WASP structure or subcellular localization, possibly affecting the action of proteins associated at other regions of N-WASP. Among the proteins known to bind N-WASP, profilin has been shown to suppress Rho-induced stress fibers (Suetsugu et al. 1999), and, thus, could be a good candidate effector for mediating the loss of actin bundles. Despite the open questions, the data shows that N-WASP mediates not only PDGF-induced Cdc42-dependent filopodium formation, but also contributes to induce the decrease in actin bundles and focal adhesions observed in cells treated with PDGF, illustrating an elegant mechanism by which N-WASP reorganizes F-actin. These results were obtained in NIH-3T3 cells. Similar results showing the ability of PDGF to activate the Cdc42/N-WASP pathway were obtained using murine embryonic fibroblasts (not shown), suggesting that this pathway is used by normal murine fibroblasts.
We present data showing that Cdc42/N-WASP activation is essential for PDGF-induced cell migration on collagen. The ability of inhibitory mutants of the Cdc42/N-WASP pathway to decrease PDGF-induced migration on collagen correlated with their ability to block PDGF-induced filopodia formation, a decrease in fibers, and a reduction in adhesion complexes. This suggests that Cdc42/N-WASP may regulate cell migration not only by inducing migratory structures such as filopodium, but also by diminishing strong adhesion. The relevance of the Cdc42 pathway in cell migration was also examined using p85 stable cell lines. It has been previously shown that under high overexpression conditions, p85 inhibits PI3K activity and membrane ruffling (Rodríguez-Viciana et al. 1997). However, the twofold p85 overexpression level in the p85 stable cell lines did not inhibit PI3K activation (Jiménez et al. 1998), and the constitutive induction of the Cdc42 pathway in these cells resulted in an increased cell migration in response to PDGF (Fig. 9). Stably transfected p110-CAAX cells, in contrast, exhibited a slightly decreased PDGF-induced migration. It is possible that the increased number of structures mediating strong adhesion in these cells (Fig. 4), impairs cell migration. Apart from the relevance of the Cdc42 pathway to control PDGF-induced migration, transient activation of the Rac pathway is essential for PDGF-induced migration (Fig. 9; Ridley et al. 1992). Thus, we propose that Cdc42-regulated pathways may cooperate with Rac-derived pathways to promote PDGF-induced migration. Cooperation has been observed at the level of migratory structures since interference with the Cdc42 pathway partially inhibited PDGF-induced Rac-dependent membrane ruffling (Fig. 2 and Fig. 8), showing that Cdc42 activation facilitates lamellipodium formation, in agreement with Nobes and Hall 1995. In addition, Cdc42 can cooperate with Rac to establish a polarized cell phenotype (Small et al. 1999; Nobes and Hall 1999). Finally, Cdc42 and Rac pathways cooperate to reduce Rho A–mediated cellular adhesion (discussed below). In conclusion, PDGF-induced migration is controlled both by the Cdc42 pathway and the Rac pathway, and both cascades probably cooperate to drive cell migration.
A number of recent observations point to the activation of Cdc42 and/or Rac as a general mechanism to inhibit Rho A–triggered cell responses. A recent report has demonstrated that a constitutively active Cdc42 mutant (F28ACdc42) inhibited the GTP loading of Rho A and that PDGF also inhibited Rho A GTP loading (Sander et al. 1999). Our study showing that PDGF activates Cdc42 offers a link between these observations, suggesting that PDGF inhibits Rho A via Cdc42. We propose that Cdc42 inhibits Rho A not only at the level of GTP loading, as proposed by Sander et al. 1999, but also through the action of effectors such as N-WASP that control actin polymerization. This is supported by the observation that p85α overexpression, via Cdc42, blocks actin stress fiber formation induced by the constitutively active mutant v-Rho. Cdc42 activation after bradykinin receptor ligation also inhibited LPA-induced stress fibers (data not shown). In addition, Rac activation by Tiam-1 inhibits Rho A-GTP loading and Rho A–mediated phosphorylation of the myosin II heavy chain (Sander et al. 1999; van Leeuwen et al. 1999). Finally, the effector of v-Cdc42 and v-Rac, Pak-1, also contributes to mediating inhibition of actin stress fibers (Manser et al. 1997; Zhao et al. 1998). Together, all these observations indicate that Rho-mediated cellular actions are counteracted by Cdc42/Rac pathways using several mechanisms. The inhibition of Rho pathways when Cdc42 and Rac pathways are activated may result in a decrease in strong adhesion, when migratory structures, such as filopodia and lamellipodia, are induced, thereby facilitating cell migration.
In conclusion, PDGF activates Cdc42 and its downstream effector N-WASP, which in turn influences actin dynamics not only by triggering filopodia formation, but also by decreasing stress fibers and focal adhesions. In addition, our data support that the p85α regulatory subunit of PI3K is an upstream regulator of Cdc42 in PDGF-R signaling. These observations demonstrate that PDGF activates Cdc42 and ascribe a novel function to the PI3K regulatory subunit in actin cytoskeleton regulation. Induction of the PDGF/p85/Cdc42/N-WASP pathway contributes to the regulation of adhesion and motility, which are processes required for physiological migration and metastatic spread of tumor cells.
Acknowledgments
The authors wish to thank Drs. H. Miki and T. Takenawa for donating N-WASP Ab and cDNA, Dr. J. Downward for the v-Rho A cDNA, Dr. D. Waterfield for the anti-p110β Ab, Dr. A. Hall for the gift of the Rho family members cDNAs, Drs. P. Hawkins and L. Claesson-Welsh for the PAE cells expressing wt and Phe740/Phe751 PDGF-R, and Dr. J. Cooper for the plasmids encoding wt and Phe740/Phe751 PDGF-R. We also thank Dr. V. Calvo, Dr. A. Eguinoa, and C. Mark for critical reading.
This work was supported by grants from the Spanish Dirección General de Ciencia y Desarrollo Tecnológico, Pharmacia & Upjohn, Community of Madrid and from the Spanish Dirección General de Formación y Promoción del Conocimiento. The Department of Immunology and Oncology was founded and is supported by the Spanish Research Council (CSIC) and the Pharmacia Corporation.
Footnotes
Abbreviations used in this paper: Ab, antibody; DN, dominant negative; ECM, extracellular matrix; F-actin, filamentous actin; GEF, guanine nucleotide exchange factors; N-WASP, N-Wiskott-Aldrich Syndrome family protein; LPA, lysophosphatidic acid; PDGF, platelet-derived growth factor; PDGF-R, PDGF receptor; PI3K, phosphoinositide 3-kinase.
References
- Aspenstrom P. Effectors for the Rho GTPases. Curr. Opin. Cell. Biol. 1999;11:95–102. doi: 10.1016/s0955-0674(99)80011-8. [DOI] [PubMed] [Google Scholar]
- Bockus B.J., Stiles CD. Regulation of cytoskeletal architecture by platelet-derived growth factor, insulin and epidermal growth factor. Exp. Cell. Res. 1984;153:186–197. doi: 10.1016/0014-4827(84)90460-9. [DOI] [PubMed] [Google Scholar]
- Burridge K., Fath K., Kelly T., Nuckolls G., Turner C. Focal adhesionstransmembrane junctions between the extracellular matrix and the cytoskeleton. Annu. Rev. Cell Biol. 1988;4:487–525. doi: 10.1146/annurev.cb.04.110188.002415. [DOI] [PubMed] [Google Scholar]
- Coghlan M.P., Chou M.M., Carpenter C.L. A typical protein kinase Cλ and ζ associate with the GTP-binding protein Cdc42 and mediate stress fiber loss. Mol. Cell. Biol. 2000;20:2880–2889. doi: 10.1128/mcb.20.8.2880-2889.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutartre H., Davoust J., Gorvel J.P., Chavrier P. Cytokinesis and redistribution of actin-cytoskeleton regulatory components in cells expressing the Rho GTPase CDC42Hs. J. Cell Sci. 1996;109:367–377. doi: 10.1242/jcs.109.2.367. [DOI] [PubMed] [Google Scholar]
- Evan G.I., Lewis G.K., Ramsay G., Bishop J.M. Isolation of monoclonal antibodies specific for human c-myc protooncogene product. Mol. Cell. Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D.A., Cantley L.C., Carpenter C.L. Structural organization and alternative splicing of the murine phosphoinositide 3-kinase gene. Genomics. 1996;37:113–121. doi: 10.1006/geno.1996.0527. [DOI] [PubMed] [Google Scholar]
- Fruman D.A., Meyers R.E., Cantley L.C. Phosphoinositide kinases. Annu. Rev. Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Gillham H., Golding M., Pepperkok R., Gullick W.J. Intracellular movement of green fluorescent protein-tagged phosphatidylinositol 3-kinase in response to growth factor receptor signaling. J. Cell Biol. 1999;146:869–880. doi: 10.1083/jcb.146.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hawkins P.T., Eguinoa A., Qiu R.-G., Stokoe D., Cooke F.T., Walters R., Wennström S., Claesson-Welsh L., Evans T., Symons M., Stephens L. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr. Biol. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- Heldin C.M., Ostman A., Ronnstrand L. Signal transduction via platelet derived growth factor receptors. Biochem. Biophys. Acta. 1998;1378:79–113. doi: 10.1016/s0304-419x(98)00015-8. [DOI] [PubMed] [Google Scholar]
- Hooshmand-Rad R., Hájkova L., Klint P., Karlsson R., Vanhaesebroek B., Claesson-Welsh L., Heldin C.-H. The PI3-kinase isoforms p110α and p110β have differential roles in PDGF- and insulin-mediated signaling. J. Cell Sci. 2000;113:207–214. doi: 10.1242/jcs.113.2.207. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A., Sandborg R.R., Horwitz A.F. Adhesion in cell migration. Curr. Opin. Cell Biol. 1995;7:697–706. doi: 10.1016/0955-0674(95)80112-x. [DOI] [PubMed] [Google Scholar]
- Hynes R.O. Integrinsversatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Jiménez C., Jones D.R., Rodríguez-Viciana P., González A., Leonardo E., Wennström S., von Kobbe C., Torán J.L., L.R-Borlado V., Calvo Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:743–753. doi: 10.1093/emboj/17.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A., Escobedo J.A., Fantl W.J., Williams L.T. The C-terminal SH2 domain of p85 accounts for the high affinity and specificity of the binding of phosphatidylinositol 3-kinase to phosphorylated platelet-derived growth factor beta receptor. Mol. Cell. Biol. 1992;12:1451–1459. doi: 10.1128/mcb.12.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R., Ahmed S., Best A., Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol. 1995;15:1942–1952. doi: 10.1128/mcb.15.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundra V., Escobedo J.A., Kazlauskas A., Kim H.K., Rhee S.G., Williams L.T., Zetter B.R. Regulation of chemotaxis by the platelet-derived growth factor receptor β. Nature. 1994;367:474–476. doi: 10.1038/367474a0. [DOI] [PubMed] [Google Scholar]
- Lim L., Manser E., Leung T., Hall C. Regulation of phosphorylation pathways by p21 GTPases. The p21 Ras-related Rho subfamily and its role in phosphorylation signalling pathways. Eur. J. Biochem. 1996;242:171–185. doi: 10.1111/j.1432-1033.1996.0171r.x. [DOI] [PubMed] [Google Scholar]
- Loisel T.P., Boujemaa R., Pantaloni D., Carlier M.-F. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- Machesky L.M., Insall R.H. Signaling to actin dynamics. J. Cell Biol. 1999;146:267–272. doi: 10.1083/jcb.146.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser E., Huang H.-Y., Loo T.-H., Chen X.-Q., Dong J.-M., Leung T., Lim L. Expression of constitutively active α-Pak reveals effects of the kinase on actin and focal complexes. Mol. Cell. Biol. 1997;17:1129–1143. doi: 10.1128/mcb.17.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H., Miura K., Takenawa T. N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:5326–5335. [PMC free article] [PubMed] [Google Scholar]
- Miki H., Sasaki T., Takai Y., Takenawa T. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-Wasp. Nature. 1998;391:93–96. doi: 10.1038/34208. [DOI] [PubMed] [Google Scholar]
- Mullins R.D., Heuser J.A., Pollard T.D. The interaction of Arp2/3 complex with actinnucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes C.D., Hall A. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Nobes C.D., Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayotou G., Waterfield M.D. Phosphatidylinositol 3-kinase a key enzyme in diverse signaling pathways. Trends Cell Biol. 1992;2:358–360. doi: 10.1016/0962-8924(92)90042-l. [DOI] [PubMed] [Google Scholar]
- Reif K., Nobes C.D., Thomas G., Hall A., Cantrell D.A. Phosphatidylinositol 3-kinase signals activate a selective subset of Rac/Rho-dependent effector pathways. Curr. Biol. 1996;6:1445–1455. doi: 10.1016/s0960-9822(96)00749-x. [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Hall A. Signal transduction pathways regulating Rho-mediated stress fibre formationrequirement for a tyrosine kinase. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:2600–2610. doi: 10.1002/j.1460-2075.1994.tb06550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A.J., Paterson H.F., Johnston C.L., Diekmann D., Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Viciana P., Warne P.H., Khwaja A., Marte B.M., Pappin D., Das P., Waterfield M.D., Ridley A., Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M.C. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- Sander E.E., van Delft S., ten Klooster J.P., Reid T., van der Kammen R.A., Michiels F., Collard J.G. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell–cell adhesion or cell migration and is regulated by phosphoinositol 3-kinase. J. Cell Biol. 1998;143:1385–1398. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander E.E., ten Klooster J.P., van Delft S., van der Kammen R.A., Collard J.G. Rac downregulates rho activityreciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 1999;147:1009–1021. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small J.V., Rottner K., Kaverina I. Functional design in the actin cytoskeleton. Curr. Opin. Cell. Biol. 1999;11:54–60. doi: 10.1016/s0955-0674(99)80007-6. [DOI] [PubMed] [Google Scholar]
- Suetsugu S., Miki H., Takenawa T. Distinct roles of profilin in cell morphological changesmicrospikes, membrane ruffles, stress fibers and cytokinesis. FEBS (Fed. Exp. Biochem. Soc.) Lett. 1999;457:470–474. doi: 10.1016/s0014-5793(99)01086-8. [DOI] [PubMed] [Google Scholar]
- Symons M., Derry J.M., Karlak B., Jiang S., Lemahieu V., McCormick F., Francke U., Abo A. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. 1996;84:723–734. doi: 10.1016/s0092-8674(00)81050-8. [DOI] [PubMed] [Google Scholar]
- Tolias K.F., Cantley L.C., Carpenter C.L. Rho family GTPases bind to phosphoinositide kinases. J. Biol. Chem. 1995;270:17656–17659. doi: 10.1074/jbc.270.30.17656. [DOI] [PubMed] [Google Scholar]
- Van Aelst L., Joneson T., Bar-Sagi D. Identification of a novel Rac-1-interacting protein involved in membrane ruffling. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3778–3786. [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroek B., Jones G.E., Allen W.E., Zicha D., Hoosman-Rad R., Sawyer C., Wells C., Waterfield M.D., Ridley A.J. Distinct PI(3)Ks mediate mitogenic signalling and cell migration in macrophages. Nat. Cell Biol. 1999;1:69–71. doi: 10.1038/9045. [DOI] [PubMed] [Google Scholar]
- van Leeuwen F.N., van Delft S., Kain H.E., van der Kammen R.A., Collard J.G. Rac regulates phosphorylation of the myosin-II heavy chain, actinomyosin disassembly and cell spreading. Nat. Cell Biol. 1999;1:242–248. doi: 10.1038/12068. [DOI] [PubMed] [Google Scholar]
- Watton S.J., Downward J. Akt/PKB localisation and 3-phosphoinositide generation at sites of epithelial cell-matrix and cell-cell interaction. Curr. Biol. 1999;9:433–436. doi: 10.1016/s0960-9822(99)80192-4. [DOI] [PubMed] [Google Scholar]
- Wennström S., Hawkins P., Cooke F., Hara K., Yonezawa K., Kasuga M., Jackson T., Claesson-Welsh L., Stephens L. Activation of phosphoinositide 3-kinase is required for PDGF-stimulated membrane ruffling Curr. Biol 4 1994. 385 396a [DOI] [PubMed] [Google Scholar]
- Wennström S., Siegbahn A., Yokote K., Arvidsson A.-K., Heldin C.-H., Mori S., Claesson-Welsh L. Membrane ruffling and chemotaxis transduced by the PDGF β-receptor require the binding site for phosphatidylinositol 3-kinase Oncogene. 9 1994. 651 660b [PubMed] [Google Scholar]
- Zhao A.S., Manser E., Chen X.Q., Chong C., Leung T., Lim L. A conserved negative regulatory region in alpha PAKinhibition of PAK kinases reveals their morphological roles downstream of Cdc42 and Rac. Mol. Cell. Biol. 1998;18:2153–2163. doi: 10.1128/mcb.18.4.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Bagrodia S., Cerione R.A. Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85. J. Biol. Chem. 1994;269:18727–18739. [PubMed] [Google Scholar]
- Zigmond S.H. Actin cytoskeletonthe Arp2/3 complex gets to the point. Curr. Biol. 1998;8:R654–R657. doi: 10.1016/s0960-9822(07)00415-0. [DOI] [PubMed] [Google Scholar]