

Abstract
The earliest contact between antigen and the innate immune system is thought to direct the subsequent antigen-specific T cell response. We hypothesized that cells of the innate immune system, such as natural killer (NK) cells, NK1.1+ T cells (NKT cells), and γ/δ T cells, may regulate the development of allergic airway disease. We demonstrate here that depletion of NK1.1+ cells (NK cells and NKT cells) before immunization inhibits pulmonary eosinophil and CD3+ T cell infiltration as well as increased levels of interleukin (IL)-4, IL-5, and IL-12 in bronchoalveolar lavage fluid in a murine model of allergic asthma. Moreover, systemic allergen-specific immunoglobulin (Ig)E and IgG2a levels and the number of IL-4 and interferon γ–producing splenic cells were diminished in mice depleted of NK1.1+ cells before the priming regime. Depletion of NK1.1+ cells during the challenge period only did not influence pulmonary eosinophilic inflammation. CD1d1 mutant mice, deficient in NKT cells but with normal NK cells, developed lung tissue eosinophilia and allergen-specific IgE levels not different from those observed in wild-type mice. Mice deficient in γ/δ T cells showed a mild attenuation of lung tissue eosinophilia in this model. Taken together, these findings suggest a critical role of NK cells, but not of NKT cells, for the development of allergen-induced airway inflammation, and that this effect of NK cells is exerted during the immunization. If translatable to humans, these data suggest that NK cells may be critically important for deciding whether allergic eosinophilic airway disease will develop. These observations are also compatible with a pathogenic role for the increased NK cell activity observed in human asthma.
Keywords: natural killer cells, NK1.1+ T cells, γ/δ T cells, eosinophils, allergic asthma
Airway mucosal inflammation in allergic asthma is thought to be dependent on T lymphocytes producing proinflammatory cytokines (1, 2). The production of IL-4 and IL-5 by these cells is considered to be pivotal for the recruitment of eosinophils to the airways, a hallmark of asthma (3, 4). The development of T cell effectors from naive T cells may depend on many factors, including the nature of antigen presentation and the local cytokine milieu during the period of T cell priming (5, 6). It has been suggested that rapid generation of key cytokines by cells of the innate immunity regulates the subsequent antigen-specific T cell response (7–9). Thus, cells such as NK cells, NK1.1+ T cells (NKT cells),1 and γ/δ T cells have been given tentative roles in determining the nature of the acquired immune response. This possibility has been discussed particularly with regard to host response to infection (8, 9).
NK cells are recognized as an important component in immune responses against a number of pathogens (10, 11). Their cytokine production rather than cytolytic activity contributes to the resistance against infectious agents (10, 11). NK cells can rapidly produce IFN-γ. However, these cells may also produce a variety of other immunoregulatory mediators, including TGF-β, TNF-α, TNF-β, GM-CSF, macrophage inflammatory protein (MIP)-1α, IL-1, IL-2, IL-3, IL-5, IL-8, and IL-10 (10–12). In addition to NK cells, mouse NK1.1+ cells comprise a small population of cells that coexpress NK1.1 and TCR, i.e., NKT cells (13– 15). A human counterpart of the mouse NKT cell, coexpressing IL-4 and IFN-γ, has recently been identified (16).
Little is known about the role of NK cells in induction of immune responses to allergens. Bogen et al. (17) have demonstrated that the earliest detectable response to subcutaneous administration of a protein antigen (OVA) in adjuvant was the appearance of IFN-γ–producing NK1.1+ cells at the site of immunization. NK cells are normally present in considerable numbers in human lung interstitium, suggesting involvement of these cells in pulmonary immunity (18). Furthermore, it has been demonstrated that patients with asthma show increased numbers of NK cells and stronger NK activity in peripheral blood than normal healthy blood donors (19–21). NK cell activity may also be increased after bronchial allergen challenge in asthmatic subjects (22). However, the focus in these previous clinical studies has been on the possible association of asthma with a reduced risk for tumor disease development.
γ/δ T cells represent another lymphocyte population that can produce various cytokines early in an immune response. A recent study has shown that γ/δ T cells differentially produce IFN-γ and IL-4 in response to Th1- and Th2-inducing pathogens (23). However, the role of γ/δ T cells in allergic diseases such as asthma is still controversial. McMenamin et al. (24) have shown that CD8+ γ/δ T cells in mice may specifically downregulate IgE responses to soluble OVA. Recently, Zuany-Amorim et al. (25) reported that γ/δ T cells are required for inducing allergen-specific IgE and IgG1 responses and Th2-mediated airway inflammation in a mouse model of asthma.
We hypothesized that innate immunity, particularly NK cells, NKT cells, and γ/δ T cells, may be involved in the processes that cause eosinophilic airway inflammation in immunized and allergen-exposed mice. Specifically, this study asks whether mice that are depleted of NK1.1+ cells, i.e., NK cells and NKT cells (26), and whether mice that genetically lack NKT cells (27) or γ/δ T cells (28), develop pulmonary inflammation in an established model of allergic asthma (29, 30).
Materials and Methods
Animals and Study Design.
Male C57BL/6 mice (n = 127, 8–9 wk of age in experiment I and II, 4–6 mo of age in experiment IV) were purchased from Bomholtgaard (Table I). CD1d1 mutant (129/Sv × C57BL/6) mice and wild-type littermates (n = 20, 7 wk of age) were used in experiment III (27). TCR δ gene mutant (C57BL/6) mice (n = 6, 6 mo of age) were purchased from The Jackson Laboratory (28). All mice were kept in well-controlled animal housing facilities and had free access to tap water and pelleted food throughout the experimental period. Animals were killed by intraperitoneal injection of pentobarbital 8, 24, or 30 h after the last aerosol exposure. Each group consisted of 5–11 animals. Groups of mice were subjected to bronchoalveolar lavage (BAL) and/or blood sampling before lungs and spleen were dissected out.
Table I.
Study Design
| Groups | Treatment/ genotype | Start of treat- ment | Challenge | Termina- tion of experi- ment | ||||
|---|---|---|---|---|---|---|---|---|
| d | h | |||||||
| Experiment I | ||||||||
| 1 | IgG | −2 | SAL ×2 | 8 | ||||
| 2 | mAb NK1.1 | −2 | SAL ×2 | 8 | ||||
| 3 | IgG | −2 | OVA ×2 | 8 | ||||
| 4 | mAb NK1.1 | −2 | OVA ×2 | 8 | ||||
| 5 | IgG | −2 | SAL ×7 | 8 | ||||
| 6 | mAb NK1.1 | −2 | SAL ×7 | 8 | ||||
| 7 | IgG | −2 | OVA ×7 | 8 | ||||
| 8 | mAb NK1.1 | −2 | OVA ×7 | 8 | ||||
| Experiment II | ||||||||
| 9 | IgG | −2 | OVA ×7 | 8 | ||||
| 10 | mAb NK1.1 | −2 | OVA ×7 | 8 | ||||
| 11 | mAb NK1.1 | −7 | OVA ×7 | 8 | ||||
| 12 | IgG | 13 | OVA ×7 | 8 | ||||
| 13 | mAb NK1.1 | 13 | OVA ×7 | 8 | ||||
| 14 | IgG | −2 | OVA ×7 | 30 | ||||
| 15 | mAb NK1.1 | −2 | OVA ×7 | 30 | ||||
| Experiment III | ||||||||
| 16 | Wild-type (CD1+/+) | – | OVA ×7 | 8 | ||||
| 17 | CD1d1 mutant (CD1−/−) | – | OVA ×7 | 8 | ||||
| Experiment IV | ||||||||
| 18 | Wild-type | – | SAL ×7 | 24 | ||||
| 19 | Wild-type | – | OVA ×7 | 24 | ||||
| 20 | γ/δ T cell– deficient | – | OVA ×7 | 24 |
Four independent experiments were performed and are shown separately. All mice were immunized with OVA on day 0.
Immunization and Challenge.
We have used a protocol slightly modified from that developed by Brusselle and colleagues (29). On the first day of the experiment (day 0), all mice were actively immunized by injection of 10 μg i.p. chicken OVA (Grade III; Sigma), adsorbed to 1 mg of alum adjuvant. From days 14–15 (groups 1–4) or from days 14–20 (all other groups) after immunization, the mice were exposed daily to aerosolized saline (SAL) or OVA over a 30-min period by placing groups of 5–12 awake mice in an exposition chamber (Table I). The aerosols were generated into the chamber using a nebulizer (500 ml Inline Micronebulizer driven at 4 bar; Bird Co.). The concentration of OVA in the nebulizer was 1% wt/vol.
Depletion of NK1.1+ Cells.
To deplete NK1.1+ cells in vivo, mice were injected starting 2 d before immunization with 100 μg i.p. of anti-NK1.1 mAb (26), and every 5 d thereafter with 25 μg i.p. of the anti-NK1.1 mAb until termination of the experiment. One group of mice was depleted of NK1.1+ cells in a similar manner, but the treatment regimen started 7 d before immunization (group 11). A third group of animals was also depleted of NK1.1+ cells, but the treatment started 1 d before first allergen aerosol challenge (group 13). Control mice were injected with a similar volume (0.2 ml) and dose of mouse IgG antibody (Sigma) as the appropriate isotype control. The efficacy of depletion of NK1.1+ cells was monitored by flow cytometric analysis of spleen cells at the end of the experimental period. Animals exhibiting >1.0% NK1.1+ cells in the spleen at the end of the experiment were excluded from the study (n = 5).
Histochemistry.
Lung tissue specimens obtained 8, 24, and 30 h after the last OVA or SAL exposure were immersed overnight in Stefanini's fixative (2% paraformaldehyde and 0.2% picric acid in 0.1 M phosphate buffer, pH 7.2), rinsed repeatedly in buffer (Tyrode buffer supplemented with 10% sucrose), frozen in mounting medium (Tissue-Tek; Miles, Inc.), and stored at −80°C until sectioning. Eosinophils were detected by histochemical visualization of cyanide-resistant eosinophil peroxidase activity (31). In brief, cryosections (10 μm) were incubated for 8 min at room temperature in PBS buffer (pH 7.4) supplemented with 3,3-diaminobenzidine tetrahydrochloride (60 mg/100 ml; Sigma), 30% H2O2 (0.3 ml/100 ml), and NaCN (120 mg/100 ml). Slides were then rinsed in water and mounted in Kaiser's medium (Merck). Eosinophils were identified by their dark brown reaction product. For histochemical detection of mucus-containing cells in lungs, 10-μm cryosections were stained with periodic acid-Schiff reagent (PAS). Distinct purple-red granules could be observed in the mucus-containing cells. For assessment of general airway morphology, sections were stained with hematoxylin and erythrosin.
Immunohistochemistry.
Cryosections (10 μm) were fixed in cold acetone diluted 1:2 in distilled water for 30 s, followed by final fixation in cold acetone (100%) for 5 min. Subsequent incubations were carried out sequentially for 30 min, with 5 min in PBS between each step. Unspecific antibody binding was blocked by incubation with PBS containing 10% (vol/vol) normal rabbit serum for 5 min (X0902; Dako). Incubation with an mAb recognizing the mouse CD3 antigen (clone KT3; Serotec) was followed by rabbit anti–rat IgG antibody (Z494; Dako) diluted 1:50 in PBS containing 10% (vol/vol) normal rabbit serum. After a final incubation with a monoclonal rat alkaline phosphatase–anti-alkaline phosphatase reagent (D488; Dako), the alkaline phosphatase reaction was developed using BCIP/NBT/ INT mixed with Levamisole (K599 and X3021; Dako) for 10 min.
Analysis of Cytokines in BAL Fluid.
All mice in experiments I and III and groups 12 and 13 in experiment II were subjected to BAL at 8 h after last aerosol exposure (Table I). After the animals had been anesthetized with an intraperitoneal injection of pentobarbital, a tracheal cannula was inserted via a midcervical incision and the airways were lavaged twice with 0.9 ml of PBS (Life Technologies). The BAL fluid (BALF) was immediately centrifuged (10 min, 4°C, 160 g), and the supernatant was rapidly frozen. Commercial ELISA kits were used to measure levels of IL-4 (Biosource International), IL-5 (Nycomed Amersham plc), IL-12 (Genzyme), and IFN-γ (Biosource International) in the BALF. The limit of detection was 5 pg/ml for IL-4 and IL-5, 10 pg/ml for IL-12, and 1 pg/ml for IFN-γ.
Measurement of OVA-specific IgE and IgG2a in Plasma.
Blood was drawn from all mice in experiments II–IV (Table I) by cardiac puncture and placed in EDTA tubes. After centrifugation, the plasma samples were rapidly frozen. OVA-specific IgE and IgG2a levels were determined by sandwich ELISA in 96-well ELISA plates. Sample wells were coated overnight with OVA grade III (100 μg/ml, 100 μl/well; Sigma). Wells were then blocked with 3% BSA in PBS (200 μl/well) at 37°C for 2 h. Diluted samples and standard (100 μl/well) were incubated overnight at 4°C. Plates were washed with 0.05% Tween 20/PBS and incubated with biotin-conjugated anti–mouse IgE (diluted 1:500) or IgG2a (diluted 1:100, 100 μl/well; PharMingen) at room temperature for 90 min. After another wash procedure, plates were incubated for 90 min at room temperature with 100 μl/well of ExtrAvidin-HRP (Sigma) diluted 1:600 in 1% BSA/PBS. Plates were developed with TMB Microwell Peroxidase Substrate (100 μl/well; Kirkegaard & Perry Labs). Plates were read at 450 nm. A plasma pool of OVA-immunized mice was used as internal laboratory standard. A 1:100 dilution of this pool was chosen as arbitrary unit.
Enzyme-linked Immunospot Analysis of Cytokine-producing Spleen Cells.
The number of spleen cells producing IL-4 and IFN-γ was evaluated in groups 9 and 10, 14 and 15, and 16 and 17 (Table I). Spleens were rapidly dissected out and placed in ice-cooled RPMI 1640 medium (Life Technologies). The enzyme-linked immunospot (ELISPOT) method was carried out the following day essentially as described elsewhere (32). Purified primary antibodies for IFN-γ (R4-6A2) and for IL-4 (BVD4-1D11) obtained from Nordic Biosite were diluted to 15 μg/ml in PBS and coated to plastic plates (Dynatech). The plates were washed, and Con A–stimulated (2.5 μg/ml; Sigma), OVA-stimulated (10 μg/ml, Grade III; Sigma), or unstimulated cells were added at suitable concentrations and incubated overnight at 37°C in 5% CO2. After washing, the plates were incubated with secondary biotinylated antibodies diluted in PBS to 1 μg/ml specific for IFN-γ (XMG1.2; Nordic Biosite) and IL-4 (BVD6-24G2; Nordic Biosite), respectively. After incubation at 4°C overnight, the plates were washed, avidin–alkaline phosphatase (Dako) was added, and after 2 h in room temperature and further washing, BCIP phosphatase substrate solution (Sigma) was added and the plates were developed. Spots in each well were counted with a reversed microscope. The number of CD3+ spleen cells was determined, and the cytokine response was expressed as the number of spot-forming cells (SFCs)/CD3+ cells.
Quantification and Statistics.
For evaluation of the number of eosinophils and CD3+ cells in pulmonary tissue, 40 randomly selected areas (0.04 mm2 each) in 1 lung section from each animal were examined. The number of eosinophils and CD3+ cells in the 40 areas was counted at a magnification of 400, and the mean was expressed as eosinophils or CD3+ cells per unit area. All quantifications were performed blind. Data are expressed as mean ± SEM unless otherwise indicated. To calculate significance levels between treatment groups, the Student's t test was used throughout the study. Values below detection limits were assigned the value of the detection limit. To achieve comparable SDs, ELISA values were transformed to logarithms before statistical analysis. Probabilities <0.05 were used as the generally accepted level of statistical significance for differences between mean values.
Results
Early Allergen-induced Airway Changes in Mice Depleted of NK1.1+ Cells before Immunization.
Histologic analysis of lungs taken 8 h after last (second) aerosol exposure from both IgG-treated and NK1.1+ cell–depleted mice receiving two OVA challenges (groups 3 and 4) exhibited a slight pulmonary eosinophilia (3.9 ± 1.0 and 3.8 ± 0.7 cells/unit area, respectively). Although a few small eosinophilic infiltrates were detected in lung tissue in the majority of these animals, the eosinophilia was not significantly increased compared with corresponding immunized and SAL-exposed animals (2.2 ± 0.4 and 2.3 ± 0.4 cells/unit area, respectively). To detect the emerging Th2 response in the lungs of these animals after allergen exposure, we measured IL-4 in BALF using ELISA. The levels of IL-4 in BALF were increased in immunized and OVA-challenged IgG-treated mice compared with corresponding immunized and SAL-exposed animals (142.7 ± 14.8 vs. 89.7 ± 19.7 pg/ml; P < 0.05). A tendency towards higher IL-4 levels in NK1.1+ cell–depleted animals compared with IgG-treated mice was demonstrated. Thus, IL-4 levels in BALF from OVA-challenged and NK1.1+ cell–depleted mice did not differ significantly from those in the corresponding SAL-exposed group (159.0 ± 25.2 vs. 131.4 ± 7.4 pg/ml).
Late Allergen-induced Airway Changes in Mice Depleted of NK1.1+ Cells before Immunization.
Immunized and IgG-treated mice receiving seven OVA challenges exhibited at the 8 h time point a marked eosinophilia perivascularly and peribronchially in the lung (groups 7 and 9; Fig. 1, a and b, and Fig. 2 a). In contrast, corresponding mice depleted of NK1.1+ cells (groups 8 and 10) showed a clearly inhibited eosinophilia in lung tissue (Fig. 1, a and b). These mice exhibited a few scattered eosinophilic infiltrates, or a complete absence of pulmonary inflammation (Fig. 2 b). Eosinophilic infiltrates were not observed in lung tissue from SAL-exposed mice of either the IgG-treated or the NK1.1+ cell–depleted groups (groups 5 and 6; Fig. 1 a). The marked attenuation of lung tissue eosinophilia in mice depleted of NK1.1+ cells was still observed 30 h after the last OVA exposure (group 15; Fig. 1 b). At this time point, one outlier (determined by Q test) was obvious, without which the difference between IgG-treated and depleted animals had been statistically significant (P < 0.05). It is possible that this single animal (exhibiting the second most pronounced pulmonary eosinophilia in the experiment) was not successfully depleted of NK1.1+ cells after the first injection of mAb NK1.1, 2 d before immunization.
Figure 1.

(a and b) Effect of depleting NK1.1+ cells before immunization on lung tissue eosinophilia. Mice were immunized on day 0 and challenged daily with aerosolized OVA or SAL on days 14–20. The animals were killed 8 or 30 h after last challenge. In two independent experiments, mice received mAb NK1.1 treatment. The first experiment (a) involved BAL, which may change the cellular composition in the lung tissue. Therefore, the two experiments are shown separately. Solid bars, mean (n = 5–8 in each group); OVA, OVA-challenged animals; SAL, SAL-challenged animals; IgG, IgG-treated animals; NK1.1, mAb NK1.1– treated animals. In a, *P < 0.05 between OVA- and SAL-challenged mice. In b, ***P < 0.001 between OVA-challenged IgG-treated and NK1.1+ cell–depleted mice. At the 30-h time point, one outlier in the NK1.1+ cell–depleted group was demonstrated (determined by Q test), without which the difference between IgG-treated and depleted animals had been statistically significant (P < 0.05).
Figure 2.
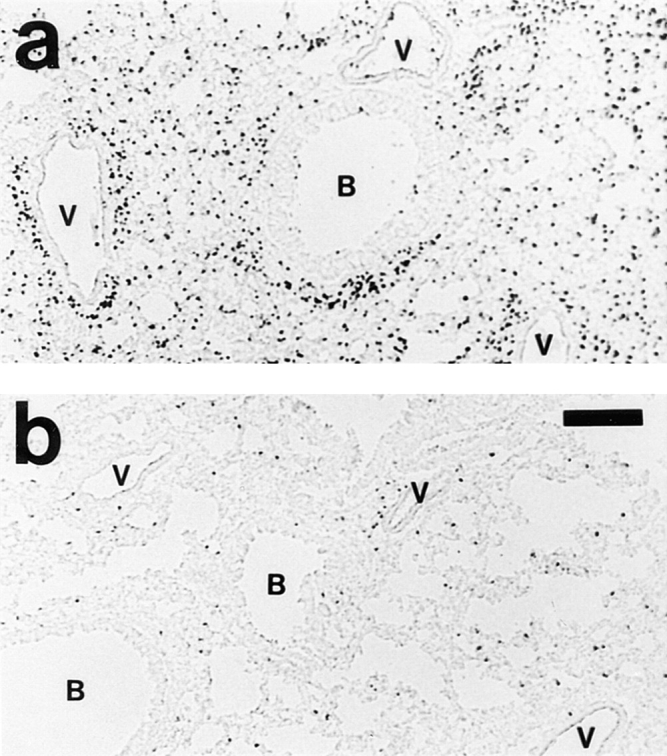
Effect of depleting NK1.1+ cells before immunization on the development of eosinophilic pulmonary inflammation. Animals were immunized and repeatedly challenged to aerosolized OVA (seven times). 8 h after last exposure, lungs were processed for histologic analysis. Eosinophils are visualized by histochemical demonstration of cyanide-resistant eosinophil peroxidase activity. Multifocal perivascular and peribronchial eosinophilic distribution in the lung tissue is seen in immunized and IgG-treated mice 8 h after last OVA exposure (a). Corresponding mice depleted of NK1.1+ cells exhibited a few scattered eosinophilic infiltrates only (b), or a complete absence of pulmonary inflammation (not shown). B, bronchus; V, blood vessel. Bar, 150 μm.
The number of CD3+ cells in lung tissue from immunized and IgG-treated animals receiving seven OVA challenges was increased compared with corresponding immunized and SAL-exposed animals at the 8-h time point (12.0 ± 2.0 vs. 5.1 ± 0.4 cells/unit area; P < 0.05). In contrast, the number of CD3+ cells in lung tissue from immunized and NK1.1+ cell–depleted animals remained low after allergen challenges (6.7 ± 1.0 vs. 6.8 ± 1.4 cells/unit area in corresponding SAL-exposed animals).
Sections stained with hematoxylin and erythrosin or PAS further demonstrated that the pulmonary eosinophilia was accompanied by a dense infiltration of mononuclear cells and an evident increase in airway epithelial mucus cells in lungs of OVA-exposed IgG-treated mice. Also, these morphological changes were markedly reduced in OVA-challenged mice depleted of NK1.1+ cells.
To determine the type of immune response (Th1 and/or Th2) being induced in the airways of immunized mice after multiple allergen aerosol exposures, we measured cytokines in BALF taken 8 h after last aerosol exposure. The levels of IL-4 in BALF were similar in both IgG-treated and NK1.1+ cell–depleted mice receiving seven OVA challenges compared with corresponding SAL-exposed animals (Fig. 3). Thus, the increased levels demonstrated after two OVA exposures in immunized IgG-treated mice (but not in corresponding mice depleted of NK1.1+ cells) were not detected under these more chronic conditions. BALF from IgG-treated mice receiving seven allergen challenges contained measurable amounts of IL-5 (P < 0.05, compared with SAL-exposed IgG-treated animals; Fig. 3). In contrast, NK1.1+ cell–depleted animals failed to release IL-5 after OVA exposure (Fig. 3). The levels of IL-12 in BALF increased after allergen exposure in IgG-treated animals (P < 0.001, compared with corresponding SAL-challenged animals; Fig. 3). In contrast, IL-12 levels in BALF from NK1.1+ cell–depleted mice remained low in response to allergen exposure, similar to the levels in corresponding SAL-exposed animals. The levels of IFN-γ in BALF decreased after allergen challenge to values very near or below the detection limit of the assay in five out of seven IgG-treated animals (Fig. 3). Interestingly, the two animals exhibiting high IFN-γ values showed undetectable IL-5 levels in BALF and no pulmonary eosinophilia. OVA challenge of mice depleted of NK1.1+ cells caused a moderate decrease in levels of IFN-γ (P < 0.05, compared with corresponding SAL-exposed mice; Fig. 3).
Figure 3.

Effect of depleting NK1.1+ cells before immunization on BALF cytokine levels 8 h after last OVA or SAL exposure. Mice were immunized on day 0 and challenged daily with aerosolized OVA or SAL on days 14–20. Solid bars, mean (n = 5–8 per group); OVA, OVA-challenged animals; SAL, SAL-challenged animals; IgG, IgG-treated animals; NK1.1, mAb NK1.1–treated animals. Dotted lines, detection limit of the assays. *P < 0.05, ***P < 0.001.
Systemic Allergen-specific IgE and IgG2a Levels in Mice Depleted of NK1.1+ Cells before Immunization.
To assess the peripheral immune response to immunization and allergen challenge, we measured OVA-specific IgE and IgG2a levels in plasma using ELISA. Immunization and allergen exposure of IgG-treated animals induced both OVA-specific IgE and IgG2a (Fig. 4). In corresponding NK1.1+ cell– depleted animals, this induction of OVA-specific IgE and IgG2a was significantly suppressed (P < 0.01, P < 0.05, compared with immunized and OVA-challenged mice treated with IgG).
Figure 4.

Effect of depleting NK1.1+ cells before immunization on systemic levels of OVA-specific IgE and IgG2a. Mice were immunized on day 0 and challenged daily with aerosolized OVA on days 14–20. Plasma was collected 8 or 30 h after last OVA challenge. Immunization and allergen exposure of IgG-treated animals induced both allergen-specific IgE and IgG2a. In corresponding NK1.1+ cell–depleted animals, this induction of allergen-specific IgE and IgG2a was significantly suppressed (**P < 0.01 and *P < 0.05, respectively). Solid bars, mean (n = 13–14 per group); IgG, IgG-treated animals; NK1.1, mAb NK1.1–treated animals.
Cytokine Production by Spleen Cells after Allergen Challenge in Mice Depleted of NK1.1+ Cells before Immunization.
To estimate the systemic T cell cytokine response in spleen after immunization and OVA challenge, we used ELISPOT. The number of IL-4–producing spleen cells (both unstimulated and OVA-stimulated cells) was overall lower in mice depleted of NK1.1+ cells compared with IgG-treated animals (Fig. 5, a and b). The number of OVA-stimulated IL-4–producing cells at the 8-h time point was significantly lower in NK1.1+ cell–depleted mice compared with IgG-treated animals (P < 0.05). Similar patterns were obtained for IFN-γ–producing cells (Fig. 5, a and b). Significant differences were obtained at the 8- and 30-h time points between the number of unstimulated IFN-γ–producing spleen cells from IgG-treated and NK1.1+ cell–depleted animals (P < 0.05).
Figure 5.
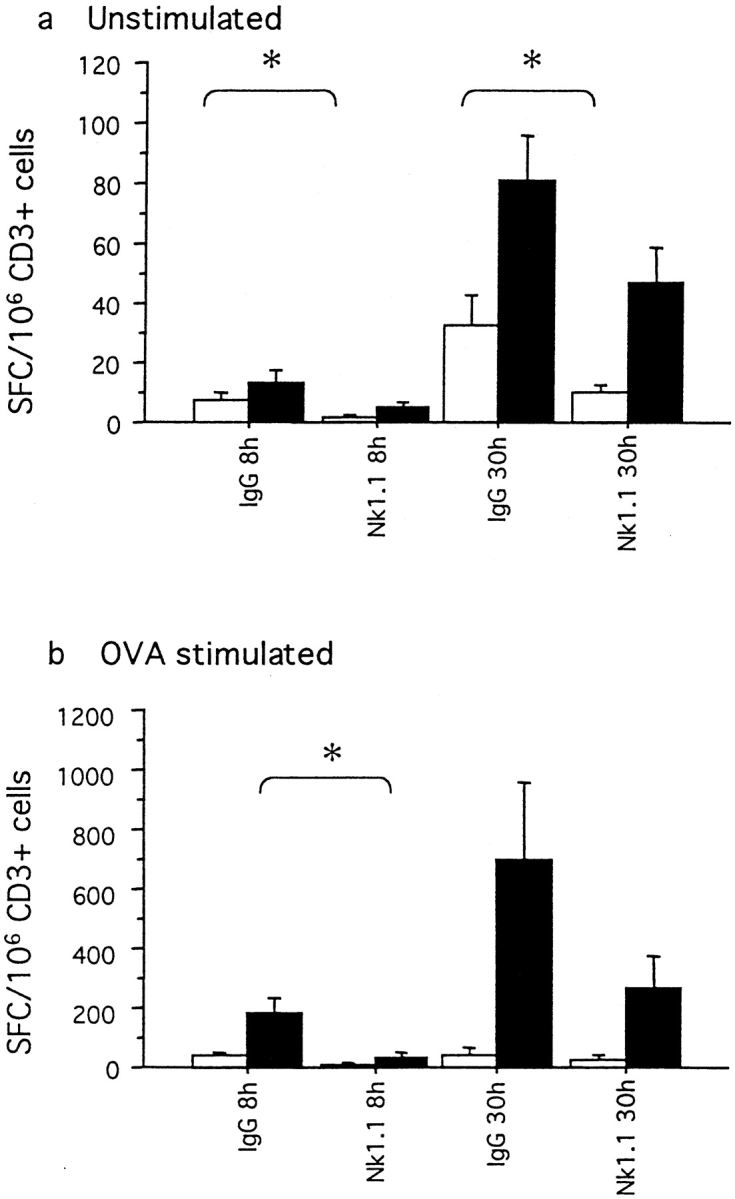
(a and b) Cytokine production by splenic cells after allergen challenge in IgG-treated and NK1.1+ cell–depleted mice. Mice were immunized on day 0 and challenged daily with aerosolized OVA on days 14–20. Mean (± SEM) numbers of IFN-γ (white bars) and IL-4 (black bars) spot-forming spleen cells (SFCs)/106 cells. The numbers of SFCs for unstimulated cells (a) and the numbers of SFCs for cells stimulated with OVA (b) are shown. IgG, IgG-treated animals; NK1.1, animals depleted of NK1.1+ cells. *P < 0.05.
Allergen-induced Changes in Mice Depleted of NK1.1+ Cells during the Challenge Period Only.
To determine if NK1.1+ cells are required for the initiation of the immune response at the time of immunization or for the onset of pulmonary inflammation secondary to allergen challenge, mice were depleted of NK1.1+ cells during the challenge period only (group 13). Immunization and OVA exposure (seven times) of mice depleted of NK1.1+ cells during the challenge period only led to the development of a pulmonary eosinophil-rich inflammation similar to that observed in corresponding IgG-treated animals (26.3 ± 3.9 and 19.5 ± 2.5 eosinophils/unit area, respectively). These groups of mice also exhibited similar levels of IL-4 (70.9 ± 8.8 and 85.6 ± 20.9 pg/ml, respectively), IL-12 (1,524.5 ± 137.1 and 1,184.5 ± 202.1 pg/ml, respectively), and IFN-γ (levels < detection limit in both groups) in BALF. Furthermore, systemic levels of OVA-specific IgE were not altered (4,132.3 ± 1,661.3 versus 6,281.0 ± 2,563.6 U/ml in the corresponding IgG-treated group).
Allergen-induced Changes in Mice Depleted of NK1.1+ Cells 7 d before Immunization.
To confirm that no acute nonspecific effects (such as cytokine release) of depletion with mAb NK1.1 caused the demonstrated effects on the immune response, mice were depleted of NK1.1+ cells from day –7 before immunization (group 11). This group of mice exhibited a reduction of lung tissue eosinophilia compared with mice treated with IgG from day −2 before immunization (group 9) (14.2 ± 2.4 vs. 25.9 ± 3.7 cells/unit area; P < 0.05). Also, these mice exhibited suppressed levels of OVA-specific IgE compared with mice treated with IgG from day –2 before immunization (group 9) (1,133.7 ± 428.8 vs. 6,971.1 ± 4,735.1 U/ml; P < 0.05). Depletion of NK1.1+ cells from day −7 before immunization also suppressed OVA-specific IgG2a levels. In this group, all mice had levels of IgG2a below detection limit (P < 0.05, compared with mice treated with IgG from day –2 before immunization).
Allergen-induced Changes in Mice Deficient in NKT Cells.
To elucidate if the suppression of the allergic immune response seen after NK1.1+ cell depletion requires NK cells or NKT cells, CD1d1 mutant mice, which selectively lack NKT cells (group 17), were immunized and OVA challenged. Immunization and OVA exposure (seven times) of mice deficient in NKT cells led to the development of a pulmonary eosinophil-rich inflammation similar to that observed in corresponding wild-type animals (Fig. 6). CD1d1 mutant and wild-type mice also exhibited similar levels of OVA-specific IgE (4,342.9 ± 1,054.3 and 6,814.8 ± 2,702.9 U/ml, respectively). The numbers of IL-4– and IFN-γ–producing spleen cells in immunized and allergen-challenged CD1d1 mutant and wild-type mice were determined by the ELISPOT method. There was a tendency, although statistically insignificant, of reduced numbers of IL-4–producing unstimulated and OVA-stimulated spleen cells from CD1d1 mutant mice compared with wild-type mice (5.4 ± 1.2 vs. 16.2 ± 6.0 SFCs/106 CD3+ unstimulated cells and 150.0 ± 27.1 vs. 418.2 ± 212.8 SFCs/106 CD3+ OVA-stimulated cells). The number of unstimulated IFN-γ–producing cells was increased in CD1d1 mutant mice compared with wild-type mice (60.0 ± 17.2 vs. 15.4 ± 7.8 SFCs/106 CD3+ cells; P < 0.05). There was also a tendency of increased numbers of OVA-stimulated IFN-γ–producing cells from mutant mice compared with wild-type animals (134.4 ± 54.1 vs. 40.9 ± 11.9 SFCs/106 CD3+ cells).
Figure 6.

CD1d1 (CD1−/−) mutant mice and wild-type controls (CD1+/+) were immunized on day 0 and challenged daily with aerosolized OVA on days 14–20. The number of eosinophils in lung tissue was similar in both groups of mice. Solid bars, mean (n = 9–11 per group).
Allergen-induced Changes in Mice Deficient in γ/δ T Cells.
A moderate reduction, although statistically insignificant, of lung tissue eosinophilia was observed in OVA-challenged γ/δ T cell–deficient animals compared with corresponding wild-type mice (Fig. 7). Similarly, no significant difference in systemic levels of OVA-specific IgE was observed between OVA-challenged γ/δ T cell–deficient animals and corresponding wild-type mice (1,066.3 ± 314.5 and 2,567.4 ± 1,497.1 U/ml, respectively).
Figure 7.

Number of eosinophils in lung tissue from immunized wild-type and γ/δ T cell– deficient animals receiving seven exposures with OVA or SAL. Solid bars, mean (n = 6 per group); SAL γ/δ+/+, SAL-challenged wild-type mice; OVA γ/ δ+/+, OVA-challenged wild-type mice; OVA γδ−/−, OVA-challenged γ/δ T cell–deficient animals. An increased number of eosinophils in lung tissue was observed in OVA-challenged wild-type mice compared with corresponding SAL-exposed animals (**P < 0.01). No significant difference was observed between OVA-challenged wild-type mice and γ/δ T cell–deficient mice.
Discussion
This study demonstrates that depletion of NK1.1+ cells (NK cells and NKT cells) before the priming regime inhibits pulmonary eosinophil and CD3+ T cell infiltration as well as increased levels of IL-4, IL-5, and IL-12 in BALF in a murine model of allergic asthma. Consistent with the suppression of allergic airway inflammation seen in mice depleted of NK1.1+ cells before immunization, i.e., during the initiation of the acquired immune response, diminished systemic levels of OVA-specific IgE and IgG2a and impaired T cell cytokine production in spleen were observed in these mice. The demonstration of a marked eosinophil-rich inflammation in lung tissue of NKT cell–deficient CD1d1 mutant mice strongly supports an important role of NK cells, but not of NKT cells, for development of allergen-induced pulmonary inflammation. However, we cannot completely exclude the possibility that NK cells and NKT cells somehow interact and that depletion of both cell types may be required for inhibition of the allergic immune response in this model. A recent study has shown that allergen-specific IgE and IgG1 responses and allergic airway inflammation, induced by repeated immunization and intranasal allergen challenges, are reduced in γ/δ T cell–deficient mice (25). In our experimental model, allergen-induced changes in γ/δ T cell–deficient animals were attenuated, but this effect was moderate compared with the marked suppression of the allergic airway inflammation demonstrated in mice depleted of NK1.1+ cells. Together, these data support the hypothesis that innate immunity regulates the acquired immune response in allergic inflammation. Given the caution required in any translation of findings in murine allergic models to human asthma (33), the present observations suggest the possibility that human NK cells may govern development of allergic eosinophilic airway disease.
The present protocol for immunization and allergen aerosol challenge was adopted from Brusselle et al. (29). In this mouse model of asthma, repeated daily exposure of immunized mice to aerosolized OVA leads to a CD4+ T cell– dependent (29) and IL-4–dependent (29, 34) eosinophilic inflammation with increased secretory cell epithelial lining (30), lung and splenic Th2 cytokine production (35; this study), and systemic allergen-specific IgE production (29, 34, 35; this study), consistent with a Th2-associated immune response. However, indices not only of Th2 expansion but also of Th1 expansion are present in this model. Thus, systemic OVA-specific IgG2a production and increased levels of IL-12 in BALF were demonstrated in immunized and repeatedly allergen-challenged mice. Also in allergic asthma there may be a mixed Th2/Th1 response. Although there is a predominance of IL-4– and IL-5–producing CD4+ T cells, a population of IFN-γ–producing CD4+ T cells has thus been demonstrated in BALF from asthmatic subjects (1, 36).
It may be hypothesized that the suppressed allergic response in mice depleted of NK1.1+ cells is caused by deficient IL-4 production at the time of immunization. Studies have failed to detect IL-4 production by NK cells (12). However, it has been hypothesized that NKT cells may be an important source of IL-4 and, according to the paradigm of Th1 and Th2 (37), for the development of Th2 immune responses (38–40). Accordingly, in this study, spleens of NKT cell–deficient CD1d1 mutant mice exhibited a tendency of reduced numbers of IL-4–producing CD3+ T cells and an associated increase in IFN-γ–producing CD3+ T cells. However, since CD1d1 mutant mice developed lung tissue eosinophilia and allergen-specific IgE levels not different from those observed in wild-type mice, we conclude that NKT cells and their swift production of IL-4 are not critical for the allergic responses in this model. Also, previous studies using β2-microglobulin–deficient animals have shown that NKT cells are dispensable for allergen- induced Th2 responses in the airways (41, 42).
Our data on systemic levels of OVA-specific IgE and IgG2a in NK1.1+ cell–depleted mice are in contrast to those reported recently by Wang et al. (43). In their study, depletion of NK1.1+ cells altered neither OVA-specific IgE and IgG2a levels nor OVA-stimulated cytokine production by spleen cells in bulk culture in immunized mice (43). There are no obvious explanations for this discrepancy. However, since they delivered the mAb NK1.1 on the day of immunization, it can be speculated that the lack of immune suppression may in part have been caused by antibody-induced nonspecific production of cytokines during the initiation of the immune response. Thus, Asea et al. (44) demonstrated recently that a single delivery (50 μg i.v.) of mAb NK1.1 triggered a population of NKT cells to produce IL-4 within 90 min. This production returned to baseline levels by 24 h after antibody treatment. To confirm that no acute nonspecific effects (such as cytokine release) of depletion with mAb NK1.1 on day –2 caused the effects on immunization in this study, mice were depleted of NK1.1+ cells from day –7 before immunization. These mice also exhibited suppressed pulmonary eosinophilia and decreased systemic levels of IgE and IgG2a. In another control experiment, the efficacy of depletion of NK1.1+ cells from day –7 and from day –2 was monitored by flow cytometric analysis of spleen cells at the day of immunization. All antibody-treated mice (n = 10) exhibited <1% NK1.1+ cells in the spleen at this time point (our unpublished data).
The critical role of NK cells in this model of allergic asthma is limited to the immunization phase, as depletion of NK1.1+ cells during the challenge period only did not influence the magnitude of pulmonary eosinophilia, levels of OVA-specific IgE, and cytokines in BALF. Thus, it appears likely that NK cells influence the initial antigen presentation to T cells and/or the differentiation of naive T cells into effector cells, but not the secondary activation of antigen-specific T cells. Recently, Zhang et al. (45) reported that depletion of NK cells augments disease severity, T cell proliferation, and production of Th1 cytokines in experimental autoimmune encephalomyelitis, a prototype Th1-mediated condition. These novel data are in contrast to the general paradigm that NK cells are required for optimal activation of Th1-like immune responses. It can be speculated that depletion of NK cells has similar effects in our experimental model, causing a switch from Th2 to Th1. However, our data on systemic IgE and IgG2a levels, BALF cytokine levels, and CD3+ splenic T cell cytokine production fail to indicate this. The early appearance of NK cells at the site of immunization (17) and their ability to produce a variety of cytokines implicate these cells in several critical steps in the development of the acquired immune response to allergens. One possibility is that NK cells provide mediators that critically influence differentiation of resting APCs to an activated phenotype with the T cell costimulatory capacity that is required for the exaggerated T cell activation in allergic pulmonary inflammation (5, 46). Indeed, both isoforms of B7 on APCs are upregulated by IFN-γ (47), a cytokine which may derive in part from NK cells. Other immunoregulatory functions of NK cells may be attributed to their capacity to produce IL-5 (12). Indeed, Walker et al. are now demonstrating that IL-5 produced by NK cells may contribute to eosinophil infiltration in a mouse model of allergic peritonitis (48). Although it appears likely that NK cells exert their effects by cytokine production, it cannot be excluded that NK cells influence the immunization phase by cytolytic mechanisms. It has been shown that APCs, i.e., dendritic cells and macrophages, are highly susceptible to lysis by NK cells, which thus might modulate the antigen presentation (49). Further studies are warranted to elucidate details of cellular and molecular interplays that may explain the present observations.
The present data suggest the possibility that the increased NK cell activity that has been demonstrated in asthma (19– 21) reflects a predisposition of individuals with high NK cell activity to develop exaggerated T cell responses to inhaled antigens and hence to be at risk of developing asthma. In epidemiological investigations, a predisposition to develop asthma has been strongly associated with elevated levels of serum IgE (50). Interestingly, a correlation between NK cell activity and total serum IgE levels has been observed in healthy subjects (51).
In conclusion, the present data indicate that NK cells may have a critical role in immunization and subsequent development of allergen-induced airway inflammation. These new data suggest the possibility of a pathogenic role for the increased number of NK cells observed in asthma and suggest that NK cell activity may be one of the factors governing the development of allergic eosinophilic airway disease.
Acknowledgments
This work was supported by the Swedish Medical Research Council (V1180, 06P-11813, 16X-12219, 8308, 4499), Vårdalstiftelsen, the Swedish Heart and Lung Foundation, the Swedish Cancer Society, and Astra Draco, Lund, Sweden.
Abbreviations used in this paper
- BAL
bronchoalveolar lavage
- BALF
BAL fluid
- ELISPOT
enzyme-linked immunospot
- NKT cells
NK1.1+ T cells
- SAL
saline
- SFC
spot-forming cell
References
- 1.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 2.Watanabe A, Mishima H, Renzi PM, Xu L-J, Hamid Q, Martin JG. Transfer of allergic airway responses with antigen-primed CD4+ but not CD8+T cells in brown Norway rats. J Clin Invest. 1995;96:1303–1310. doi: 10.1172/JCI118165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukacs NW, Stieter RM, Chensue SW, Kunkel SL. Interleukin-4-dependent pulmonary eosinophil infiltration in a murine model of asthma. Am J Respir Cell Mol Biol. 1994;10:526–532. doi: 10.1165/ajrcmb.10.5.8179915. [DOI] [PubMed] [Google Scholar]
- 4.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsuyuki S, Tsuyuki J, Einsle K, Kopf M, Coyle AJ. Costimulation through B7-2 (CD86) is required for the induction of a lung mucosal T helper cell 2 (TH2) immune response and altered airway responsiveness. J Exp Med. 1997;185:1671–1679. doi: 10.1084/jem.185.9.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swain SL, Weinberg AD, English M, Huston G. IL-4 directs the development of Th2-like helper effectors. J Immunol. 1990;145:3796–3806. [PubMed] [Google Scholar]
- 7.Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response. Science. 1996;272:50–54. doi: 10.1126/science.272.5258.50. [DOI] [PubMed] [Google Scholar]
- 8.Bendelac A, Fearon DT. Innate immunity. Innate pathways that control acquired immunity. Curr Opin Immunol. 1997;9:1–3. doi: 10.1016/s0952-7915(97)80151-3. [DOI] [PubMed] [Google Scholar]
- 9.Medzhitov R, Janeway CA., Jr Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997;9:4–9. doi: 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 10.Biron CA. Activation and function of natural killer cell responses during viral infections. Curr Opin Immunol. 1997;9:24–34. doi: 10.1016/s0952-7915(97)80155-0. [DOI] [PubMed] [Google Scholar]
- 11.Scharton-Kersten TM, Sher A. Role of natural killer cells in innate resistance to protozoan infections. Curr Opin Immunol. 1997;9:44–51. doi: 10.1016/s0952-7915(97)80157-4. [DOI] [PubMed] [Google Scholar]
- 12.Warren HS, Kinnear BF, Phillips JH, Lanier LL. Production of IL-5 by human NK cells and regulation of IL-5 secretion by IL-4, IL-10, and IL-12. J Immunol. 1995;154:5144–5152. [PubMed] [Google Scholar]
- 13.Bix M, Locksley RM. Natural T cells. Cells that co-express NKRP-1 and TCR. J Immunol. 1995;155:1020–1022. [PubMed] [Google Scholar]
- 14.Vicari AP, Zlotnik A. Mouse NK1.1+ T cells: a new family of T cells. Immunol Today. 1996;17:71–76. doi: 10.1016/0167-5699(96)80582-2. [DOI] [PubMed] [Google Scholar]
- 15.Bendelac A, Rivera MN, Park S-H, Roark JH. Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu Rev Immunol. 1997;15:535–562. doi: 10.1146/annurev.immunol.15.1.535. [DOI] [PubMed] [Google Scholar]
- 16.Prussin C, Foster B. TCR Vα24 and Vβ11 coexpression defines a human NK1 T cell analog containing a unique Th0 subpopulation. J Immunol. 1997;159:5862–5870. [PubMed] [Google Scholar]
- 17.Bogen SA, Fogelman I, Abbas AK. Analysis of IL-2, IL-4, and IFN-γ-producing cells in situ during immune responses to protein antigens. J Immunol. 1993;150:4197–4205. [PubMed] [Google Scholar]
- 18.Weissler JC, Nicod LP, Lipscomb MF, Toews GB. Natural killer cell function in human lung is compartmentalized. Am Rev Respir Dis. 1987;135:941–949. doi: 10.1164/arrd.1987.135.4.941. [DOI] [PubMed] [Google Scholar]
- 19.Timonen T, Stenius-Aarniala B. Natural killer cell activity in asthma. Clin Exp Immunol. 1985;59:85–90. [PMC free article] [PubMed] [Google Scholar]
- 20.Jira M, Antosova E, Vondra V, Strejcek J, Mazakova H, Prazakova J. Natural killer and interleukin-2 induced cytotoxicity in asthmatics. I. Effect of acute antigen-specific challenge. Allergy. 1988;43:294–298. doi: 10.1111/j.1398-9995.1988.tb00903.x. [DOI] [PubMed] [Google Scholar]
- 21.Krejsek J, Kral B, Vokurkova D, Derner V, Touskova M, Parakova Z, Kopechy O. Decreased peripheral blood γδ T cells in patients with bronchial asthma. Allergy. 1998;53:73–77. doi: 10.1111/j.1398-9995.1998.tb03776.x. [DOI] [PubMed] [Google Scholar]
- 22.Vesterinen E, Timonen T. Natural killer cell activity in specific and non-specific bronchial challenge. Ann Allergy. 1988;60:247–249. [PubMed] [Google Scholar]
- 23.Ferrick DA, Schrenzel MD, Mulvania T, Hsieh B, Ferlin WG, Lepper H. Differential production of interferon-γ and interleukin-4 in response to Th1- and Th2-stimulating pathogens by γδ T cells in vivo. Nature. 1995;373:255–257. doi: 10.1038/373255a0. [DOI] [PubMed] [Google Scholar]
- 24.McMenamin C, Pimm C, McKersey M, Holt PG. Regulation of IgE responses to inhaled antigen in mice by antigen-specific γδ T cells. Science. 1994;265:1869–1871. doi: 10.1126/science.7916481. [DOI] [PubMed] [Google Scholar]
- 25.Zuany-Amorim C, Ruffie C, Haile S, Vargaftig BB, Pereira P, Pretolani M. Requirement for γδ T cells in allergic airway inflammation. Science. 1998;280:1265–1267. doi: 10.1126/science.280.5367.1265. [DOI] [PubMed] [Google Scholar]
- 26.Koo GC, Peppard JR. Establishment of monoclonal anti-Nk-1.1 antibody. Hybridoma. 1984;3:301–303. doi: 10.1089/hyb.1984.3.301. [DOI] [PubMed] [Google Scholar]
- 27.Mendiratta SK, Martin WD, Hong S, Boesteanu A, Joyce S, Van Kaer L. CD1d1 mutant mice are deficient in natural T cells that promptly produce IL-4. Immunity. 1997;6:469–477. doi: 10.1016/s1074-7613(00)80290-3. [DOI] [PubMed] [Google Scholar]
- 28.Itohara S, Mombaerts P, Lafaille J, Iacomini J, Nelson A, Clarke AR, Hooper ML, Farr A, Tonegawa S. T cell receptor δ gene mutant mice: independent generation of αβ T cells and programmed rearrangements of γδ TCR genes. Cell. 1993;72:337–348. doi: 10.1016/0092-8674(93)90112-4. [DOI] [PubMed] [Google Scholar]
- 29.Brusselle GJ, Kips JC, Tavernier JH, Van der Heyden JG, Cuvelier CA, Pauwels RA, Bluethmann H. Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin Exp Allergy. 1994;24:73–80. doi: 10.1111/j.1365-2222.1994.tb00920.x. [DOI] [PubMed] [Google Scholar]
- 30.Korsgren M, Erjefält JS, Korsgren O, Sundler F, Persson CGA. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell–deficient mice. J Exp Med. 1997;185:885–892. doi: 10.1084/jem.185.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ten RM, Pease LR, McKean DJ, Bell MP, Gleich GJ. Molecular cloning of the eosinophil peroxidase: evidence for the existence of a peroxidase multigene family. J Exp Med. 1989;169:1757–1769. doi: 10.1084/jem.169.5.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rönnelid J, Klareskog L. A comparison between ELISPOT methods for the detection of cytokine producing cells: greater sensitivity and specificity using ELISA plates as compared to nitrocellulose membranes. J Immunol Methods. 1997;200:17–26. doi: 10.1016/s0022-1759(96)00170-6. [DOI] [PubMed] [Google Scholar]
- 33.Persson CGA, Erjefält JS, Korsgren M, Sundler F. The mouse trap. Trends Pharmacol Sci. 1997;18:465–467. doi: 10.1016/s0165-6147(97)01142-5. [DOI] [PubMed] [Google Scholar]
- 34.Brusselle G, Kips J, Joos G, Bluethmann H, Pauwels R. Allergen-induced airway inflammation and bronchial responsiveness in wild-type and interleukin-4-deficient mice. Am J Respir Cell Mol Biol. 1995;12:254–259. doi: 10.1165/ajrcmb.12.3.7873190. [DOI] [PubMed] [Google Scholar]
- 35.Lambrecht BN, Salomon B, Klatzmann D, Pauwels RA. Dendritic cells are required for the development of chronic eosinophilic airway inflammation in response to inhaled antigen in sensitized mice. J Immunol. 1998;160:4090–4097. [PubMed] [Google Scholar]
- 36.Krug N, Madden J, Redington AE, Lackie P, Djukanovic R, Schauer U, Holgate ST, Frew AJ, Howarth PH. T-cell cytokine profile evaluated at the single cell level in BAL and blood in allergic asthma. Am J Respir Cell Mol Biol. 1996;14:319–326. doi: 10.1165/ajrcmb.14.4.8600935. [DOI] [PubMed] [Google Scholar]
- 37.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 38.Yoshimoto T, Paul WE. CD4+, NK1.1+T cells promptly produce interleukin 4 in response to in vivo challenge with anti-CD3. J Exp Med. 1994;179:1285–1295. doi: 10.1084/jem.179.4.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bendelac A, Lantz O, Quimby ME, Yewdell JE, Bennink JR, Brutkiewicz RR. CD1 recognition by mouse NK1+ T lymphocytes. Science. 1995;268:863–865. doi: 10.1126/science.7538697. [DOI] [PubMed] [Google Scholar]
- 40.Bendelac A, Hunziker RD, Lantz O. Increased interleukin 4 and immunoglobulin E production in transgenic mice overexpressing NK1 T cells. J Exp Med. 1996;184:1285–1293. doi: 10.1084/jem.184.4.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Rogers KH, Davis DB. β2-microglobulin–dependent T cells are dispensable for allergen- induced T helper 2 responses. J Exp Med. 1996;184:1507–1512. doi: 10.1084/jem.184.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown DR, Fowell DJ, Corry DB, Wynn TA, Moskowitz NH, Cheever AW, Locksley RM, Reiner SL. β2-microglobulin–dependent NK1.1+T cells are not essential for T helper cell 2 immune responses. J Exp Med. 1996;184:1295–1304. doi: 10.1084/jem.184.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang M, Ellison CA, Gartner JG, Hayglass KT. Natural killer cell depletion fails to influence initial CD4 T cell commitment in vivo in exogenous antigen-stimulated cytokine and antibody responses. J Immunol. 1998;160:1098–1105. [PubMed] [Google Scholar]
- 44.Asea A, Stein-Streilein J. Signalling through NK1.1 triggers NK cells to die but induces NK T cells to produce interleukin-4. Immunology. 1998;93:296–305. doi: 10.1046/j.1365-2567.1998.00422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T. Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J Exp Med. 1997;186:1677–1687. doi: 10.1084/jem.186.10.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keane-Myers A, Gause WC, Linsley PS, Chen S-H, Wills-Karp M. B7-CD28/CTLA-4 costimulatory pathways are required for the development of T helper cell 2-mediated allergic airway responses to inhaled antigens. J Immunol. 1997;158:2042–2049. [PubMed] [Google Scholar]
- 47.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-γ. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 48.Walker C, Checkel J, Cammisuli S, Leibson PJ, Gleich GJ. IL-5 production by NK cells contributes to eosinophil infiltration in a mouse model of allergic inflammation. J Immunol. 1998;161:1962–1969. [PubMed] [Google Scholar]
- 49.Chambers BJ, Salcedo M, Ljunggren H-G. Triggering of natural killer cells by the costimulatory molecule CD80 (B7-1) Immunity. 1996;5:311–317. doi: 10.1016/s1074-7613(00)80257-5. [DOI] [PubMed] [Google Scholar]
- 50.Burrows B, Martinez FD, Halonen M, Barbee RA, Cline MG. Association of asthma with serum IgE levels and skin-test reactivity to allergens. N Engl J Med. 1989;320:271–277. doi: 10.1056/NEJM198902023200502. [DOI] [PubMed] [Google Scholar]
- 51.Kusaka Y, Sato K, Zhang Q, Morita A, Kasahara T, Yanagihara Y. Association of natural killer cell activity with serum IgE. Int Arch Allergy Immunol. 1997;112:331–335. doi: 10.1159/000237476. [DOI] [PubMed] [Google Scholar]