

Abstract
Many viral fusion proteins exhibit a six-helix bundle as a core structure. HIV Env–induced fusion was studied to resolve whether membrane merger was due to the transition into the bundle configuration or occurred after bundle formation. Suboptimal temperature was used to arrest fusion at an intermediate stage. When bundle formation was prevented by adding inhibitory peptides at this stage, membranes did not merge upon raising temperature. Inversely, when membrane merger was prevented by incorporating lysophosphatidylcholine (LPC) into cell membranes at the intermediate, the bundle did not form upon optimizing temperature. In the absence of LPC, the six-helix bundle did not form when the temperature of the intermediate was raised for times too short to promote fusion. Kinetic measures showed that after the temperature pulse, cells had not advanced further toward fusion. The latter results indicate that bundle formation is the rate-limiting step between the arrested intermediate and fusion. Electrical measures showed that the HIV Env–induced pore is initially large and grows rapidly. It is proposed that bundle formation and fusion are each contingent on the other and that movement of Env during its transition into the six-helix bundle directly induces the lipid rearrangements of membrane fusion. Because peptide inhibition showed that, at the intermediate stage, the heptad repeats of gp41 have become stably exposed, creation of the intermediate could be of importance in drug and/or vaccine development.
Keywords: fusion pore, fusion intermediates, inhibitory peptides, fusion kinetics, hemifusion
Introduction
It has been observed that many viral fusion proteins share a common crystallographic structure: a six-helix bundle with each monomer of a trimer set in a hairpin bend (Skehel and Wiley 1998). This observation leads to the possibility that many fusion proteins induce fusion by essentially the same mechanism. The Env protein of HIV is among the class of proteins that form six-helix bundles.
HIV Env is a trimer, with each monomer consisting of two subunits, gp120 and gp41 (Skehel and Wiley 1998; Wyatt and Sodroski 1998). gp120 binds to a CD4 receptor on target membranes and undergoes conformational changes that allow gp120 to then interact with chemokine receptors, the coreceptors on the target membrane (Berger et al. 1999). A cascade of conformational changes ensues, culminating in gp41 inducing fusion of the viral envelope with the plasma membrane, thereby initiating infection. Each gp41 contains an NH2-terminal leucine/isoleucine heptad repeat (HR) segment (HR1; Fig. 1) that has been crystallographically shown to form a central triple-stranded α-helical coiled coil core (Chan et al. 1997; Weissenhorn et al. 1997). The coiled coil creates highly conserved grooves that are transiently exposed during the fusion process (Chan and Kim 1998; Furuta et al. 1998; Munoz-Barroso et al. 1998). Exposure permits a COOH-terminal HR segment (HR2; Fig. 1) to pack into the grooves in an antiparallel orientation, forming the core gp41 configuration; at this point, HR1 and HR2 do not undergo any further conformational changes and a six-helix bundle with each gp41 in a hairpin bend has been created.
Figure 1.

Schematic of location of HR1 and HR2 within HIV-1 gp41 and the hairpin bend of a monomer within a six-helix bundle. FP, NH2-terminal fusion peptide; TM, transmembrane domain.
Mutations that weaken the interactions between HR1 and HR2 hinder fusion, and synthetic peptides with amino acid sequences of the NH2- and COOH-terminal HRs prevent the formation of the six helix-bundle and thereby abolish fusion for HIV and other viruses (Wild et al. 1994; Chan et al. 1998; Joshi et al. 1998). Development of in vitro resistance to the synthetic peptide T20 has been demonstrated to be due to mutations in HR1 (Rimsky et al. 1998). Thus it has been shown that the six-helix bundle is in some way critical for fusion to proceed for several viruses. The significance of fusion proteins assembled into coiled coil bundles may even extend to eukaryotic cells: a complex of soluble N-ethylmaleimide–sensitive factor (NSF) attachment protein receptors (SNAREs), thought to be responsible for intracellular fusion, has been crystallographically shown to form bundle structures (Poirier et al. 1998; Sutton et al. 1998).
Membrane merger is the central and defining event of fusion. In viral fusion, it has often been assumed that the six-helix bundle forms before fusion and serves to bring the membranes into close apposition (Weissenhorn et al. 1997; Chan and Kim 1998). An alternate possibility, however, is that the free energy released as fusion proteins reconfigure into the six-helix bundle induces fusion, and by the time the bundle has formed, fusion pores have resulted (Baker et al. 1999). It is pivotal to pinpoint the role of the six-helix bundle in fusion to understand the process of membrane merger, but direct determination of the precise order of events is difficult because the lipid and protein rearrangements during fusion are transient and local. We devised an experimental approach to determine the temporal order of formation of the six-helix bundle and the fusion pore based on the following logic: if the six-helix bundle itself causes fusion, the bundle must be able to form before fusion pore formation; if the bundle cannot form before the membranes fuse, it cannot be the cause of merger. Through this approach we demonstrate that for HIV Env protein the six-helix bundles do not form before membrane fusion. Therefore, fusion is caused by the movement of protein into the six-helix bundle rather than by the six-helix bundle itself.
Materials and Methods
Materials
Soluble CD4 (sCD4, from Dr. R. Sweet, SmithKline Beecham, Department of Surgical Biology, King of Prussia, PA) and anti-CD4 mAb Q4120 (from Dr. Q. Sattentau, Centre d'Immunologie de Marseille-Luminy, Marseillle, France; Healey et al. 1990) were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health. All fluorescent dyes were purchased from Molecular Probes, Inc. Oleic acid (OA), stearoyl-lysophosphatidylcholine (LPC), BSA, and poly-l-lysine were purchased from Sigma-Aldrich. The T22 peptide was obtained from Bachem Bioscience. To inhibit fusion between cells, we used the peptides T20 (corresponding to residues 643–678 of gp41) and T21 (residues 558–595). T20 and T21 (also known as DP178 and DP107, respectively) were synthesized as described previously (Lawless et al. 1996). The purity of the peptides was determined by analytical HPLC to be >95%. Concentrations of stock solutions containing the peptides were measured by the method of Edelhoch 1967.
Maintenance and Fluorescent Labeling of the Cells
TF228.1.16 cell line constitutively expressing gp160 of the T-tropic BH10 strain of HIV-1 (Jonak et al. 1993) was a gift from Dr. Z.L. Jonak (SmithKline Beecham, Philadelphia, PA). These cells, designated as effector cells, were grown in RPMI 1640 supplemented with 10% FBS (both from GIBCO BRL). The HelaT4+ cells expressing CD4 and chemokine receptor (CXCR4) (Maddon et al. 1986) were obtained through the AIDS Research and Reference Reagent Program. HelaT4+ cells were maintained in DMEM (GIBCO BRL) supplemented with 10% Cosmic™ serum (HyClone Laboratories) and 0.5 mg/ml of geneticin (GIBCO BRL) and were used as target cells for fusion experiments. The effector and target cells were labeled essentially as described (Munoz-Barroso et al. 1998; Melikyan et al. 2000). In brief, ∼4 × 106 TF228 cells/ml were labeled with 1.3 μM calcein AM. HelaT4+ cells (∼2 × 106) were colabeled with 30 μM of the cytoplasmic marker 7-amino-4-chloromethylcoumarin (CMAC) and, when required, with 15 μg/ml of the membrane dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI).
Fluorescence Microscopy Assay for Cell–Cell Fusion and Use of Inhibitory Agents
The fluorescently labeled effector and target cells were bound to each other in one of two ways. In the first protocol, 7–10 × 104 of both target and effector cells were mixed together in small microfuge tubes containing 0.2 ml of Hepes-buffered DMEM (pH 7.2) supplemented with 1 mg/ml of BSA and transferred into the wells of an 8-chambered glass slide (Lab-Tek®; Nunc) that was precoated with poly-l-lysine. The precoating allowed the cells to quickly and efficiently adhere to the glass and establish side by side contacts with each other. Side by side placement facilitated identification of bound cells and quantification of dye spread. In the second protocol, ∼8 × 104 target HelaT4+ cells per well were grown overnight, to ∼80–90% confluency, on noncoated glass in the 8-chambered slides. Cells were then loaded with CMAC but could not, as a practical matter, be labeled with DiI. About 2 × 105 effector TF228 cells were placed on top of the flattened target cells, and within 30 min the effector cells had adhered sufficiently well that they were not removed by multiple washings. This procedure yielded an ∼1:1 ratio of target to effector cells. The second protocol was used whenever the fusion-inhibitory peptide T20 had to be removed by washing. (T20 bound to both the uncoated and polylysine-coated glass slide and could not be effectively washed out, whereas T21 was efficiently removed from coated glass by washing.) Using the second protocol, both T20 and T21 were efficiently removed from the chamber by washing. For both protocols, once effector and target cells were bound to each other, the chambered slides were either incubated for 3 h at 22–24°C in the dark, followed by a 30–45-min incubation at 37°C or the temperature was immediately raised to 37°C and maintained for 2 h.
The extent of fusion was assessed by visual microscopic examination. As required, the fluorescence images were captured with an intensified (KS1380; Video Scope) CCD video camera (model 72; Dage-MTI, Inc.), were recorded on S-VHS format videotape, and the images were analyzed off-line (Qiao et al. 1999). Images of fluorescent cells were acquired separately for each fluorescent dye, digitized, and pseudocolored according to their emission wavelength. For cells bound to polylysine-coated glass, three images were then superimposed to detect whether fluorescent dyes had redistributed; fused cells had a lighter (pinkish) appearance. Cells partially overlapping in the bound state also present a lighter appearance, but are readily distinguishable from cells that have actually fused. The fraction of effector and target cells in contact that became stained with all 3 dyes (screening >100 cell pairs per well) provided the extent of fusion (Melikyan et al. 2000). For example, ∼50% of the contacting cells have fused in Fig. 2 B. When effector cells were placed upon target cells grown on untreated glass, the two images (one for each aqueous dye) were superimposed. The extent of fusion was quantified as the number of target cells that acquired calcein (from the effector cells) normalized by the total number of target cells in the field of view. When high-time resolution kinetic measurements for the onset of calcein spread between effector and target cells were required, a temperature jump (T-jump) technique was used (see below) to determine the cells that fused within 5 min of raising temperature (∼20% of the total cell pairs).
Figure 2.

A three-color fluorescent assay of fusion. The cells were adhered to polylysine-coated glass. Effector cells are green (calcein) and target cells are purple (a mixture of the red DiI and the blue CMAC). Fused cells are pinkish. (A) Fusion did not occur at TAS. (B) After raising the temperature to 37°C for 30 min, fusion occurred and all three dyes redistributed (arrowheads).
Agents that inhibited HIV Env–mediated fusion (antibodies, soluble CD4, and peptides) were added in three different ways: (a) to effector cells, followed by removal of the agent, before binding the effector cells to the target cells; (b) at the time the effector and target cells were brought together; or (c) after the intermediate of the temperature-arrested stage (TAS; see Results) was created. In separate experiments, these additions were performed singly or in combinations. Lipophilic agents (LPC, SDS, or OA freshly dissolved in PBS containing Ca2+ and Mg2+) were added at TAS and allowed to incorporate into cell membranes for 5–10 min at room temperature, followed by two washings with a phosphate-buffered saline just before incubation at 37°C. Unless stated otherwise, T20 or T21 was allowed to bind for 30–60 min at 23°C.
Inducing Rapid Increases in Temperature
We employed a water-cooled Peltier device (20/20 Technology) to maintain the temperature in the experimental chamber between 4°C and 37°C. A T-jump, an increase of temperature of a small volume (∼1 μl) of the chamber, was achieved by illuminating with an infrared (IR) laser diode (Opto Power Corp.) as described (Melikyan et al. 2000). To selectively heat a cell pair of interest and its surrounding volume, we mounted IR-absorbing glass to the bottom of the experimental chamber. Cells were placed on the IR-absorbing glass, and a cell pair was positioned in the center of illumination of the fixed laser beam. The temperature of the solution immediately bathing an illuminated cell was determined by measuring the changes in the resistance of a second patch pipette manipulated within 10 μm of the cell. The changes of the pipette resistance were converted into temperature with use of a calibration curve obtained from the relation between the pipette resistances and temperatures of a bulk solution in the temperature-controlled chamber. A steady state temperature was established within 2–4 s upon illumination. We validated the use of pipette resistance by using hydrocarbons with known melting temperature. A steady state temperature of 37°C achieved by IR illumination, according to pipette resistance, melted eicosane (melting point 36–38°C; Sigma-Aldrich), but not henecosane (melting point 40–42°C), over an area with diameter ∼300 μm.
Electrical Measurements of the Fusion Pore
A coverslip with cells at TAS was transferred into a chamber and maintained at 12–14°C. The effector cells were patch clamped in the whole-cell mode, and time-resolved admittance (i.e., “capacitance”) measurements were performed and pore conductance was calculated as described previously (Melikyan et al. 1999; Qiao et al. 1999). Cell–cell fusion was triggered by quick, local increase of temperature to 37°C by our T-jump method. Average conductances were determined by aligning pores at their moment of opening and calculating the mean pore conductance every 5 ms. Pore conductance was converted to approximate pore diameter by using the conductivity of the bulk solutions and assuming a pore length of 15 nm.
Online Supplemental Material
Supplemental figures are supplied in which both the images (Figure S1) and the graphs (Figure S2) demonstrate that if T20 or T21 blocked pore formation, it also blocked lipid dye transfer. All supplementary materials are available at http://www.jcb.org/cgi/content/full/151/2/413/DC1.
Results
We used a suboptimal temperature to arrest fusion at an intermediate stage preceding (i.e., upstream of) lipid mixing and pore formation. Our strategy was to arrest the fusion process as close as we could to the point of pore formation and then manipulate conditions from this stage in order to separate six-helix bundle formation and membrane merger from a multitude of prior steps. We showed that when temperature was subsequently increased, the intermediate proceeded on to fusion. We tested for bundle formation at the temperature-arrested intermediate with peptides that inhibit fusion when they bind to the grooves on the surface of the central coiled coil core (T20, also known as DP178) and to HR2 (T21, also known as DP107). Whether the bundle could still form when lipid merger was prevented was tested by inserting LPC into membranes after establishing the intermediate and then raising the temperature. We also tested in another manner (without the need to add exogenous lipid) whether the six-helix bundle might form before the creation of a fusion pore by examining whether temperature increases maintained for short times could induce bundle formation without causing fusion. The relation between bundle formation and membrane merger could be determined by this logical design of experiments, whichever temporal sequence proved to be the case.
Creating a Temperature-arrested Intermediate of Fusion
We used a stable cell line constitutively expressing an HIV Env (from BH10 strain) that requires CXCR4 chemokine receptors on CD4+ target cells in order for fusion to occur. Calcein-labeled Env-expressing cells (we define these as effector cells) and CD4+/CXCR4+ target cells labeled with DiI and CMAC were plated onto polylysine-coated glass, or alternatively, the effector cells were laid on top of target cells that had been grown overnight and then fluorescently labeled with CMAC. We used the temperature sensitivity of Env-induced fusion (Frey et al. 1995; Hart et al. 1996) to arrest fusion at an intermediate stage. Cells maintained at 23°C showed neither aqueous nor membrane dye transfer (Fig. 2 A) even up to 5 h. However, upon raising the temperature to 37°C, both aqueous and membrane dye mixed efficiently (Fig. 2 B). We refer to the stage reached after maintaining 23°C (usually for 3 h) as the temperature-arrested stage, TAS, of fusion.
To establish a reference to which TAS could be compared, cells were maintained at 37°C and at a set time were placed on ice to stop further fusion activity. (In control experiments, we verified that this method did arrest further fusion: dye did not spread between additional cells for 1 h after lowering the temperature to 4°C; not shown.) After fusion was stopped, the kinetics of fusion was obtained by microscopically counting the percentage of cell pairs that had exchanged fluorescent dye at a given time (Fig. 3 A, open squares). Fusion was slow: a significant lag time, ∼15–20 min (in agreement with previous studies; Frey et al. 1995; Weiss et al. 1996), preceded the onset of dye spread, and it took ∼2 h to reach its maximum extent.
Figure 3.
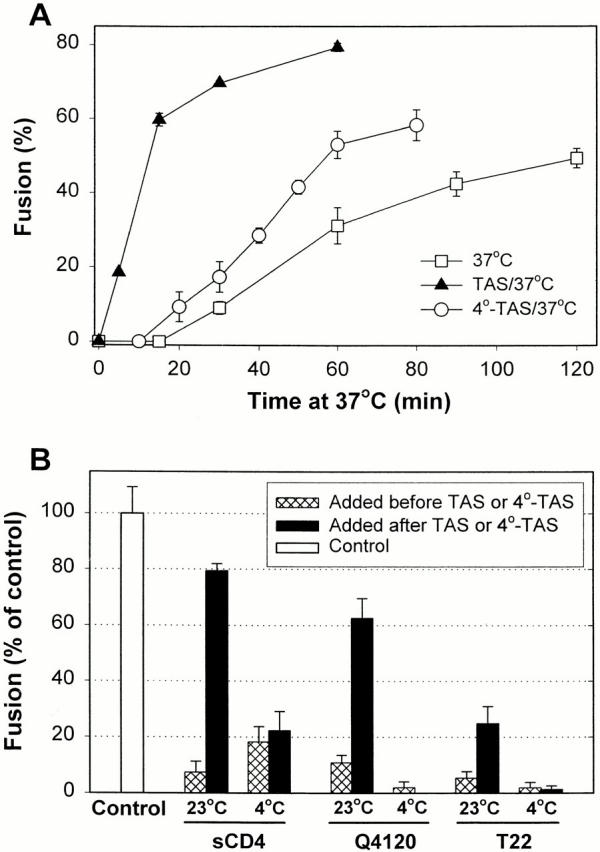
(A) The fraction of cells in contact that fuse as a function of time at 37°C. Cells were either coincubated directly at 37°C (□), or TAS (▴) or 4°-TAS (○) was first established and temperature was then raised to 37°C. Error bars show the standard error of four to eight experiments. (B) The ability of agents to block fusion through binding to gp120, CD4, or CXCR4. In control experiments (first bar), TAS was created and temperature was then brought to 37°C for 45 min. sCD4, a neutralizing antibody against CD4 (Q4120), or a peptide that binds to CXCR4 (T22) were added either at the beginning of coincubating the effector and target cells for 3 h at 23°C (23°C, cross-hatched bars) or after establishing TAS by a 2-h coincubation and then allowing 1 h at 23°C for the agents to bind (black bars). Alternatively, the inhibitory agents were added either before (4°C, cross-hatched bars) or after a 2-h coincubation (establishing 4°-TAS) of cells at 4°C (black bars). The extent of fusion was normalized by the control experiments without inhibitory agents. The concentrations of the agents were: 50 μg/ml of sCD4, 40 μg/ml of Q4120, and 20 nM of T22 peptide.
In contrast, when temperature was raised to 37°C at the point of TAS, fusion occurred quickly, by the earliest time measured (5 min) and reached its maximal level in <60 min (Fig. 3 A, filled triangles; actual percentage of bound cells that fused is shown). Coincubating cells at 23°C for a longer time (i.e., 5 h) did not yield faster fusion (data not shown). When cells were preincubated at 4°C for 3 h (establishing 4°-TAS), the fusion that occurred upon raising temperature to 37°C was much slower than fusion from TAS (Fig. 3 A, open circles). The lag time until fusion was comparable to that for cells maintained at 37°C from the point of binding, but kinetics was somewhat faster after the lag period. The absence of a substantial lag time in the case of TAS compared with that when temperature was maintained at 37°C throughout clearly shows that for TAS, the steps causing the appreciable lag time must have already been traversed. (Lowering temperature of cells at TAS to 4°C did not cause them to revert to an earlier state in the fusion process: the kinetics of fusion upon raising temperature to 37°C was the same as from TAS; data not shown.)
Membrane and Protein Conformational Changes En Route to TAS
Although the fusion process had been advanced by creating TAS, lipid mixing had not yet occurred (Fig. 2 A). Adding 0.5 mM chlorpromazine (known to promote fusion between hemifused pairs of influenza virus hemagglutinin (HA)-expressing cells and erythrocytes; Melikyan et al., 1997; Chernomordik et al., 1998) at the point of TAS did not promote lipid or aqueous content mixing (not shown). The complete absence of lipid dye spread even after the addition of chlorpromazine shows that hemifusion most probably has not been established at TAS.
We assessed how far gp120/gp41 has proceeded toward fusion by the point of TAS. Adding sCD4, or an mAb against CD4 that competes with gp120 binding (Q4120), or a peptide (T22) that binds to CXCR4, at the start of coincubating the effector and target cells at 23°C strongly suppressed Env-mediated fusion (Fig. 3 B, cross-hatched bars, 23°C). The results we found in this control were as expected, because these agents are known to protect against infection by HIV-1 and inhibit Env-induced cell–cell fusion (Healey et al. 1990; Endres et al. 1996; Murakami et al. 1999). However, sCD4 and Q4120 inhibited fusion much less when added at TAS (Fig. 3 B, black bars, 23°C), demonstrating that either TAS is a post–CD4-binding stage or the binding sites of CD4 and/or gp120 become less accessible. Adding T22 at TAS inhibited fusion more effectively than did sCD4 or Q4120, but not as well as when added before TAS. This indicates that CXCR4-dependent steps may not have been completed or the association between gp120 and the coreceptor is still reversible (Doranz et al. 1999) at TAS. When effector and target cells were maintained at 4°C for 3 h to create 4°-TAS, all three agents abolished fusion regardless of whether they were added at the onset of the coincubation (cross-hatched bars, 4°C) or at 4°-TAS (black bars, 4°C). The greater inhibition by each of these agents at 4°-TAS than at TAS demonstrates that 4°-TAS is upstream of TAS and has not significantly advanced in the fusion process; this is in agreement with the kinetic measurements (Fig. 3 A).
The HRs of gp41 Are Stably Exposed at TAS
The peptides T20 and T21 cannot bind to either the native structure of gp41 or the final six-helix bundle; they can only bind to an intermediate conformation(s) of gp41 (Chen et al. 1995; Furuta et al. 1998; Kliger and Shai 2000). When T20 or T21 was added to effector cells at 23°C (in the absence of target cells) and then unbound peptides were washed away, fusion was not inhibited when the effector cells were subsequently bound to target cells (grown on glass slides) and the temperature was raised to 37°C (Fig. 4 A, second and fourth bars). Neither CD4 nor CXCR4 was present when the peptides were added to effector cells, and as expected gp41 remained in its native state. For many strains (including BH10), binding sCD4 to gp120 is sufficient to expose the grooves created by HR1 to T20, and HR2 to T21 (Furuta et al. 1998). We refer, for convenience, to states of HIV Env that will allow T20 and T21 to bind as “activated.” For our system, adding the peptides to effector cells (again, without target cells) in the presence of sCD4 at 23°C significantly inhibited subsequent fusion to the target cells after the sCD4 and unbound peptides were washed away (Fig. 4 A, third and fifth bars). We showed that this inhibition was caused by binding of the peptides rather than by sCD4-induced shedding of gp120 or other adverse effects: adding (and then washing out) a high concentration of sCD4 alone to effector cells did not suppress fusion to target cells (Fig. 4 A, sixth bar). Thus, sCD4 alone is sufficient to activate gp41 at 23°C.
Figure 4.
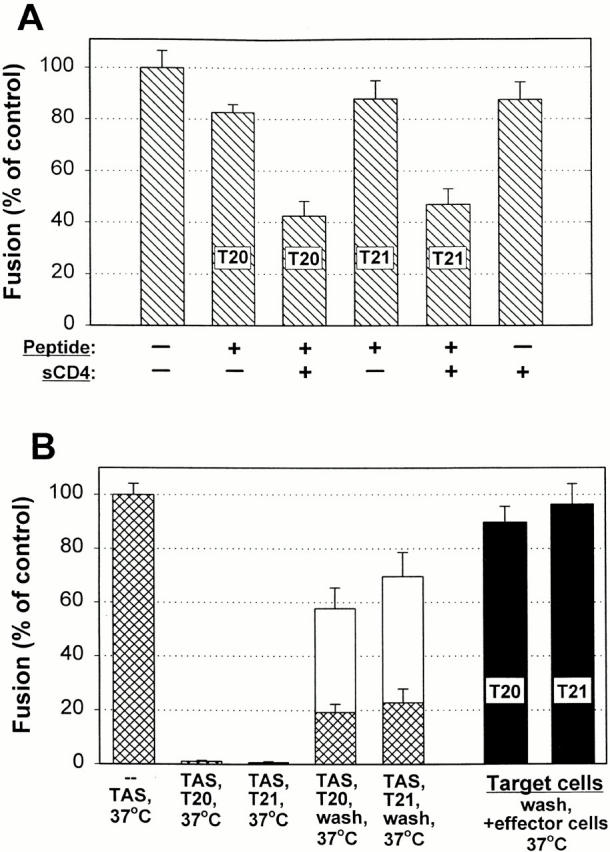
Using T20 and T21 to assess formation of six-helix bundles. (A) T20 and T21 inhibit fusion after sCD4 binding in the absence of target cells. Effector TF228 cells were treated with agents as indicated and the agents were removed by washing; the effector cells were then bound to target HelaT4 cells grown on untreated glass, and fusion was measured after raising temperature to 37°C for 2 h. When adding T20 and T21 to effector cells alone, fusion occurred to the same extent as for the control (first bar). Adding sCD4 alone did not affect fusion (sixth bar), but the addition did expose the HRs to T20 (third versus second bar) and T21 (fifth versus fourth bar). The peptides and sCD4 were added simultaneously. (B) T20 and T21 inhibited fusion after creating TAS and the peptides remained bound to gp41 for long times. Effector cells were laid on top of target cells grown on slides and TAS was created. After a mock wash (without adding peptides), raising temperature to 37°C for 30 min led to fusion (first bar). (The extent of fusion was the same when the mock wash was omitted.) Adding T20 or T21 after TAS was created abolished fusion (second and third bars). Fusion was significantly reduced even after washing out unbound T20 or T21 and immediately raising temperature to 37°C (fourth and fifth cross-hatched bars). Raising temperature 1 h after washing led to a greater, but still significantly impeded, extent of fusion (white bars above cross-hatches). In control experiments, T20 or T21 (black bars) were incubated for 1 h at 23°C with target cells grown on slides, the unbound peptides were removed by washing, the effector cells were bound, and temperature was raised to 37°C for 2 h, fusion was not inhibited. The concentrations of agents were: 50 μg/ml of sCD4, 40 nM T20, and 220 nM T21.
As a control, T20 and T21 were added at the beginning of a 3-h period of effector and target cell coincubation at 23°C. As expected (Munoz-Barroso et al. 1998), both peptides efficiently inhibited fusion upon subsequent elevation of the temperature to 37°C (data not shown). These peptides completely inhibited fusion when added after the 3-h effector and target cell incubation at 23°C (i.e., at TAS) before raising temperature to 37°C (Fig. 4 B, second and third bars). Adding the peptides at TAS and then washing to remove peptides free in solution led to only a small extent of fusion when temperature was immediately raised to 37°C (Fig. 4 B, fourth and fifth cross-hatched bars). Waiting for 1 h after washing to allow the peptides to further dissociate before raising temperature led to a greater extent of fusion, but still appreciably less than if the peptides had not been added (Fig. 4 B, white bars). We performed an additional control against the peptides inhibiting fusion by means other than binding to gp41: T20 or T21 were added to target cells grown on slides, the peptides were washed out, and only then were the effector cells placed on the target cells. Full fusion resulted upon raising temperature (Fig. 4 B, black bars). Thus, the peptides were effectively removed from target cells and effector cells (Fig. 4 A) by our washing procedures. In summary, even though fusion does not occur at 23°C, after gp120 binds to either target cells or sCD4, HR1 and HR2 of gp41 are exposed to T20 and T21, respectively, and the peptides dissociate from these sites, but slowly.
We found that if T20 and T21 blocked pore formation, they also abolished lipid dye mixing. That is, for all concentrations of inhibitory peptides, they always blocked the process before any lipid dye spread (see Figures S1 and S2 at http://www.jcb.org/cgi/content/full/151/2/413/DC1), which would have signaled hemifusion of cell membranes. (This contrasts to the findings of Munoz-Barroso et al. 1998, who reported that low concentrations of T20, but not T21, blocked aqueous dye mixing while still permitting lipid dye transfer.) Hemifusion is the merger of outer monolayer membrane leaflets with the inner leaflets contacting each other to form a hemifusion diaphragm that continues to separate aqueous contents.
The Formation of the Six-Helix Bundle Requires Membrane Merger
Preventing Membrane Merger with LPC Abolishes Bundle Formation.
LPC inhibits fusion in a wide variety of systems by preventing the merger of contacting lipid monolayer leaflets of membranes (i.e., hemifusion). It is thought to do so because of its positive spontaneous curvature (Chernomordik et al. 1995). Whether or not hemifusion does indeed occur before fusion, in general LPC arrests fusion at the point of membrane merger (Chernomordik et al. 1998) as a lipid constituent of membranes and not by binding to proteins. Stearoyl-LPC (LPC) is particularly useful because it is sufficiently hydrophobic to remain in membranes after any LPC still in solution is removed, or, if necessary, it can be removed from the membranes with a BSA wash (Chernomordik et al. 1997). We added LPC after TAS was established and removed the unbound fraction, using cells labeled with only aqueous dyes so that lipid composition would not be affected by incorporation of lipid dyes. Temperature was raised to 37°C for 15 min, by which time the majority of cell pairs would have fused if LPC had not been added (Fig. 3 A). We then lowered temperature to 23°C, and found that pore formation was meager (Fig. 5 A, second bar). We refer to the stage reached after lowering temperature back to 23°C as a lipid-arrested stage (LAS) of fusion (Fig. 5 A, inset). Fusion still did not occur when the membrane-bound LPC was removed at the point of LAS (data not shown). But fusion occurred efficiently when temperature was again brought to 37°C (Fig. 5 A, third bar); the extent was comparable to the control (first bar) in which LPC had not been added but cells were washed in the same way as if it had. This high level of fusion demonstrates that the action of LPC was reversible and that gp41 was not inactivated when LAS was created. Significantly, blocking fusion (probably at the point of merger of outer leaflets) by the incorporation of LPC into the membrane prevented gp41 from folding into the six-helix bundle: adding T20 (Fig. 5 A, fourth bar) or T21 (fifth bar) after removing the LPC prevented fusion when temperature was raised to 37°C. Mechanistically, the presence of membrane-bound LPC prevented gp41 from transiting into its final and stable six-helix bundle. Thus, we showed that in order for the bundle to form, membrane merger must take place. Therefore, the bundle itself cannot be the cause of membrane merger.
Figure 5.
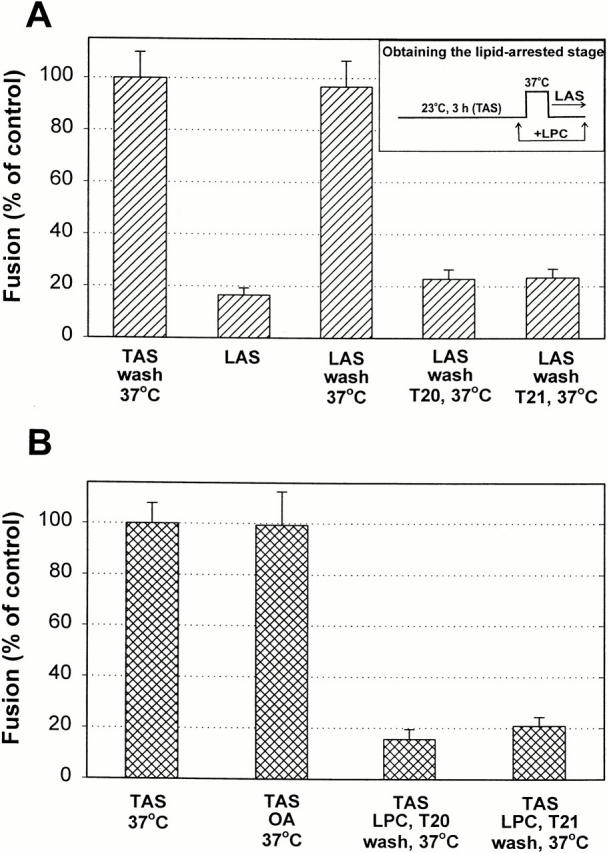
The formation of the six-helix bundle requires membrane merger. (A) Adding 285 μM LPC to cells at TAS, followed by removal of the unbound LPC 5 min later, suppressed fusion when temperature was raised to 37°C (LAS, second bar). But removing the membrane-bound LPC allowed fusion to occur at 37°C (third bar). Addition of 40 nM T20 or 440 nM T21 after removing all LPC inhibited fusion when temperature was again increased to 37°C (fourth and fifth bars). Effector cells were bound to target cells that had been grown overnight on untreated glass. (B) Exposure of cells at TAS to 265 μM OA yielded the same extent of fusion (second bar) as the control (first bar). LPC was added to cells at TAS and the unbound fraction was removed (with the cells retaining the membrane-bound LPC); the addition of T20 or T21 (concentrations as in A) followed by removal of the membrane-bound LPC and unbound peptide suppressed fusion (third and fourth bars).
It remained possible that LPC did not inhibit fusion by preventing lipid merger as is generally thought, but through some binding to Env (Gunther-Ausborn and Stegmann 1997). To ensure that the positive spontaneous curvature of LPC was the cause of the inhibition, we performed two sets of control experiments. In one set, we substituted SDS (at 70 μM) for LPC because it has a different structure and charge, but should have positive spontaneous curvature, and alternatively substituted OA because it has negative spontaneous curvature. The addition of OA should not prevent lipid merger, and in fact it did not inhibit fusion when added at TAS (Fig. 5 B, second column). The same results as with LPC were obtained with SDS despite their different structures. When fusion had been inhibited by SDS, the subsequent addition of T20 or T21 eliminated the resumption of fusion at 37°C after removing the SDS (data not shown). In a second set of controls, we showed that LPC did not compete with T20 or T21 for binding sites on gp41. When LPC was added at TAS and then any LPC still in solution was washed out, the addition of T20 or T21 inhibited fusion at 37°C even after removing both the membrane-bound LPC and the unbound peptide with a BSA wash (Fig. 5 B, third and fourth columns). This control shows that the peptides were tightly bound to the HRs of gp41 even in the presence of membrane-bound LPC (Shu et al. 2000). Thus, the HRs of gp41 should have been able to bind among themselves into a six-helix bundle. Because they did not (Fig. 5 A), LPC (and SDS) inhibited the association of HR1 and HR2 into a six-helix bundle by its presence in cell membranes rather than by binding to HRs. That is, LPC prevented membrane merger by altering spontaneous curvature. It is unlikely that LPC bound to gp41 and in this way adversely affected fusion.
Raising Temperature at TAS Does Not Induce Bundle Formation without Also Causing Fusion.
We devised an alternative approach to test whether the six-helix bundle could form before lipid merger without fusion then occurring. This approach avoided the need to add exogenous agents such as LPC. We reasoned that if the bundle formed before fusion and this structure then induced fusion, one should be able to increase temperature to 37°C for times too short to cause lipid or aqueous dye spread, but sufficiently long to induce the formation of the bundle. (Once formed, the six-helix bundle is extremely stable [Blacklow et al. 1995; Weissenhorn et al. 1996] and hence will not convert back to a prior configuration.) Cells at TAS were quickly brought to 37°C with a T-jump method we devised (see Materials and Methods); with several cell pairs in a field of view, temperature was returned to 4°C as soon as one or two cell pairs were observed to fuse (Fig. 6 A). The fused cells acted as an indicator that temperature had been elevated for a sufficient time to have promoted bundle formation for those cells remaining (“preconditioned cells”). T20 or T21 was added and after maintaining 4°C for 5 min, aqueous dye did not spread when the temperature was again jumped to 37°C (Fig. 6 B, middle and right bars). However, when the peptides had not been added, fusion did occur (Fig. 6 B, left bar). Thus, the bundle had not formed for the preconditioned cells. We tested whether the preconditioned cells were advanced at all beyond TAS to a further stage still before fusion, if in fact such a stage existed. If it did, the kinetics of fusion for the preconditioned cells should be faster than from the point of TAS. The times from the start of the second jump in temperature and fusion of the preconditioned cells were ranked to generate cumulative distributions (Fig. 6 B, inset, triangles). We found that this distribution was close to and no faster than the distribution that occurred when temperatures of cells at TAS were raised and maintained at 37°C (Fig. 6 B, inset, open circles). To verify the accuracy of kinetics obtained by dye spread in our experiments, we used electrical capacitance measurements to monitor the moment that a pore formed between individual cell pairs. Electrical measures provide the greatest temporal resolution and sensitivity for detecting pores. The electrically determined kinetics (Fig. 6 B, inset, filled circles) was statistically identical to those obtained by calcein redistribution (open circles), demonstrating that the rate of Env-induced fusion is reliably monitored by fluorescence microscopy. The kinetic measures thus indicate that any further reconfigurations of gp41 beyond TAS but before bundle formation are less rate limiting than either conversion of gp41 to the bundle and/or lipid rearrangements required for pore formation. If lipid rearrangements were rate limiting, we should have been able to capture preconditioned cells with bundles already formed. Because we could not, we propose that bundle formation is the rate-limiting step. The fact that fusion kinetics was independent of whether temperature had previously been raised to 37°C also suggests that fusion from TAS was stochastic, without “memory” of past temperatures.
Figure 6.
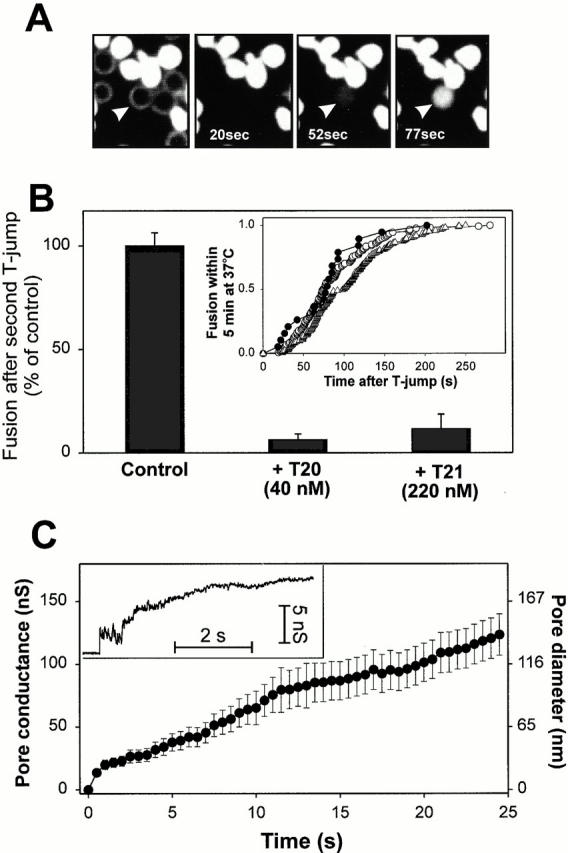
Fusion evoked from TAS by a T-jump to 37°C. (A) Fluorescence images of fusion between calcein-labeled effector cells and unlabeled target cells (the bright field image is superimposed on the first panel). The times after the T-jump are shown. Arrowhead indicates the first target cell that acquired aqueous dye. Effector and target cells were mixed together and plated on polylysine-coated coverslips. (B) The extents of fusion for preconditioned cells in the absence and presence of peptides. TAS was created and temperature was then decreased to 4°C. Preconditioned cells were generated by increasing the temperature of several cells in a microscopic view to 37°C and once one or two cells fused, quickly lowering the temperature to 4°C. T20 (middle bar) or T21 (right bar) were added or not added (left bar, control) and fusion of the remaining preconditioned cells was induced by again raising temperature to 37°C. Inset: kinetics of fusion when temperature was maintained at 37°C was continuously monitored either by the onset of calcein redistribution (○) or by measuring the increments in electrical capacitance of cell membranes due to fusion pore opening (•). Fusion kinetics of preconditioned cells measured by calcein redistribution is shown by ▵. (C) Electrical recording of fusion pores formed by Env. A representative pore is shown in the inset. The average pore conductance was determined from eight individual records (•). The right-hand scale shows the approximate pore diameter. Bars indicate the standard error.
HIV Env–induced Fusion Pores Readily Enlarge
The identical kinetics obtained by spread of aqueous dye (calcein, M r 623) and electrical measurements immediately indicate that after formation, pores quickly grew large enough to readily permit passage of calcein. Direct electrical measurements showed that the conductances of individual gp41-induced fusion pores did, in point of fact, quickly and steadily enlarge after formation (Fig. 6 C, inset). The behavior of gp41-induced fusion pores quantitatively varied between individual experiments, as do fusion pores for all viral (Melikyan and Chernomordik 1997) and cellular systems (Lindau and Almers 1995). Therefore, we calculated the average pore conductance over time (Fig. 6 C) and found that it reached 7 nS (∼16-nm diameter) within 15 ms and ∼100 nS (∼116 nm) within the first 20 s after opening. This rapid enlargement of the pore is the reason the more convenient detection of dye spread yields quantitatively accurate fusion kinetics.
Discussion
The Formation of the Six-Helix Bundle and Membrane Merger Is Tightly Linked
It is generally agreed that the formation of the six-helix bundle or the structure of the bundle itself must be intimately involved in the fusion process because six-helix bundles are formed for so many viral fusion proteins. However, an opposite view has been argued, that the six-helix bundle forms long after fusion has occurred and is unrelated to the fusion process (Shangguan et al. 1998). But if T20 and T21 prevent HIV Env from folding into a six-helix bundle at the point of bundle formation itself, rather than by preventing a prior upstream conformational change, this latter view could not be correct.
The precise function of the bundle, however, had not been experimentally identified. Most commonly it has been assumed that the purpose of the movement of a fusion protein into a six-helix bundle is to force membranes into close proximity (Weissenhorn et al. 1997; Chan and Kim 1998), in which case subsequent processes such as oligomerization of fusion proteins or structures outside the bundle would promote fusion. The six-helix bundle portions of viral fusion proteins are extremely stable, and therefore after formation should not undergo further conformational changes that would directly induce fusion. The assumption that bundle formation brings membranes into contact is reasonable because the fusion peptide of gp41 probably inserts into the target membrane before fusion (as has been shown for HA; Durrer et al. 1996; Stegmann et al. 1991), and the transmembrane (TM) domain that is located within the viral membrane would, logically, pull the two membranes together as the hairpin bend forms. Investigators generated fragments of ectodomains of fusion proteins that contained the preassembled six-helix bundle as well as other portions of the protein in order to determine how the bundle and regions outside the bundle could interact with membranes to promote hemifusion and/or fusion from a state of close membrane apposition (LeDuc et al. 2000). Alternatively, it has been suggested that the formation of the six-helix bundle does not simply bring membranes into close apposition, but that conformational changes of fusion proteins into the bundle directly couple to membrane fusion (Baker et al. 1999): the fusion peptides and TM domains of some fusion proteins form a continuous structure with their adjacent HRs (Baker et al. 1999), which immediately suggests that these two domains should come into close proximity as the six-helix bundle forms. However, for other fusion proteins such continuous structures do not exist (Chen et al. 1999).
We have shown that the addition of LPC to outer leaflets of membranes prevents gp41 from folding into a six-helix bundle: even though all other fusion conditions, including temperature, were optimal and should have allowed bundles to form, they did not form. LPC has prevented bundle formation, and has done so through some effect on the lipid bilayer portion of the membrane. Since the incorporation of LPC into membranes prevents hemifusion, we can logically conclude that preventing hemifusion is that effect. If we assume that LPC has no further effect than to prevent hemifusion, we can further conclude that upon its removal, the bundle could not have formed before hemifusion and then induce hemifusion, or the bundle would have been able to form in the presence of LPC. We cannot rule out the remote possibility that LPC prevents bundle formation through some effect other than hemifusion, some unknown effect that blocks bundle formation. But there is no evidence that LPC in membranes could inhibit the kind of large scale conformational changes that occur in the ectodomain by any means other than hemifusion and/or fusion. In fact, to the limit of the time resolution of the kinetic experiments, the finding that gp41 of preconditioned cells had not yet folded into a six-helix bundle provides independent support for the hypothesis that without hemifusion (or fusion) the bundle does not form. If bundle formation is essential for membrane merger, and membrane merger is required for bundle formation, then membrane merger occurs as some of the fusion proteins are assuming the six-helix bundle.
Mechanistically, fusion could take place as follows. Fusion peptides insert into the target membrane, and as the fusion proteins bend into six-helix bundles, the fusion peptides and TM domains pull the two membranes locally toward each other (Weissenhorn et al. 1997; Chan and Kim 1998). For every viral fusion protein in which the six-helix bundle has been observed, each of the COOH-terminal helices has been shown to pack on the outside of and antiparallel to the NH2-terminal coiled coil (Skehel and Wiley 1998). For this crystallographically identified bundle to actually form, the fusion peptides (arrows of Fig. 7) and TM domains should come close to each other, and because the bundle exhibits threefold symmetry, the two membrane-inserted regions may even intermingle. Membrane continuity would be a necessary concomitant for these two domains, inserted in different membranes, to come into contact. Hemifusion could occur before folding of gp41 into the bundle (this is consistent with our LPC results), and the formation of the bundle could disrupt a small hemifusion diaphragm and thereby create the pore. This could also be the case for many other viral fusion proteins. In fact, for HA of influenza virus, a large body of evidence indicates that fusion is initiated through hemifusion (Kemble et al. 1994; Melikyan et al. 1997; Chernomordik et al. 1998). It has been advocated that the TM domain (Kemble et al. 1994; Melikyan et al. 1999; Markosyan et al. 2000) and the fusion peptide (Qiao et al. 1999) effect fusion by breaking the hemifusion diaphragm.
Figure 7.
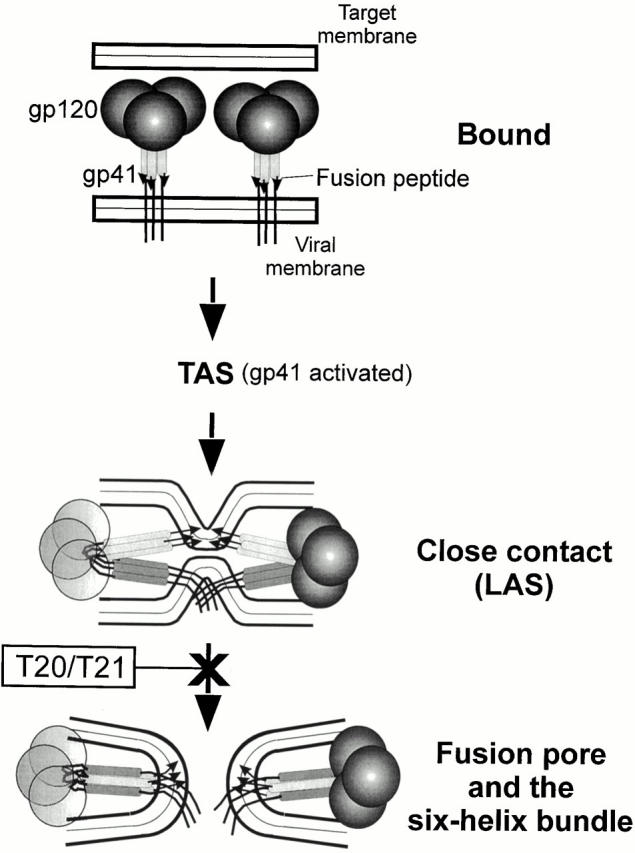
A proposed sequence of events for Env-mediated membrane fusion. The relative positions of HR1 and HR2 in the bound state are not known and only HR1 is depicted. (Neither CD4 nor the coreceptors are shown.) When gp41 is activated, the grooves of the central coiled coil (light gray bars with fusion peptide indicated by arrows) and the COOH-terminal helices (dark gray bars attached to the TM domain and cytoplasmic tail) have become exposed. The gp120 on the left is made transparent, for clarity. When gp41 further reconfigures into a six-helix bundle, the fusion peptide and TM domain (in different membranes) are forced toward each other and induce pore formation (probably by rupturing the hemifusion diaphragm, not shown).
The Free Energies Released during Formation of the Six-helix Bundle Are Sufficient to Induce Fusion
The change in free energy accompanying bundle formation that leads to the forces that induce fusion can be estimated. Because T20 inhibits fusion by a dominant-negative mechanism (Chen et al. 1995; Furuta et al. 1998), and several fusion proteins may act cooperatively to create a pore, T20 may block Env-mediated fusion when it binds to one or only a few HR1 domains. T20 binds to individual monomers of soluble trimeric forms of the central core with an affinity of at least ∼1 μM (Chen et al. 1995) and inhibits fusion at a half-maximal concentration of ∼1 nM (Chen et al. 1995; Munoz-Barroso et al. 1998). A binding constant of 1 μΜ for T20 to an individual groove yields ∼14 kT of energy upon binding to a monomer; an affinity of 1 nM yields ∼20 kT (where k is Boltzmann's constant and T is temperature in degrees Kelvin). Thus, the free energy of binding the native COOH-terminal α-helices to each of the three grooves of the central coiled coil provides 40–60 kT that could be used for pore formation. With several trimers participating in fusion, the energy of transition to six-helix bundles would be more than sufficient for a fusion pore to form according to all quantitative models of fusion (Kozlov et al. 1989; Siegel 1993).
Whereas ectodomains free in solution readily form six-helix bundles, when the ectodomain is membrane anchored the transition would be physically far more difficult. Steric constraints should make it rather difficult for individual HR2 segments within a gp41 trimer to move away from each other and into close contact with their respective HR1 segments, since the HR2 segments are continuous with the TM domains and there is only a short stretch of amino acids between them. Thus, although considerable free energy is released by six-helix bundle formation, the process may not be spontaneous and a large activation barrier may have to be surmounted. A large activation barrier would account for the steep temperature dependence of fusion from TAS.
The Energy Barriers Upstream of TAS Are Smaller Than Those between Tas and Fusion
The longer time before the start of fusion (the lag) from 4°-TAS than from TAS (upon raising temperature to 37°C) shows that the steps contributing to the delay are temperature dependent. A step that intervenes between TAS and fusion was more highly temperature dependent than the steps preceding TAS: fusion does not occur at 23°C but does at 37°C. In terms of energetics this could mean that the barriers between cell–cell contact and TAS are lower than between TAS and fusion. This difference in barriers could be the reason we were able to capture TAS as a stable intermediate state just before fusion. If the energy barriers before TAS were not large, many steps would occur before TAS because delays until fusion were long when cells were maintained at 37°C (Fig. 3 A). Multiple copies of Env (and possibly CD4 and/or CXCR4) may have moved into and associated with each other at a site where the pore could be created (Frey et al. 1995; Weissenhorn et al. 1997; Munoz-Barroso et al. 1998). It is unlikely that the association of Env into a complex is so highly temperature dependent that it does not occur at 23°C: the preconditioned cells fused no faster than when temperature was originally raised and maintained (Fig. 6 B).
Possible Utility of Creating TAS
Although many fusion proteins reconfigure into six-helix bundle structures, the fusion pores created by these different proteins exhibit distinct characteristics. The fusion pores formed by HIV Env had not been previously measured electrically. By first creating TAS and then patch clamping cells, we developed a means to routinely electrically measure HIV Env–induced fusion pores. These pores have large sizes within milliseconds of formation, grow rapidly, and do not flicker. Each of these features contrasts with pores induced by influenza virus HA, the most extensively characterized fusion pore. The fusion pores induced by Moloney murine leukemia virus Env protein, the only other retroviral fusion pore electrically measured (Melikyan et al. 2000), also does not flicker and grows rapidly, but the initial HIV Env pore is larger than that of Moloney.
As a practical matter, the remarkable stability of exposure of a highly conserved region of gp41 and the convenience of releasing TAS on to fusion may be potentially useful for testing drugs that bind to this region (Eckert et al. 1999; Ferrer et al. 1999) and for the development of a broadly effective HIV vaccine. CD4 and coreceptor-triggered HIV Env can elicit antibodies that neutralize infectivity of diverse HIV isolates (LaCasse et al. 1999). For the immunogen to have elicited broadly effective antibodies, conserved regions of HIV Env must have been exposed (LaCasse et al. 1999); these may be the same regions that are readily and reliably exposed for long times by creating TAS.
In conclusion, we have shown that the six-helix bundle does not form in the absence of membrane merger and verified that fusion does not occur if the bundle cannot form. Thus, it is not the six-helix bundle as a stable protein configuration that causes fusion, but rather the free energy released when gp41 undergoes this conversion that is directly and immediately used for pore formation. We propose that all viral fusion proteins that form six-helix bundles will likely achieve fusion through the same or similar mechanisms as HIV Env. There may even be some parallels in eukaryotic fusion: intracellular membrane fusion is thought to be mediated by SNAREs (Südhof 1995; Hay and Scheller 1997; Weber et al. 1998), and SNAREs form a heterotrimeric, parallel four-helix bundle (Poirier et al. 1998; Sutton et al. 1998). The energy released during this bundle assembly may contribute to intracellular fusion.
Supplemental Material
Acknowledgments
We thank the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, for providing materials, and Dr. Z. Jonak of SmithKline Beecham for supplying the TF228.1.16 cell line. We are grateful to Drs. L.V. Chernomordik, R.A. Lamb, and J. Rucker for critical readings of the manuscript.
Supported by National Institutes of Health grants GM27367 and GM54787.
Footnotes
The online version of this article contains supplemental material.
Abbreviations used in this paper: CMAC, 7-amino-4-chloromethylcoumarin; CXCR4, chemokine receptor; DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; HA, hemagglutinin; HR, heptad repeat; IR, infrared; LAS, lipid-arrested stage; LPC, lysophosphatidylcholine; OA, oleic acid; SNARE, soluble N-ethylmaleimide–sensitive factor attachment protein receptor; TAS, temperature-arrested stage; TM, transmembrane.
References
- Baker K.A., Dutch R.E., Lamb R.A., Jardetzky T.S. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell. 1999;3:309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- Berger E.A., Murphy P.M., Farber J.M. Chemokine receptors as HIV-1 coreceptorsroles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- Blacklow S.C., Lu M., Kim P.S. A trimeric subdomain of the simian immunodeficiency virus envelope glycoprotein. Biochemistry. 1995;34:14955–14962. doi: 10.1021/bi00046a001. [DOI] [PubMed] [Google Scholar]
- Chan D.C., Kim P.S. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- Chan D.C., Fass D., Berger J.M., Kim P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- Chan D.C., Chutkowski C.T., Kim P.S. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. USA. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.H., Matthews T.J., McDanal C.B., Bolognesi D.P., Greenberg M.L. A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivativesimplication for viral fusion. J. Virol. 1995;69:3771–3777. doi: 10.1128/jvi.69.6.3771-3777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Skehel J.J., Wiley D.C. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. USA. 1999;96:8967–8972. doi: 10.1073/pnas.96.16.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L., Kozlov M.M., Zimmerberg J. Lipids in biological membrane fusion. J. Membr. Biol. 1995;146:1–14. doi: 10.1007/BF00232676. [DOI] [PubMed] [Google Scholar]
- Chernomordik L.V., Leikina E., Frolov V., Bronk P., Zimmerberg J. An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J. Cell Biol. 1997;136:81–93. doi: 10.1083/jcb.136.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L.V., Frolov V.A., Leikina E., Bronk P., Zimmerberg J. The pathway of membrane fusion catalyzed by influenza hemagglutininrestriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 1998;140:1369–1382. doi: 10.1083/jcb.140.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doranz B.J., Baik S.S., Doms R.W. Use of a gp120 binding assay to dissect the requirements and kinetics of human immunodeficiency virus fusion events. J. Virol. 1999;73:10346–10358. doi: 10.1128/jvi.73.12.10346-10358.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrer P., Galli C., Hoenke S., Corti C., Gluck R., Vorherr T., Brunner J. H+-induced membrane insertion of influenza virus hemagglutinin involves the HA2 amino-terminal fusion peptide but not the coiled coil region. J. Biol. Chem. 1996;271:13417–13421. doi: 10.1074/jbc.271.23.13417. [DOI] [PubMed] [Google Scholar]
- Eckert D.M., Malashkevich V.N., Hong L.H., Carr P.A., Kim P.S. Inhibiting HIV-1 entrydiscovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- Endres M.J., Clapham P.R., Marsh M., Ahuja M., Turner J.D., McKnight A., Thomas J.F., Stoebenau-Haggarty B., Choe S., Vance P.J. CD4-independent infection by HIV-2 is mediated by fusin/CXCR4. Cell. 1996;87:745–756. doi: 10.1016/s0092-8674(00)81393-8. [DOI] [PubMed] [Google Scholar]
- Ferrer M., Kapoor T.M., Strassmaier T., Weissenhorn W., Skehel J.J., Oprian D., Schreiber S.L., Wiley D.C., Harrison S.C. Selection of gp41-mediated HIV-1 cell entry inhibitors from biased combinatorial libraries of non-natural binding elements. Nat. Struct. Biol. 1999;6:953–960. doi: 10.1038/13324. [DOI] [PubMed] [Google Scholar]
- Frey S., Marsh M., Gunther S., Pelchen-Matthews A., Stephens P., Ortlepp S., Stegmann T. Temperature dependence of cell–cell fusion induced by the envelope glycoprotein of human immunodeficiency virus type 1. J. Virol. 1995;69:1462–1472. doi: 10.1128/jvi.69.3.1462-1472.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta R.A., Wild C.T., Weng Y., Weiss C.D. Capture of an early fusion-active conformation of HIV-1 gp41. Nat. Struct. Biol. 1998;5:276–279. doi: 10.1038/nsb0498-276. [DOI] [PubMed] [Google Scholar]
- Gunther-Ausborn S., Stegmann T. How lysophosphatidylcholine inhibits cell–cell fusion mediated by the envelope glycoprotein of human immunodeficiency virus. Virology. 1997;235:201–208. doi: 10.1006/viro.1997.8699. [DOI] [PubMed] [Google Scholar]
- Hart T.K., Truneh A., Bugelski P.J. Characterization of CD4-gp120 activation intermediates during human immunodeficiency virus type 1 syncytium formation. AIDS Res. Hum. Retroviruses. 1996;12:1305–1313. doi: 10.1089/aid.1996.12.1305. [DOI] [PubMed] [Google Scholar]
- Hay J.C., Scheller R.H. SNAREs and NSF in targeted membrane fusion. Curr. Opin. Cell Biol. 1997;9:505–512. doi: 10.1016/s0955-0674(97)80026-9. [DOI] [PubMed] [Google Scholar]
- Healey D., Dianda L., Moore J.P., McDougal J.S., Moore M.J., Estess P., Buck D., Kwong P.D., Beverley P.C., Sattentau Q.J. Novel anti-CD4 monoclonal antibodies separate human immunodeficiency virus infection and fusion of CD4+ cells from virus binding. J. Exp. Med. 1990;172:1233–1242. doi: 10.1084/jem.172.4.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonak Z.L., Clark R.K., Matour D., Trulli S., Craig R., Henri E., Lee E.V., Greig R., Debouck C. A human lymphoid recombinant cell line with functional human immunodeficiency virus type 1 envelope. AIDS Res. Hum. Retroviruses. 1993;9:23–32. doi: 10.1089/aid.1993.9.23. [DOI] [PubMed] [Google Scholar]
- Joshi S.B., Dutch R.E., Lamb R.A. A core trimer of the paramyxovirus fusion proteinparallels to influenza virus hemagglutinin and HIV-1 gp41. Virology. 1998;248:20–34. doi: 10.1006/viro.1998.9242. [DOI] [PubMed] [Google Scholar]
- Kemble G.W., Danieli T., White J.M. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 1994;76:383–391. doi: 10.1016/0092-8674(94)90344-1. [DOI] [PubMed] [Google Scholar]
- Kliger Y., Shai Y. Inhibition of HIV-1 entry before gp41 folds into its fusion-active conformation. J. Mol. Biol. 2000;295:163–168. doi: 10.1006/jmbi.1999.3368. [DOI] [PubMed] [Google Scholar]
- Kozlov M.M., Leikin S.L., Chernomordik L.V., Markin V.S., Chizmadzhev Y.A. Stalk mechanism of vesicle fusion. Intermixing of aqueous contents. Eur. Biophys. J. 1989;17:121–129. doi: 10.1007/BF00254765. [DOI] [PubMed] [Google Scholar]
- LaCasse R.A., Follis K.E., Trahey M., Scarborough J.D., Littman D.R., Nunberg J.H. Fusion-competent vaccinesbroad neutralization of primary isolates of HIV. Science. 1999;283:357–362. doi: 10.1126/science.283.5400.357. [DOI] [PubMed] [Google Scholar]
- Lawless M.K., Barney S., Guthrie K.I., Bucy T.B., Petteway S.R., Jr., Merutka G. HIV-1 membrane fusion mechanismstructural studies of the interactions between biologically-active peptides from gp41. Biochemistry. 1996;35:13697–13708. doi: 10.1021/bi9606962. [DOI] [PubMed] [Google Scholar]
- LeDuc D.L., Shin Y.K., Epand R.F., Epand R.M. Factors determining vesicular lipid mixing induced by shortened constructs of influenza hemagglutinin. Biochemistry. 2000;39:2733–2739. doi: 10.1021/bi992457v. [DOI] [PubMed] [Google Scholar]
- Lindau M., Almers W. Structure and function of fusion pores in exocytosis and ectoplasmic membrane fusion. Curr. Opin. Cell Biol. 1995;7:509–517. doi: 10.1016/0955-0674(95)80007-7. [DOI] [PubMed] [Google Scholar]
- Maddon P.J., Dalgleish A.G., McDougal J.S., Clapham P.R., Weiss R.A., Axel R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 1986;47:333–348. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- Markosyan R.M., Cohen F.S., Melikyan G.B. The lipid-anchored ectodomain of influenza virus hemagglutinin (GPI-HA) is capable of inducing nonenlarging fusion pores. Mol. Biol. Cell. 2000;11:1143–1152. doi: 10.1091/mbc.11.4.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan G.B., Chernomordik L.V. Membrane rearrangements in fusion mediated by viral proteins. Trends Microbiol. 1997;5:349–355. doi: 10.1016/S0966-842X(97)01107-4. [DOI] [PubMed] [Google Scholar]
- Melikyan G.B., Brener S.A., Ok D.C., Cohen F.S. Inner but not outer membrane leaflets control the transition from glycosylphosphatidylinositol-anchored influenza hemagglutinin–induced hemifusion to full fusion. J. Cell Biol. 1997;136:995–1005. doi: 10.1083/jcb.136.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan G.B., Lin S., Roth M.G., Cohen F.S. Amino acid sequence requirements of the transmembrane and cytoplasmic domains of influenza virus hemagglutinin for viable membrane fusion. Mol. Biol. Cell. 1999;10:1821–1836. doi: 10.1091/mbc.10.6.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan G.B., Markosyan R.M., Brener S.A., Rozenberg Y., Cohen F.S. Role of the cytoplasmic tail of ecotropic moloney murine leukemia virus Env protein in fusion pore formation. J. Virol. 2000;74:447–455. doi: 10.1128/jvi.74.1.447-455.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Barroso I., Durell S., Sakaguchi K., Appella E., Blumenthal R. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J. Cell Biol. 1998;140:315–323. doi: 10.1083/jcb.140.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T., Zhang T.Y., Koyanagi Y., Tanaka Y., Kim J., Suzuki Y., Minoguchi S., Tamamura H., Waki M., Matsumoto A. Inhibitory mechanism of the CXCR4 antagonist T22 against human immunodeficiency virus type 1 infection. J. Virol. 1999;73:7489–7496. doi: 10.1128/jvi.73.9.7489-7496.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier M.A., Xiao W., Macosko J.C., Chan C., Shin Y.K., Bennett M.K. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat. Struct. Biol. 1998;5:765–769. doi: 10.1038/1799. [DOI] [PubMed] [Google Scholar]
- Qiao H., Armstrong R.T., Melikyan G.B., Cohen F.S., White J.M. A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Mol. Biol. Cell. 1999;10:2759–2769. doi: 10.1091/mbc.10.8.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimsky L.T., Shugars D.C., Matthews T.J. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 1998;72:986–993. doi: 10.1128/jvi.72.2.986-993.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangguan T., Siegel D.P., Lear J.D., Axelsen P.H., Alford D., Bentz J. Morphological changes and fusogenic activity of influenza virus hemagglutinin. Biophys. J. 1998;74:54–62. doi: 10.1016/S0006-3495(98)77766-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu W., Liu J., Ji H., Radigen L., Jiang S., Lu M. Helical interactions in the HIV-1 gp41 core reveal structural basis for the inhibitory activity of gp41 peptides. Biochemistry. 2000;39:1634–1642. doi: 10.1021/bi9921687. [DOI] [PubMed] [Google Scholar]
- Siegel D.P. Energetics of intermediates in membrane fusioncomparison of stalk and inverted intermediate mechanisms. Biophys. J. 1993;65:2124–2140. doi: 10.1016/S0006-3495(93)81256-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C. Coiled coils in both intracellular vesicle and viral membrane fusion. Cell. 1998;95:871–874. doi: 10.1016/s0092-8674(00)81710-9. [DOI] [PubMed] [Google Scholar]
- Stegmann T., Delfino J.M., Richards F.M., Helenius A. The HA2 subunit of influenza hemagglutinin inserts into the target membrane prior to fusion. J. Biol. Chem. 1991;266:18404–18410. [PubMed] [Google Scholar]
- Südhof T.C. The synaptic vesicle cyclea cascade of protein–protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Sutton R.B., Fasshauer D., Jahn R., Brunger A.T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Weber T., Zemelman B.V., McNew J.A., Westermann B., Gmachl M., Parlati F., Sollner T.H., Rothman J.E. SNAREpinsminimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Weiss C.D., Barnett S.W., Cacalano N., Killeen N., Littman D.R., White J.M. Studies of HIV-1 envelope glycoprotein-mediated fusion using a simple fluorescence assay. AIDS. 1996;10:241–246. doi: 10.1097/00002030-199603000-00001. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W., Wharton S.A., Calder L.J., Earl P.L., Moss B., Aliprandis E., Skehel J.J., Wiley D.C. The ectodomain of HIV-1 env subunit gp41 forms a soluble, alpha-helical, rod-like oligomer in the absence of gp120 and the N-terminal fusion peptide. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:1507–1514. [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W., Dessen A., Harrison S.C., Skehel J.J., Wiley D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- Wild C.T., Shugars D.C., Greenwell T.K., McDanal C.B., Matthews T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt R., Sodroski J. The HIV-1 envelope glycoproteinsfusogens, antigens, and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}