

Abstract
It has been proposed that the generally low affinities of T cell receptors (TCRs) for their peptide–major histocompatibility complex (pMHC) ligands (Kd ∼10−4 to 10−7 M) are the result of biological selection rather than an intrinsic affinity limitation imposed by the TCR framework. Using a soluble version of the 2C TCR, we have used complementarity determining region (CDR)-directed mutagenesis to investigate whether the affinity of this receptor for its allogeneic pMHC ligand can be improved upon. We report that several mutants at positions lying within CDR3α and CDR2β showed increased affinities for pMHC compared with the wild-type receptor. Additionally, we have investigated whether Vα mutations that have been implicated in the phenomenon of CD8+ repertoire skewing achieve this skewing by means of generalized increases in affinity for MHC-I molecules. Two mutants (S27F and S51P), which each promote skewing toward a CD8+ phenotype, exhibited significantly reduced affinity for pMHC-I, consistent with a quantitative-instructional model of CD4/CD8 lineage commitment. This model predicts that CD8 is downregulated on thymocytes that have TCR–ligand interactions above a minimal energy threshold. Together, the results (a) demonstrate that engineering higher affinity TCRs is feasible, and (b) provide TCR–pMHC energy values associated with CD4/CD8 repertoire skewing.
Keywords: T cell receptor, peptide–major histocompatibility complex, affinity, CD4/ CD8, complementarity determining region
Antibodies exhibit structural similarity to TCRs and undergo similar gene rearrangement processes, yet they generally exhibit much higher affinities for their antigens (Kd ∼10−7 to 10−12 M) than do TCRs for their peptide–MHC complex (pMHC)1 ligands (Kd ∼10−4 to 10−7 M) (1). Although the molecular explanation for these differences could be that somatic mutation does not operate on the TCR, there are likely functional explanations also. For example, negative selection acts to eliminate high TCR affinities. In addition, it has been hypothesized that there may be a ceiling on useful TCR–pMHC affinity (∼10−7 M), above which there is no functional advantage (2, 3) (and above which are possibly disadvantages, due to impaired serial triggering of TCRs [4]). Antibodies, which undergo affinity maturation, may have different functional ceilings on their affinity optimization (5).
Another possibility is that the framework regions of antibody V regions are more optimally suited as a scaffold on which optimal-affinity CDR regions may be constructed, and that TCR framework regions are handicapped in this regard. Although the three-dimensional structures are similar, important differences have been observed in the overall surface topology of antibodies compared with at least the 2C TCR (6). If the low affinities observed for TCR–pMHC interactions are a result of a selection process rather than inherent structural differences, then it should be possible to improve this affinity through CDR-directed mutagenesis.
Using the 2C TCR system, we have explored the possibility of engineering soluble TCR molecules with improved affinities for the allogeneic ligand QL9/Ld. Previous work involving alanine scanning mutagenesis of the 2C CDR regions revealed four residues that yielded moderate improvements in affinity when changed to alanine (7). In this report, these mutations have been combined into double and triple mutant TCRs to test whether the affinity of the 2C TCR for its allogeneic ligand QL9/Ld can be further improved. This interaction has the highest affinity of TCR–pMHC interactions that have been measured (Kd = 10−7 M [8]). Engineering a higher affinity TCR–QL9/Ld interaction would support the notion that a ceiling on measured TCR–pMHC interactions is imposed by biological processes rather than an intrinsic inability of the TCR framework to support higher affinities. Additionally, soluble TCR molecules with improved affinities could theoretically serve as antagonists for detrimental T cell responses. We report here that a two- to threefold increase in TCR affinity could be achieved through CDR-directed mutagenesis.
Another aspect of TCR–pMHC affinity relates to the process of CD4/CD8 phenotype selection. The 2C TCR is invariably skewed toward the CD8 phenotype, consistent with findings that this TCR is positively selected by a class I MHC product, Kb (9). However, the 2C TCR is somewhat unusual in that it expresses the Vα3.1 region, which has been shown by Gascoigne and colleagues to skew polyclonal populations of T cells toward the CD4 phenotype (13). In fact, various TCR Vα region genes have now been associated with CD4/CD8 repertoire selection (10–15), presumably because these Vα chains preferentially bind to either an MHC class II or class I restricting element. In the case of the most thoroughly studied family, Vα3, the phenomenon is apparently dependent on two amino acid residues, at position 27 in CDR1 and position 51 in CDR2. Either residue is sufficient to affect the CD4/CD8 balance, and together they have an additive effect (13, 14). Vα3.2 is strongly skewed toward the CD8 phenotype and has Phe27α and Pro51α at these positions, whereas Vα3.1 is reciprocally skewed toward CD4 and has Ser27α and Ser51α. Thus, it was proposed that Vα3.2 interacts “preferentially” with MHC class I molecules and that the skewing attributed to positions 27 and 51 might result from increased positive selection (13). It is interesting to note that Vα11.3 also contains a proline at position 51 and, like Vα3.2, is skewed toward the CD8 phenotype (15).
Structural analysis of the 2C TCR places both Ser27α and Ser51α in contact with the α helices of MHC class I. This has led to the suggestion that they may play a key role in orienting the TCR–pMHC interaction (16). One of these interactions (Ser51α with the α2 helix) is also present in the A6/HLA-A2/Tax structure (17). The binding energetics associated with the preferential interaction of these two residues with either MHC class I or II molecules has yet to be elucidated. While the intuitive explanation might predict that the skewing phenomenon resulted from an increase in affinity (e.g., of Vα3.2 for MHC class I) that favored positive selection, this has not been proven. In this study, we have tested the hypothesis that the CD8/CD4 repertoire skewing attributed to these two residues is accomplished by increases in affinity for MHC class I molecules. Unexpectedly, we found that single or double mutants at these two positions exhibited affinities that were not consistent with a simple affinity model of increased positive selection, but rather support the quantitative-instructional model of lineage commitment.
Materials and Methods
Single-chain TCR Mutagenesis.
CDR mutant single-chain (sc)TCRs were constructed using a PCR-based technique (18). In brief, a short mutagenic primer and a Vα- or Vβ-specific primer were used in the first PCR step to generate a “megaprimer,” which was then isolated and used in a second PCR with the opposing Vα- or Vβ-specific primer. All PCR reactions were carried out using cloned Pfu DNA polymerase (Stratagene). After the second PCR, the KpnI-BamHI–digested product was ligated into pUC-19M and transformed into DH5α or NM522 Escherichia coli strains for sequencing. After confirmation of the sequence, the scTCR gene was subcloned into the pTRXFus vector (Invitrogen) and transformed into the GI698 E. coli strain for expression.
scTCR Expression and Purification.
Proteins were expressed in E. coli as described previously (7, 19) and purified from inclusion bodies by denaturing metal affinity chromatography and G-200 size exclusion chromatography. Protein purity was assessed by SDS-PAGE as well as by electrospray mass spectrometry for several of the mutants (performed at the University of Illinois Mass Spectrometry Facility).
mAbs.
KJ16 (20) is a rat IgG mAb that is specific for the mouse Vβ8.1 and Vβ8.2 regions. F23.1 (21) is a mouse IgG2a mAb that is specific for the mouse Vβ8.1, Vβ8.2, and Vβ8.3 regions. F23.2 (21) is a mouse IgG1 mAb that is specific for the mouse Vβ8.2 region. 1B2 is an anticlonotypic mouse mAb that is specific for the α/β TCR found on the T cell clone 2C (22). 30-5-7 (23) is a mouse IgG2a mAb that is specific for the α2 domain of H2-Ld. Antibodies were purified from either culture supernatants or ascites. Fab fragments of 30-5-7 were produced by digestion with papain (Sigma) for 5 min at 37°C followed by size exclusion chromatography through a Superdex G-200 column. Residual intact mAb or Fc regions were removed by passage over a protein A–agarose column (GIBCO BRL).
ELISAs.
Vβ8-specific competition ELISAs were performed by adsorption of KJ16, F23.1, or F23.2 to the wells of Immulon 2 ELISA plates (Dynatech Labs). Wells were blocked with PBS, 0.25% BSA, 0.05% Tween and after washing, 100 μl of mutant or wild-type TCR and 50 μl of biotinylated wild-type TCR at a 1:1,000–1:3,000 dilution were added to the wells. After 30 min, wells were washed, and binding was detected using horseradish peroxidase–streptavidin and TMB peroxidase substrate (Kirkegaard & Perry Labs). Inhibition curves were plotted, and the IC50 for each mutant and wild-type were calculated using linear regression analysis. For 1B2 capture ELISAs, 1B2 was adsorbed to wells and after washing, 50 μl of mutant or wild-type TCR was added to wells. Bound TCR was detected with KJ16 and goat anti–rat horseradish peroxidase conjugate (Kirkegaard & Perry Labs).
Peptide–MHC Binding Assays.
A competition, cell binding assay was used to monitor binding of TCR to QL9/Ld complexes, as described previously (7, 24, 25). To form peptide–Ld complexes on the surfaces of T2-Ld target cells, cells were incubated with ∼10 μM of the specific peptide for 3–5 h. For the competitive binding assay, peptide-upregulated cells (3 × 105/ well) were incubated with 0.7 nM 125I-labeled 30-5-7 Fab and various concentrations of scTCR for 1 h on ice in the presence of 0.7% BSA. After incubation, bound and free ligands were separated by centrifugation through dibutyl phthalate/olive oil. All assays were done in triplicate. The cell binding assay was able to detect binding that was 15–20-fold reduced compared with wild-type.
All surface plasmon resonance (SPR) measurements were performed on a BIAcore 2000 instrument (BIAcore) at 25°C in PBS. Purified molecules were immobilized on a CM5 chip at pH 5.2 in 10 mM sodium acetate buffer by classical amine coupling chemistry. Blank surfaces were made by ethanolamine deactivation of a dextran surface. Purified Ld–peptide complexes were injected at 6, 3, 1.5, 0.75, and 0.375 μM at a flow rate of 20 μl/ min. Data were normalized and analyzed using the BIAevaluation 2.1 and 3.0 software programs (BIAcore). Single Langmuir binding model was used for curve fitting analysis. Fittings were assessed by K2 function (<5.0). Baseline drift between successive injections was <2%. Scatchard analyses were plotted from subtracted sensorgrams (Ld-QL9 minus Ld-murine CMV [MCMV] traces). Binding studies were performed with immobilized TCR proteins and the reverse orientation with immobilized Ld–peptide complexes.
Statistical Analysis of Binding Data.
The percent inhibition of T2-Ld binding was corrected for the binding of 125I-labeled 30-5-7 Fab fragments to Ld molecules which do not contain the peptide of interest, as follows: % inhibition = (cpmQL9, no TCR − cpmQL9, +TCR) / (cpmQL9, no TCR − cpmno peptide) × 100. Inhibition curves were constructed, and within each experiment a sample of wild-type scTCR was included as an internal control. QL9/Ld reactivity was calculated as the IC50(mutant)/IC50(wild-type), and was normalized for Vβ8 reactivity to account for variations in the percentages of properly folded proteins.
Structural Analysis.
Structural analysis was performed on an Indigo 2 workstation (Silicon Graphics) using the Quanta software package (Molecular Simulations). The refined structure of the 2C/dEV8/Kb complex (16) was used to assign pMHC contacts for those 2C residues tested by mutagenesis.
Results
Recently, we have mapped the energy of interactions of 2C TCR residues by performing alanine scanning mutagenesis of the 2C TCR and analyzing the mutants for binding to QL9/Ld (7). The mutagenesis study revealed four residues (Glu56β, Thr55β, Ser76β, Ser102α) that, when changed to alanine, exhibited increases (up to twofold) in affinity for QL9/Ld. Two other residues (Ser27α and Ser51α) are also thought to play a significant role in the binding of the class I ligand. This prediction is based both on the contact of these residues with conserved residues in the MHC helices (16) and on their effects on skewing of T cells to a CD4 or CD8 phenotype (13, 14). These six residues are shown in Fig. 1, where the 2C TCR is modeled onto the QL9/Ld ligand based on coordinates of the 2C/dEV8/Kb complex (26). To explore various issues that involve the binding contributions of these residues, mutagenesis was performed and the two classes of mutants (alanine-substituent affinity mutants and CD4/CD8 repertoire-skewing mutants) were analyzed for binding to anti-TCR antibodies and to the pMHC ligand QL9/Ld.
Figure 1.

Location of amino acid residues in the 2C TCR that were mutated in this study. The 2C CDR loops are shown interacting with the alloantigen QL9/Ld. The 2C/QL9/Ld modeled complex is from reference 26.
TCR Mutagenesis, Expression, and Purification
The Vα3.1/Vβ8.2 TCR from CTL clone 2C has been produced as a single-chain thioredoxin fusion protein in E. coli for use in binding and mutagenesis studies (7, 24, 25, 27). Various single, double, and triple mutants involving six residues (Glu56β, Thr55β, Ser76β, Ser102α, Ser27α, and Ser51α) were generated using a two-step PCR–based approach. Mutants were subjected to automated DNA sequencing to confirm mutations. TCRs were expressed in E. coli and purified by metal affinity and size exclusion chromatography to ≥95% purity. Electrospray mass spectrometry was used to confirm the expected mass difference relative to wild-type for several of the mutant TCRs (Fig. 2). The wild-type scTCR is evident as a single peak of mass 40,538 ± 4 daltons (0.01% accuracy), whereas the various mutant TCRs exhibit mass differences equivalent to those predicted based on their amino acid differences. This sensitive method is able to confirm the mass difference attributable to the loss of as little as a single oxygen atom in an ∼40,000-dalton protein, as demonstrated with the αS27A mutant (Fig. 2).
Figure 2.

Mass spectrometry of scTCR CDR mutants. Purified wild-type (WT) and mutant TCR proteins were dialyzed into 30% acetonitrile, 0.1% formic acid and subjected to mass spectrometry. Sensitivity of measurement was generally ±4 daltons (i.e., 0.01%). Observed mass differences attributable to the amino acid substitutions are shown. Expected mass differences (in daltons) were as follows: S51P, +10; S27A, −16; S27F, +60.
Alanine-substituent Affinity Mutants
The four single-site alanine mutants of TCR residues (Glu56β, Thr55β, Ser76β, Ser102α) that exhibited modest increases in affinity for QL9/Ld (7) were examined as double and triple alanine mutants to test whether synergistic affinity increases might be achieved. In addition, residue 56β was changed to a positive charge (Lys56β) to test the possibility that this glutamic acid side chain in the wild-type is near a negatively charged residue in the ligand and thus that a positive charge at residue 56β may yield even higher affinity for QL9/Ld.
Antibody Reactivity.
The degree to which each mutant was properly refolded was analyzed using the mAbs KJ16 (Vβ8.1/8.2 specific) and F23.1 (Vβ8.1/8.2/8.3 specific). These two mAbs recognize determinants on the TCR distal from the CDR regions. Each of the mutants exhibited essentially the same reactivity as wild-type (Fig. 3 A, and data not shown), indicating that the mutant TCRs have the same degree of refolding as wild-type, and confirming that mutations have not globally disrupted the protein's tertiary conformation. Thus, any differences observed in reactivities of the mutants toward other antigens can be attributable to the role of the substituted amino acid and not to a global destabilization of the receptor.
Figure 3.

Analysis of higher affinity alanine-substituent 2C TCR double mutants. Representative binding curves are illustrated for each of the different ligands for the 102/55, 102/56, and wild-type (WT) scTCR proteins. ELISA binding curves are presented for the mAbs F23.1 (Vβ8.1, 8.2, 8.3; A) and F23.2 (Vβ8.2; B), as well as the clonotypic mAb 1B2 (C). Inhibition of 30-5-7 (anti-Ld) Fab binding to cell surface QL9/Ld is shown in D.
The mAb F23.2 is specific for Vβ8.2, and its epitope has been shown to include residue Glu56β, which is critical for binding (7). The reactivity of each mutant was analyzed in F23.2 competition ELISA experiments where it was confirmed that those mutants containing the βE56A mutation (i.e., 102/56, 102/56/76, 56/76, E56A, or E56K) reduced binding to F23.2 by at least 100-fold (≥2.5 kcal/mol). Alanine substitution at Thr55β exerts only a moderate effect (three- to fourfold reduction) on F23.2 binding (7). As expected, this moderate reduction in affinity is also observed in the T55A-containing mutants (i.e., 102/55, 55/76, 102/ 55/76, and T55A; Fig. 3 B, and Fig. 4). Thus, mAb F23.2 serves as an additional probe of the tertiary conformation of the scTCR mutants.
Figure 4.
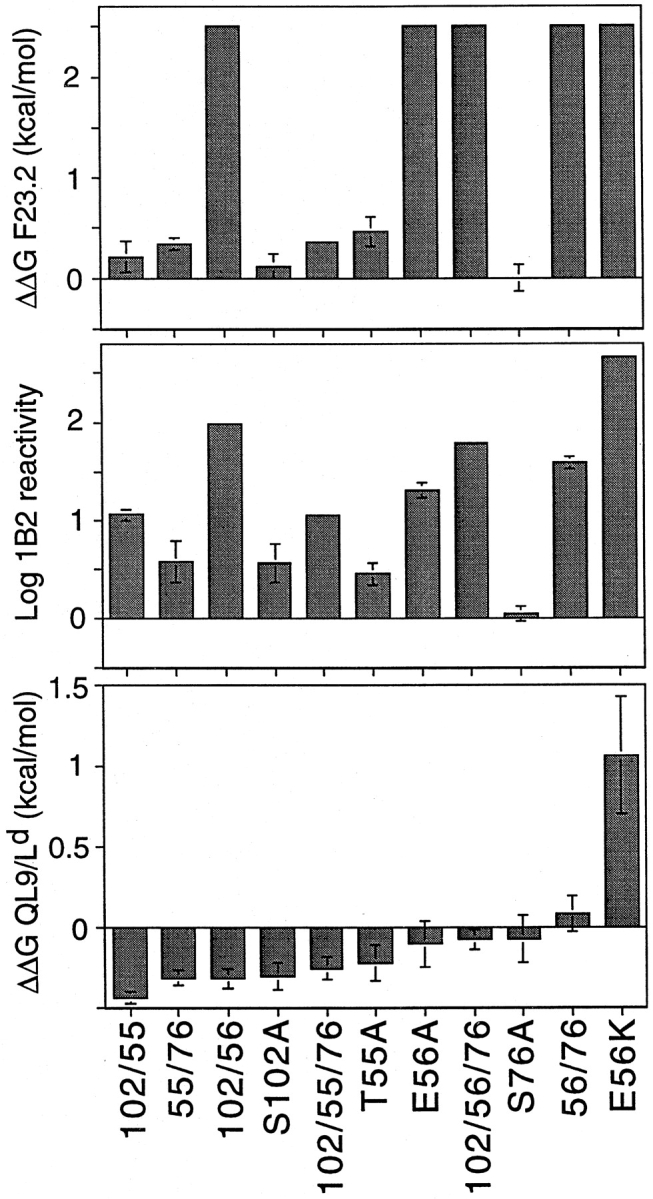
Summary binding data for alanine-substituent mutants analyzed in this study. Free energy changes for F23.2 (top) and QL9/Ld (bottom) binding were calculated from competition experiments using IC50(mutant)/IC50(wild-type) and were normalized for KJ16/F23.1 reactivity to account for slight variations in sample refolding and purity. 1B2 reactivity (middle) was determined from a capture ELISA format, with the concentration of scTCR yielding 50% maximal signal used analogously to IC50. ΔΔG values >0 indicate decreased binding.
The clonotypic antibody 1B2 recognizes an epitope on the 2C TCR which overlaps with the pMHC epitope (7). Therefore, it was of interest to determine the effect of double and triple alanine mutations on 1B2 binding. Similar to the effect observed with F23.2, each of the mutants that contained an E56 mutation had at least a 20-fold reduction in 1B2 activity (Fig. 3 C, and Fig. 4). The S102A mutant exhibited a more moderate fivefold reduced 1B2 activity, whereas T55A had threefold reduced activity. The combination of these two mutants in a single protein resulted in an approximately additive effect on binding (i.e., 11-fold reduced binding activity for either 102/55 or 102/55/76). Consistent with the observation that Ser76β is not involved in either the F23.2 or 1B2 epitopes (7), no additional effect on reactivity was observed with double or triple mutants that involved this position.
Peptide–MHC Reactivity.
To assess the effect of alanine-substituent mutations on pMHC recognition, mutants were tested for the ability to inhibit binding of 125I-labeled anti-Ld Fab fragments to the QL9/Ld alloantigen. In contrast to the significant impairments seen in F23.2 and 1B2 antibody reactivity, all of the alanine-substituent mutants bound to pMHC at least as well as the wild-type scTCR (Fig. 3 D, and Fig. 4). The greatest improvement in pMHC recognition was achieved with the 102/55 double mutant, which was 2.3-fold better than wild-type (free energy change [ΔΔG] = −0.44 kcal/mol). The effect observed with 102/55 appears to have been at least partially additive, in that S102A and T55A had ΔΔG values of −0.31 and −0.23 kcal/mol, respectively. In contrast, the effect seen with 102/56 (ΔΔG = −0.32 kcal/mol) is only a slight improvement on the binding energy of the S102A single mutant (S102A and E56A have ΔΔGs of −0.31 and −0.11 kcal/mol, respectively).
Residue Ser76β is within the HV4 loop and does not interact with antigen in the refined 2C/dEV8/Kb structure (16). Consequently, the modest improvement in pMHC affinity initially observed with S76A was attributed to indirect effects (7). In this study, the effects of the S76A mutation when paired with other mutations was variable. The 55/76 mutant appeared to bind pMHC marginally better than T55A, but incorporation of an S76A mutation reduced the affinity of 102/55, 102/56, and E56A (Fig. 4). Thus, whatever indirect conformational effects S76A makes on its own to increase affinity do not appear to be readily accommodated in the presence of other mutations.
In the model of the 2C TCR–QL9/Ld complex, residue Glu56β is positioned 9 Å opposite Glu 71 of the Ld α1 helix. We suggested that this pairing might represent an unfavorable electrostatic interaction (7). We reasoned that introduction of a positively charged lysine residue at position 56β might create a more favorable electrostatic pairing of the 2C and QL9/Ld surfaces. In contradiction to this notion, the E56K mutant exhibited significantly decreased binding to QL9/Ld (ΔΔG = 1.06 kcal/mol). Thus, simple introduction of a positive charge at this position does not create a more favorable electrostatic interaction between the TCR and pMHC.
Kinetics of Alanine-substituent Mutants.
Since the greatest improvement in binding affinity was achieved with the 102/55 double mutant, we sought to investigate the kinetic parameters involved in its binding to pMHC. Using immobilized mutant TCRs, binding to soluble pMHC complexes was analyzed by BIAcore SPR. The 102/55 double mutant was found to have on and off rates which were both slowed compared with wild-type (Fig. 5). The on and off rates for wild-type were 2.4- and 3.2-fold faster, respectively, than the rates for 102/55 (Fig. 5). The relatively larger difference observed for the off rates translates into a 23% lower overall equilibrium dissociation constant for 102/55 compared with wild-type, consistent with results from the Fab inhibition experiments. The reverse orientation, with biotinylated Ld–peptide complexes immobilized on the chip, yielded similar kinetic differences between the wild-type and 102/55 TCR (data not shown). In addition, the Kd values of the two proteins differed by 1.3–2.7-fold using three different methods of BIAcore analysis: (a) by kinetic measurements with immobilized TCR, Kd of wild-type TCR = 3.2 μM and of 102/55 TCR = 2.4 μM (Fig. 5); (b) by kinetic measurements with immobilized Ld–peptide, Kd of wild-type TCR = 1.9 μM and of 102/55 TCR = 0.7 μM (data not shown); and (c) by Scatchard analyses of binding at various Ld–peptide concentrations, Kd of wild-type TCR = 1.9 μM and of 102/55 TCR = 1.2 μM (data not shown). These results, together with data from the cell binding assay described above, indicate that the 102/55 mutant has a higher affinity that the wild-type.
Figure 5.

SPR analysis and comparison of wild-type and 102/55 TCR mutants. QL9/Ld complexes bind both TCRs compared with MCMV–Ld complexes (A–C). The higher affinity of the double mutant is due to a differential slowing of both its off rate (D and F) and its on rate (D and E) compared with the wild-type (WT). Similar results were obtained in the reverse orientation (immobilized Ld-QL9).
CD4/CD8 Repertoire-skewing Mutants
Previous work has suggested that a phenylalanine at position 27 (in CDR1) and a proline at position 51 (in CDR2) within the Vα3.2 region may each interact with MHC class I, thereby accounting for the tendency of Vα3.2+ T cells to skew toward a CD8 phenotype (13). To test the hypothesis that this preferential interaction involves an increase in affinity for MHC class I, we constructed mutant 2C scTCRs incorporating these “Vα3.2-like” substitutions into the 2C Vα3.1 region (see Table I). Additionally, we also analyzed alanine-substituent mutants at these two positions.
Table I.
Sequence Comparison of CDR1 and CDR2 Regions from 2C TCR (Vα3.1) and AV3 Gene Family Members
| CDR1 | CDR2 | |||
|---|---|---|---|---|
| 24 31 | 48 55 | |||
| Vα3.1 (2C) | YSYSATPY | KYYSGDPV | ||
| Vα3.2 (AV3S2) | ---FG--- | ---P---- | ||
| Vα3.1 (AV3S5) | ----G--- | -------- |
AV3 family sequences are from B6 mice. CTL 2C was isolated from a Balb.B mouse.
Antibody Reactivity.
Each of these Vα3.2-like mutants exhibited equivalent reactivity to wild-type in KJ16, F23.1, and F23.2 ELISAs, indicating the same degree of refolding (Fig. 6 A, and data not shown). Alanine substitution at either position did not affect recognition by 1B2, but S27F exhibited a slight increase in reactivity toward 1B2 and S51P had ∼60-fold reduced 1B2 reactivity (data not shown). The failure of these mutations to affect F23.2, KJ16, and F23.1 reactivity confirms that there has not been a global effect on TCR conformation, beyond the α chain CDR regions.
Figure 6.

Analysis of Vα3.2-like TCR mutants. Mutants exhibited equivalent reactivity toward the Vβ8-specific mAb KJ16 (A). Vα3.2-like mutants were impaired in their ability to inhibit 30-5-7 (anti-Ld) Fab binding to cell surface QL9/Ld (B). Summary data indicate the relative change in binding free energy for mutant TCR compared with wild-type (C). Free energy changes were calculated from competition experiments using IC50(mutant)/IC50(wild-type). Mutants with no detectable binding in the cell binding assay were assigned ΔΔG values = 1.5 kcal/mol, the sensitivity limit of the assay. ΔΔG values >0 indicate decreased binding.
Peptide–MHC Reactivity.
In both the 2C/dEV8/Kb structure and the 2C/QL9/Ld model, serines 27 and 51 are each involved in hydrogen bonding to conserved residues on the α helices (Fig. 1). We have previously reported that elimination of the hydroxyl moiety of Ser51α does not have an effect on recognition of the allogeneic pMHC ligand (7). Elimination of the hydroxyl at Ser27α similarly failed to disrupt binding to QL9/Ld (Fig. 6, B and C). Together, these data suggest that the amount of binding energy contributed by these two hydrogen bonds is negligible in the interaction of 2C with the Ld alloantigen.
In contrast to our expectations, substitution of a phenylalanine for the serine at position 27 in CDR1 resulted in a mutant that had 1.6-fold reduced binding to pMHC (ΔΔG = 0.24 kcal/mol; Fig. 6, B and C). Furthermore, the αS51P mutant in CDR2 exhibited ∼15-fold reduced binding to pMHC (ΔΔG = 1.5 kcal/mol). Combination of both mutations yielded approximately additive effects on both QL9/Ld binding (Fig. 6 C) and 1B2 binding (data not shown). The pMHC affinity of the S27F/S51P Vα3.2-like double mutant was below the detection limits of the cellular binding assay. Together, these results illustrate that the Vα3.2 mutations do not result in a general enhancement of binding to MHC class I molecules.
Discussion
This study addressed two questions of immunological significance regarding the binding of a TCR to its class I ligand. First, is it possible to use site-directed mutagenesis of CDR residues to increase the affinity of a TCR? Second, do various TCR mutants in Vα residues involved in repertoire skewing of a TCR toward the CD8 phenotype exhibit correlative affinities for the class I ligand?
The TCR from the alloreactive clone 2C could be used to address each of these questions. The 2C–QL9/Ld interaction represents the highest affinity TCR–pMHC interaction that has been measured (8), and our recent alanine scanning study identified several residues that might serve as the starting point for the rational design of higher affinity TCR mutants (7). To examine if TCR affinity could be improved, we concentrated on the four CDR positions initially found to have very modest increases in affinity when mutated to alanine. Thus, alanine mutants at positions Ser102α, Thr55β, Glu56β, and Ser76β were combined as either double or triple mutants. Since Thr55β and Glu56β are adjacent to each other in the refined 2C/dEV8/Kb structure, it was thought that they might be achieving their effect via similar mechanisms, and the 55/56 double mutant was not generated. Additionally, since elimination of the negative charge at position 56 seemed energetically favorable, the charge reversal mutant E56K was analyzed. Results of antibody reactivity of the various multiple alanine mutants were entirely consistent with the results from single mutants (e.g., each of the position 56 mutants had greatly reduced F23.2 binding), and also demonstrated that the mutants had equivalent refolding as measured by KJ16 and F23.1 (Figs. 3 A and Fig. 4, and data not shown).
The greatest increase in affinity for pMHC was observed with combined alanine substitution at positions Ser102α and Thr55β (i.e., 102/55), which exhibited an approximately twofold increase in affinity over wild-type and had slower association and dissociation kinetics than wild-type. The mechanism behind this increase in affinity is not clear, but based on structural analysis several possibilities exist. First, modeling of the 2C/QL9/Ld complex places Ser102α in a position of steric clash with ProP6 (26). Elimination of the hydroxyl moiety at this position might serve to eliminate or lessen this steric effect. Second, in the 2C/dEV8/Kb structure (16), Glu56β lies 9 Å opposite Glu71 of the α1 helix, a negatively charged residue common to both Kb and Ld that may represent a somewhat unfavorable electrostatic interaction surface. Alanine mutation of adjacent residue Thr55β might allow Glu56β to adopt a less electrostatically impaired conformation. However, the failure of E56K to improve pMHC affinity illustrates that the interactions that contribute to the binding energy of TCR–pMHC are subtle, context dependent, and not necessarily amenable to techniques such as charge swapping. Previous studies with other protein–protein interactions have shown similar difficulties in predicting whether charge complementation pairs might be successful in this regard.
The potential additivity of the various double and triple mutants analyzed in this study could not be predicted. This contrasts with the alanine scanning analysis of growth hormone and its receptor, in which additivity was the rule (28). This has led many to assume that additivity will be observed when noninteracting mutations are combined. However, consistent with our results with the TCR, a recent study to improve the affinity of an anti-HIV gp120 antibody observed unpredictable additivity (only one of six combinations was additive) when mutations were recombined (29). There is some evidence that a rigorous accounting of bound water molecules in protein–protein interactions can account for certain cases when double mutants behave in a less than additive manner (30). The reasons underlying the unpredictable additivity observed in this study are unclear, but could be related to the presence of water molecules and poor complementarity of fit observed in the refined structure (16).
Despite the only twofold increase in affinity achieved by directed site-specific mutagenesis, these results indicate the promise in using more sophisticated techniques to obtain even greater improvements in TCR affinity. In fact, the earliest efforts to engineer higher affinity antibodies yielded a similar magnitude of increase (31, 32). More recent approaches have greatly enhanced these efforts (33), and we predict the same will now be possible with the TCR. This expectation is also based on the observation that the TCR– pMHC interaction is marked by poor complementarity of fit (16). According to an algorithm used to calculate complementarity of fit, the 2C/dEV8/Kb and A6/HLA-A2/ Tax complexes gave values of 0.45 and 0.47, respectively, whereas most antigen–antibody interactions have values in the range of 0.66–0.68, and oligomeric proteins in the range of 0.68–0.75 (16). Clearly, there are regions of the TCR–pMHC interface where the fit can be improved through the introduction of amino acid substitutions. Thus, mutagenesis methods that explore the effects of amino acids other than alanine could yield still higher affinity soluble TCRs.
The second aspect of this report concerns the role of TCR affinity in repertoire skewing of a TCR toward the CD8 phenotype. The observation that Vα3.1 transgenic mice expressing the mutations S27F and S51P have a repertoire skewed toward a CD8+ phenotype suggested the notion that these positions contacted the class I selecting element, and the interaction was described as being preferential toward class I (13, 14). Structural analysis of the 2C TCR showed that the serines at positions 27 and 51 of the α chain contact conserved residues on the class I helices (16; depicted in Fig. 1). Together, these studies might lead one to predict (a) that the serines at positions 27 and 51 would contribute some energy to the TCR–pMHC interaction, and (b) that substitution of phenylalanine (at position 27) and proline (at position 51) would yield even higher affinity. Surprisingly, neither of these predictions was borne out in the measurements of binding affinities described here. Both alanine mutants at position 27 and 51 had negligible effects on the binding of the QL9/Ld ligand. That is, elimination of either hydroxyl group and the disruption of that hydrogen bonding capability had no effect on the equilibrium affinity of the TCR for pMHC. Substitution of the Vα3.2 residues (Phe and Pro) yielded not higher but lower affinities for the QL9/Ld ligand.
What, then, accounts for the apparent discrepancy between these results and the observed skewing? Serine is the most frequently occurring amino acid at both positions 27 and 51 in murine Vα genes (34). Threonine is also a frequent residue at both these positions, but Phe27α and Pro51α are found much less often. At position 27, the relative frequency at which these residues are found is as follows: serine 43%, threonine 22%, phenylalanine 1%. At position 51, serine occurs at 54% frequency, threonine at 25%, and proline at only 7% (34). Thus, there is conservation of hydroxyl-containing amino acids at these positions. Based on these findings and the 2C/dEV8/Kb structure, it has been suggested that these serines may help to orient the TCR diagonally on the pMHC (16). Serine residues at both positions are also found contacting MHC helices in the recent B7/Tax/HLA-A2 structure (35). Nevertheless, elimination of the hydroxyl group at either position failed to significantly effect the overall affinity of the 2C–pMHC interaction (Fig. 6). Two possible explanations may account for these observations. First, although the equilibrium affinity of the S27A and S51A mutants are unchanged from wild-type (perhaps due to compensation by bridging water molecules), it is possible that these hydroxyl residues have a favorable effect on binding kinetics (e.g., by increasing both on and off rates). This would be consistent with the kinetic differences observed for the 102/55 double alanine mutant, where elimination of two hydroxyl moieties at Ser102α and Thr55β slowed both the on and off rates. Second, it is possible that the interaction of the 2C TCR with the QL9/Ld ligand may differ fundamentally from the interaction of the 2C TCR with the dEV8/Kb ligand. Arguing against this explanation is the fact that those residues within the Kb α helices which contact Ser27α and Ser51α (Glu 58 and Arg 62 contacting Ser27α, and Ala 152, Gly 162, and Glu 166 contacting Ser51α) are identical in Ld. If the interaction with Kb and Ld was in fact fundamentally different, it is conceivable that Vα3.1 and Vα3.2 would differ in skewing toward a CD4 or CD8 phenotype if the selecting environment contained Ld as the only class I molecule.
The possibility that some class I molecules could interact with the Vα3 region in a different manner remains to be examined, and in this respect it is important to recognize that our findings involve measurements with a single TCR–pMHC system. Other TCR–pMHC interactions will no doubt need to be analyzed to support a particular model of CD4/CD8 skewing. Nevertheless, we believe our results with the alanine and Vα3.2-like substitutions at positions 27 and 51 are most consistent with the recent quantitative-instructional model for CD4/CD8 lineage commitment (36–38). According to this model, gradations in the intensity of signaling through the TCR complex of CD4+/CD8+ thymocytes determines lineage commitment. Signals of strong intensity promote CD4 differentiation, whereas weaker intensity signals promote a CD8 fate. Consistent with this model, Vα3.1 2C (Ser27α and Ser51α) binds class I with ∼15-fold higher affinity than Vα3.2-like 2C (Phe27α and Pro51α). Consequently, Vα3.1 is skewed toward CD4, whereas Vα3.2 is skewed toward CD8. Thus, it appears that energy changes amounting to as little as 0.25 kcal/mol (S27F) can skew the lineage commitment of developing thymocytes. Since 2C is a class I–restricted TCR, we are unable to address the effect of these mutations on binding to MHC class II. According to the quantitative-instructional model, one might expect that Vα3.1 (Ser27α and Ser51α) would contribute to increased affinity for MHC class II as well as class I. In this regard, the high frequency of serine residues at these positions (43 and 54%) is not surprising, since it would then act to promote general MHC reactivity. However, it is possible that interaction of thymocytes with class I molecules alone may explain Vα3 skewing, since commitment to the CD4+ lineage can occur in the absence of MHC class II molecules and does not require MHC class II–restricted TCRs (39).
A second, not mutually exclusive, possibility is that differential negative selection could also be involved in the observed CD4/CD8 skewing. According to this mechanism, CD8+ thymocytes which express Vα3.1 TCRs (i.e., possessing Ser27α and Ser51α) could bind selectively to MHC class I molecules, predisposing them to elimination by negative selection. CD8+ thymocytes which express Vα3.2 TCRs (i.e., Phe27α and Pro51α) would have an inherently lower affinity for MHC class I, thereby allowing them to escape negative selection. Presumably, this process would operate at the stage of the semimature single-positive heat stable antigen (HSA)hi thymocyte, a cell type that remains susceptible to negative selection within the thymic medulla (40). It has recently been estimated that 20–30% of thymocytes in mice are susceptible to negative selection (41), a figure which could easily accommodate a degree of repertoire skewing by differential negative selection.
In conclusion, we have provided direct evidence that the TCR framework can be engineered to support higher affinities for pMHC than are selected for during thymic development. Additionally, we have shown that the mechanism whereby two CDR mutants skew the T cell repertoire toward a CD8+ phenotype is not attributable to a general increase in affinity for MHC class I, but rather is consistent with predictions from the quantitative-instructional model of lineage commitment.
Acknowledgments
We wish to thank Drs. Carol Schlueter and Thomas Brodnicki for cloning several of the single-site mutants, Dr. Ted Hansen for providing the 30-5-7 hybridoma line, and Dr. Peter Cresswell for providing the T2-Ld cell line. We are grateful to Dr. Nicholas Gascoigne for critical reading of the manuscript. The expertise and assistance of the Mass Spectrometry and Genetic Engineering Facilities at the University of Illinois were greatly appreciated.
This work was supported by National Institutes of Health R01 grants GM55767 (to D.M. Kranz) and AI42267 (to L. Teyton).
Abbreviations used in this paper
- ΔΔG
free energy change
- MCMV
murine CMV
- pMHC
peptide–MHC complex
- scTCR
single-chain TCR
- SPR
surface plasmon resonance
References
- 1.Eisen HN, Sykulev Y, Tsomides TJ. Antigen-specific T-cell receptors and their reactions with complexes formed by peptides with major histocompatibility complex (MHC) proteins. Adv Protein Chem. 1996;49:1–56. doi: 10.1016/s0065-3233(08)60487-8. [DOI] [PubMed] [Google Scholar]
- 2.Eisen HN. Why affinity progression of antibodies during immune responses is probably not accompanied by parallel changes in the immunoglobulin-like antigen-specific receptors on T cells. Bioessays. 1986;4:269–272. doi: 10.1002/bies.950040609. [DOI] [PubMed] [Google Scholar]
- 3.Sykulev Y, Cohen RJ, Eisen HN. The law of mass action governs antigen-stimulated cytolytic activity of CD8+ cytotoxic T lymphocytes. Proc Natl Acad Sci USA. 1995;92:11990–11992. doi: 10.1073/pnas.92.26.11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 5.Foote J, Eisen HN. Kinetic and affinity limits on antibodies produced during immune responses. Proc Natl Acad Sci USA. 1995;92:1254–1256. doi: 10.1073/pnas.92.5.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, Teyton L, Wilson IA. An αβ T cell receptor structure at 2.5 Å and its orientation in the TCR-MHC complex. Science. 1996;274:209–219. doi: 10.1126/science.274.5285.209. [DOI] [PubMed] [Google Scholar]
- 7.Manning TC, Schlueter CJ, Brodnicki TC, Parke EA, Speir JA, Garcia KC, Teyton L, Wilson IA, Kranz DM. Alanine scanning mutagenesis of an αβ T cell receptor: mapping the energy of antigen recognition. Immunity. 1998;8:413–425. doi: 10.1016/s1074-7613(00)80547-6. [DOI] [PubMed] [Google Scholar]
- 8.Sykulev Y, Brunmark A, Tsomides TJ, Kageyama S, Jackson M, Peterson PA, Eisen HN. High-affinity reactions between antigen-specific T-cell receptors and peptides associated with allogeneic and syngeneic major histocompatibility complex class I proteins. Proc Natl Acad Sci USA. 1994;91:11487–11491. doi: 10.1073/pnas.91.24.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sha WC, Nelson CA, Newberry RD, Kranz DM, Russell JH, Loh DY. Positive and negative selection of an antigen receptor on T cells in transgenic mice. Nature. 1988;336:73–76. doi: 10.1038/336073a0. [DOI] [PubMed] [Google Scholar]
- 10.Utsunomiya Y, Bill J, Palmer E, Gollob K, Takagaki Y, Kanagawa O. Analysis of a monoclonal rat antibody directed to the α-chain variable region (Vα3) of the mouse T cell antigen receptor. J Immunol. 1989;143:2602–2608. [PubMed] [Google Scholar]
- 11.Jameson SC, Kaye J, Gascoigne NRJ. A T cell receptor Vα region selectively expressed in CD4+cells. J Immunol. 1990;145:1324–1331. [PubMed] [Google Scholar]
- 12.DerSimonian H, Band H, Brenner MB. Increased frequency of T cell receptor Vα12.1 expression on CD8+T cells: evidence that Vα participates in shaping the peripheral T cell repertoire. J Exp Med. 1991;174:639–648. doi: 10.1084/jem.174.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sim B-C, Zerva L, Greene MI, Gascoigne NRJ. Control of MHC restriction by TCR Vα CDR1 and CDR2. Science. 1996;273:963–966. doi: 10.1126/science.273.5277.963. [DOI] [PubMed] [Google Scholar]
- 14.Sim B-C, Wung JL, Gascoigne NRJ. Polymorphism within a TCRAV family influences the repertoire through class I/II restriction. J Immunol. 1998;160:1204–1211. [PubMed] [Google Scholar]
- 15.Sim BC, Lo D, Gascoigne NRJ. Preferential expression of TCR Vα regions in CD4/CD8 subsets: class discrimination or co-receptor recognition? . Immunol Today. 1998;19:276–282. doi: 10.1016/s0167-5699(98)01257-2. [DOI] [PubMed] [Google Scholar]
- 16.Garcia KC, Degano M, Pease LR, Huang M, Peterson P, Teyton L, Wilson IA. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science. 1998;279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
- 17.Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 1996;384:131–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 18.Landt O, Grunert H, Hahn U. A general method for rapid site-directed mutagenesis using the polymerase chain reaction. Gene. 1990;96:125–128. doi: 10.1016/0378-1119(90)90351-q. [DOI] [PubMed] [Google Scholar]
- 19.Schodin BA, Schlueter CJ, Kranz DM. Binding properties and solubility of single-chain T cell receptors expressed in E. coli. . Mol Immunol. 1996;33:819–829. doi: 10.1016/0161-5890(96)00038-7. [DOI] [PubMed] [Google Scholar]
- 20.Haskins K, Hannum C, White J, Rhoem N, Kubo R, Kappler J, Marrack K. The antigen-specific major histocompatibility complex–restricted receptor on T cells. VI. An antibody to a receptor allotype. J Exp Med. 1984;160:452–471. doi: 10.1084/jem.160.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staerz UD, Rammensee HG, Benedetto JD, Bevan MJ. Characterization of a murine monoclonal antibody specific for an allotypic determinant on T cell antigen receptor. J Immunol. 1985;134:3994–4000. [PubMed] [Google Scholar]
- 22.Kranz DM, Tonegawa S, Eisen HN. Attachment of an anti-receptor antibody to non-target cells renders them susceptible to lysis by a clone of cytotoxic T lymphocytes. Proc Natl Acad Sci USA. 1984;81:7922–7926. doi: 10.1073/pnas.81.24.7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozato K, Hansen TH, Sachs DH. Monoclonal antibodies to mouse MHC antigens. II. Antibodies to the H-2Ldantigen, the product of a third polymorphic locus of the mouse major histocompatibility complex. J Immunol. 1980;125:2473–2477. [PubMed] [Google Scholar]
- 24.Schlueter CJ, Schodin BA, Tetin SY, Kranz DM. Specificity and binding properties of a single-chain T cell receptor. J Mol Biol. 1996;256:859–869. doi: 10.1006/jmbi.1996.0132. [DOI] [PubMed] [Google Scholar]
- 25.Schlueter CJ, Manning TC, Schodin BA, Kranz DM. A residue in the center of peptide QL9 affects binding to both Ldand the T cell receptor. J Immunol. 1996;157:4478–4485. [PubMed] [Google Scholar]
- 26.Speir JA, Garcia KC, Brunmark A, Degano M, Peterson PA, Teyton L, Wilson IA. Structural basis of the 2C TCR allorecognition of H-2Ldpeptide complexes. Immunity. 1998;8:553–562. doi: 10.1016/s1074-7613(00)80560-9. [DOI] [PubMed] [Google Scholar]
- 27.Soo Hoo, W.F., M.J. Lacy, L.K. Denzin, E.W.J. Voss, K.D. Hardman, and D.M. Kranz. Characterization of a single-chain T cell receptor expressed in Escherichia coli. . Proc Natl Acad Sci USA. 1992;89:4759–4763. doi: 10.1073/pnas.89.10.4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wells JA. Additivity of mutational effects in proteins. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 29.Yang W-P, Green K, Pinz-Sweeney S, Briones AT, Burton DR, Barbas CF., III CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J Mol Biol. 1995;245:392–403. doi: 10.1006/jmbi.1995.0626. [DOI] [PubMed] [Google Scholar]
- 30.Covell DG, Wallqvist A. Analysis of protein-protein interactions and the effect of amino acid mutations on their energetics. The importance of water molecules in the binding epitope. J Mol Biol. 1997;269:281–297. doi: 10.1006/jmbi.1997.1028. [DOI] [PubMed] [Google Scholar]
- 31.Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol. 1992;226:889–896. doi: 10.1016/0022-2836(92)90639-2. [DOI] [PubMed] [Google Scholar]
- 32.Riechmann L, Weill M, Cavanagh J. Improving the antigen affinity of an antibody Fv-fragment by protein design. J Mol Biol. 1992;224:913–918. doi: 10.1016/0022-2836(92)90459-w. [DOI] [PubMed] [Google Scholar]
- 33.Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindqvist E, Schier R, Hemingsen G, Wong C, Gerhart JC, Marks JD. Efficient construction of a large nonimmune phage antibody library: the production of high- affinity human single-chain antibodies to protein antigens. Proc Natl Acad Sci USA. 1998;95:6157–6162. doi: 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arden B, Clark S, Kabelitz D, Mak TW. Mouse T-cell receptor variable gene segment families. Immunogenetics. 1995;42:501–530. doi: 10.1007/BF00172177. [DOI] [PubMed] [Google Scholar]
- 35.Ding YH, Smith KJ, Garboczi DN, Utz U, Biddison WE, Wiley DC. Two different T cell receptors bind in a similar diagonal mode to the HLA-A2/Tax peptide complex using different TCR amino acids. Immunity. 1998;8:403–411. doi: 10.1016/s1074-7613(00)80546-4. [DOI] [PubMed] [Google Scholar]
- 36.Itano A, Salmon P, Kioussis D, Tolaini M, Corbella P, Robey E. The cytoplasmic domain of CD4 promotes the development of CD4 lineage T cells. J Exp Med. 1996;183:731–741. doi: 10.1084/jem.183.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matechak EO, Killeen N, Hedrick SM, Fowlkes BJ. MHC class II-specific T cells can develop in the CD8 lineage when CD4 is absent. Immunity. 1996;4:337–347. doi: 10.1016/s1074-7613(00)80247-2. [DOI] [PubMed] [Google Scholar]
- 38.Basson MA, Bommhardt U, Cole MS, Tso JY, Zamoyska R. CD3 ligation on immature thymocytes generates antagonist-like signals appropriate for CD8 lineage commitment, independently of T cell receptor specificity. J Exp Med. 1998;187:1249–1260. doi: 10.1084/jem.187.8.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki H, Punt JA, Granger LG, Singer A. Asymmetric signaling requirements for thymocyte commitment to the CD4+ versus CD8+T cell lineages: a new perspective on thymic commitment and selection. Immunity. 1995;2:413–425. doi: 10.1016/1074-7613(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 40.Kishimoto H, Sprent J. Negative selection in the thymus includes semimature T cells. J Exp Med. 1997;185:263–271. doi: 10.1084/jem.185.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zerrahn J, Held W, Raulet DH. The MHC reactivity of the T cell repertoire prior to positive and negative selection. Cell. 1997;88:627–636. doi: 10.1016/s0092-8674(00)81905-4. [DOI] [PubMed] [Google Scholar]