

Abstract
N-formylpeptides derive from bacterial and mitochondrial proteins, and bind to specific receptors on mammalian phagocytes. Since binding induces chemotaxis and activation of phagocytes in vitro, it has been postulated that N-formylpeptide receptor signaling in vivo may be important in antimicrobial host defense, although direct proof has been lacking. Here we test this hypothesis in mice lacking the high affinity N-formylpeptide receptor (FPR), created by targeted gene disruption. FPR−/− mice developed normally, but had increased susceptibility to challenge with Listeria monocytogenes, as measured by increased mortality compared with wild-type littermates. FPR−/− mice also had increased bacterial load in spleen and liver 2 d after infection, which is before development of a specific cellular immune response, suggesting a defect in innate immunity. Consistent with this, neutrophil chemotaxis in vitro and neutrophil mobilization into peripheral blood in vivo in response to the prototype N-formylpeptide fMLF (formyl-methionyl-leucyl-phenylalanine) were both absent in FPR−/− mice. These results indicate that FPR functions in antibacterial host defense in vivo.
Keywords: chemotaxis, Listeria, inflammation, chemoattractant, neutrophil
Perhaps the most widely used probes for studying mechanisms of leukocyte chemotaxis are the N-formylpeptides, particularly the prototype formyl-methionyl-leucyl-phenylalanine (fMLF).1 However, after over 20 years of research, their biological significance is still unknown (for review see reference 1). At one extreme, they could simply mimic in vitro an endogenous leukocyte chemoattractant not yet described, and have no biological role themselves. At the other, they may be very important in antibacterial host defense. The latter view has substantial indirect support because N-formylpeptides are products of bacterial proteins (2–4), and because phagocytes are critical effectors of antibacterial host defense.
The N-formylpeptide signaling pathway has been studied in detail in vitro. Specific phagocyte receptors have been characterized biochemically, and two human genes, designated FPR1 and FPRL1, that encode specific N-formylpeptide receptor subtypes, have been cloned (5–9). Both are members of the 7-transmembrane domain receptor superfamily, and both couple to pertussis toxin–sensitive G proteins (10). They each have 69% deduced amino acid sequence identity, and are commonly referred to as FPR (formyl peptide receptor) and the FPRL1 (FPR-like 1) receptor, respectively. Both are expressed in human neutrophils and monocytes (11), both bind fMLF (5, 8), and, when activated, both induce calcium mobilization in transfected cells (6, 8, 11). However, there are several major differences: FPR binds fMLF with an ∼1,000-fold higher affinity than FPRL1 (8); FPR, but not FPRL1, has been shown to be a chemotactic receptor (12); and FPRL1, but not FPR, has been shown to be a functional lipoxin A4 receptor (13). One other related human gene, named FPRL2, has been identified by cross-hybridization, but the function of the putative receptor is undefined (9, 11). All three genes are clustered on chromosome 19q13.3 (14).
In mice, an orthologue of FPR1 and five structurally related genes have been identified by cross-hybridization with human FPR and FPRL1 probes, indicating that the FPR gene cluster has undergone differential lineage-specific expansion in mammals (15–17). This has created problems in defining orthologous genes in the two species. The mouse orthologue of human FPR1 is clearly encoded by the mouse gene Fpr1. The encoded receptor is 77% identical in amino acid sequence to human FPR and responds to fMLF in calcium flux assays, although the EC50 is ∼100-fold higher than for human FPR under comparable conditions (15). Like FPRL1, one of the related mouse genes, designated Fpr-rs1, encodes a lipoxin A4 receptor. However, neither this nor the other four related genes has been shown to encode a receptor specific for N-formylpeptides or other chemoattractant ligands (17).
Although several antagonists of fMLF have been reported (18), they have not been developed extensively as pharmacologic tools for defining N-formylpeptide biology. We have taken an alternative genetic approach, through analysis of mice lacking FPR, by targeted disruption of Fpr1.
Materials and Methods
Generation of FPR−/− Mice.
Cloning of the FPR gene (Fpr1) from a B6/CBA mouse genomic library has been described previously (15). A 7.8-kb XbaI fragment of genomic clone X was cloned into Bluescript (Stratagene). To facilitate creation of a targeting construct, three restriction enzyme sites were created in this fragment: a KpnI site 700 bp upstream of the start codon of the open reading frame (ORF), and BamHI and XhoI sites 300 and 450 bp downstream of the start codon, respectively. The 150-bp BamHI/XhoI ORF fragment, which encodes the putative first extracellular loop to the fourth transmembrane domain of FPR, was replaced by a 1.1-kb neomycin resistance cassette (neor) inserted in the opposite orientation. A 6.9-kb fragment of the resulting construct was excised by digestion of a NotI site in the polylinker at the 3′ end and the artificial KpnI site at the 5′ end, and subcloned between NotI and KpnI sites of the plasmid pPNT (19), which contains the Herpes simplex virus thymidine kinase (HSV-tk) gene. The resulting construct contained 1 and 4.8 kb of FPR sequence 5′ and 3′ from the neor gene, respectively. The construct was linearized by digestion with XbaI, and 25 μg DNA was electroporated into 107 cells from the embryonic stem cell line designated R1.211 clones resistant to G418 (350 μg/ml) and gancyclovir (2 μM) were selected and expanded on feeder cell layers in 24-well plates. Homologous recombinants were identified by Southern blot hybridization of genomic DNA digested with XbaI with a 1-kb probe located outside of the construct. Five chimeric mice were produced by microinjection into C57Bl/6 blastocysts according to standard methods (20). Chimeric mice were mated with wild-type C57Bl/6 mice to produce heterozygous (+/−) mice. Litter mates from the mating of heterozygous mice were then analyzed. This work was done under a protocol approved by the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases.
Isolation of Mouse Leukocytes.
Total leukocytes (80–85% mononuclear cells and 15–20% neutrophils) were isolated from citrated peripheral blood obtained from mouse tails; erythrocytes were lysed in ACK lysis buffer. To obtain enriched populations of neutrophils and macrophages, the peritoneal cavity was washed with PBS 3 and 72 h, respectively, after peritoneal injection with 2 ml (for neutrophils) or 1 ml (for macrophages) of thioglycollate. The purity of both populations was >90% as assessed by light microscopy of Diff-quick–stained (Sigma Chemical Co.) cytospin preparations.
Analysis of mRNA Expression.
Total RNA was prepared from peritoneal exudate neutrophils with RNA STAT-60 kit (Tel-Test, Inc.) according to the manufacturer's instructions. The cDNA was synthesized with cDNA Cycle kit (Invitrogen). Wild-type and mutated FPR mRNA was screened by PCR using primers FPR310 (5′-ATGTTCTAGGAGTCTACAAGATGG, a sense oligonucleotide located 20 bp before the start codon), AFPR660 (5′-ATATATGAATTTGCACATGAACCA, an antisense oligonucleotide located in the deleted part of the ORF in the targeting construct), and SNEO2 (5′-CGCTCCCGATTCGCAGCGCATCGC, a sense oligonucleotide of the neo gene). Primers FPR310 and AFPR660 amplify 350 bp of wild-type cDNA, and primers FPR310 and SNEO2 amplify 430 bp of mutated cDNA. The PCR conditions were 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 60°C for 30 s, and 70°C for 30 s, with a final extension at 72°C for 10 min.
[Ca2+]i Measurements.
Cells (107/ml) were loaded with FURA-2 (Molecular Probes) as previously described (21). 2 × 106 cells in 2 ml HBSS were placed in a continuously stirred cuvette at 37°C in a fluorimeter (Photon Technology Inc.). The data are presented as the relative ratio of fluorescence excited alternately at 340 and 380 nm every 0.5 s, and monitored at 510 nm, in response to fMLF (Sigma Chemical Co.) or recombinant mouse macrophage inflammatory protein (MIP)-1α (Peprotech).
Chemotaxis.
Chemotaxis was analyzed using polyvinylpyrrolidone-free polycarbonate membranes with 3-μm pores in 48-well chambers (NeuroProbe). fMLF in HBSS was placed in the lower chamber, and 50 μl of thioglycollate-elicited peritoneal neutrophils suspended in HBSS (1.5 × 106/ml) were placed in the upper chamber. After incubation for 45 min at 37°C, the membrane was removed, rinsed with PBS, fixed, and stained with Diff-Quick. Cells were counted in five randomly selected fields at 1,000-fold magnification.
Neutrophil Mobilization to Peripheral Blood.
Mice were injected subcutaneously with 200 μl of 2 μM fMLF. Complete peripheral blood leukocyte counts and differentials were determined for tail vein collections taken before injection and 1.5 h after injection.
Challenge with Listeria monocytogenes.
The experimental procedure has been described previously (22). In brief, log-phase culture of L. monocytogenes strain EGD was grown in brain–heart infusion broth (Difco Labs.), aliquoted in 1-ml volumes, and stored at −70°C. For each experiment, a vial of bacteria was thawed and diluted in PBS. In initial experiments, the approximate intravenous LD50 in first and second generation B6/129 heterozygote mice for L. monocytogenes was determined to be 2 × 104 CFU. Then, 6–8-wk-old mice were infected intravenously with 0.1 ml of a suspension containing the LD50. For the bacterial burden assay, mice were killed by cervical dislocation 48 h after infection. Liver and spleen were removed and homogenized separately in distilled water, and numbers of viable L. monocytogenes were determined by plating serial dilutions of organ homogenates on brain–heart infusion agar.
Results and Discussion
Development of Mice Lacking FPR.
To inactivate FPR, we replaced a 150-bp ORF fragment of Fpr1 with a neomycin resistance cassette (neor) by homologous recombination in 129/Sv embryonic stem cells (Fig. 1 a). The deleted region is from the first extracellular loop to the fourth transmembrane segment predicted from the FPR sequence (codons 101–150). The mutated and wild-type alleles were distinguished by an XbaI restriction fragment length polymorphism, 8.5 versus 7.8 kb respectively, of genomic DNA by Southern hybridization (Fig. 1 b). Consistent with this, mutant FPR mRNA was detected in neutrophils from +/− and −/−, but not +/+, mice, whereas normal FPR mRNA was detected in neutrophils from +/− and +/+, but not −/−, mice (Fig. 1 c).
Figure 1.

Genetic inactivation of mouse FPR. (a) Mutagenesis strategy. Maps of the wild-type and mutant alleles are shown above and below the map of the targeting construct, respectively, and corresponding sites are connected by broken lines. The neomycin resistance gene (neor) replaced 150 bp of the FPR ORF and was used to select for targeted events. The Herpes simplex virus thymidine kinase gene (HSV-tk) was used for counterselection of nontargeted events. Horizontal bars, ORFs of the indicated genes; arrows, sense orientation of the indicated ORF; X, XhoI; B, BamHI; K, KpnI. (b) Genotypic analysis of tail DNA derived from progeny of heterozygous crosses. The mutated allele (8.5-kb XbaI fragment) is distinguished from the wild-type allele (7.8-kb XbaI fragment) using a 1-kb probe, whose location is given in panel a. (c) Reverse transcription PCR analysis of total RNA from TP neutrophils from mice with the FPR genotype indicated at the top of each lane. Mutated mRNA (430-bp band) is distinguished from the wild-type mRNA (350-bp band) by reverse transcription PCR with the primers indicated in the text.
The genotypic frequencies for 264 total progeny of eight +/− mating pairs were: 24.4% +/+, 49.6% +/−, and 26.0% −/−, which is not significantly different from Mendelian expectation for an autosomal gene. FPR−/− mice were viable and fertile, and exhibited normal growth, development, anatomy, and behavior compared with +/+ littermates. In particular, no defects in the complete blood count or differential white blood cell count were detected. These mice have been observed for >17 mo, and have not exhibited defects in hemostasis or healing of tail wounds, or increased susceptibility to spontaneous infection when derived in a specific pathogen-free environment. Thus either FPR is not involved in these processes or it functions redundantly with other factors, or else, under unstressed conditions, its function is compensated by other factors in mutant mice.
Role of FPR in Neutrophil Function.
We first used the knockout mice to test whether FPR mediates fMLF signaling in primary leukocytes. First, we observed that the normal fMLF-induced calcium flux in thioglycollate-elicited peritoneal (TP) neutrophils (Fig. 2 a) and PBMCs (data not shown) from +/+ mice was absent in cells from −/− mice. As a positive control, a robust response to the CC chemokine MIP-1α was detected in the same cell types from both +/+ and −/− mice, consistent with previous observations (23). TP neutrophils from FPR−/− mice also failed to chemotax in response to fMLF (100 nM–1 μM) in vitro, whereas neutrophils from +/+ mice exhibited a typical bell-shaped dose–response curve (Fig. 2 b). Similar results were obtained from TP macrophages, but the response of +/+ macrophages to fMLF was much lower than the response of +/+ neutrophils (data not shown). These in vitro results indicate that FPR mediates mouse neutrophil chemotactic and calcium flux responses to fMLF, which has not been shown previously, and verify gene inactivation at the functional level.
Figure 2.

Loss of FPR function in neutrophils from FPR−/− mice. (a) Calcium flux response. [Ca2+]i was monitored in real time in FURA-2–loaded peritoneal neutrophils stimulated with 1 μM fMLF and 100 nM MIP-1α. Each tracing represents analysis of 3 × 106 cells from a single mouse with the FPR genotype indicated at the top (+/+, wild-type mice; −/−, knockout mice). Agonists were added at the times indicated by the arrows. Results are from a single experiment representative of at least four separate experiments. (b) Neutrophil chemotaxis. Results are the mean ± SEM of triplicate determinations in a single experiment using peritoneal neutrophils and fMLF in vitro and are representative of three separate experiments. There was no difference in MIP-1α–induced chemotaxis between neutrophils from FPR−/− and from FPR+/+ mice (data not shown). (c) Neutrophil mobilization to peripheral blood. Each pair of bars represents the peripheral blood neutrophil concentration for a single mouse before and 90 min after subcutaneous injection with 200 μl of 2 μM fMLF. Results are from a single experiment representative of three separate experiments.
We next tested the effects of fMLF in vivo in +/+ and −/− mice. We found that subcutaneous injection of +/+ mice with fMLF specifically induced a 70% increase in peripheral blood neutrophil concentrations when measured 90 min after injection (Fig. 2 c). The response was absent in −/− mice, indicating that FPR is involved. The CC chemokine MIP-1α is also able to induce this response, through the receptor CCR1 (23). We also injected 1, 10, and 100 nmoles of fMLF intraperitoneally, but did not observe local accumulation of leukocytes in either +/+ or −/− mice when the peritoneal cavity was washed 4 h after injection. Presumably, the peptide is scavenged or degraded by factors present at this site. In contrast, intraperitoneal injection with thioglycollate, a nonspecific inflammatory stimulus, induced peritoneal exudation, with neutrophil and macrophage predominance at 4 and 72 h, respectively, consistent with previous reports (23). However, no significant difference was observed for this response between FPR−/− and FPR+/+ mice. Reduced leukocyte mobilization in response to thioglycollate has been reported in several chemokine receptor knockout mice (24–28) and in C5a receptor knockout mice (29), all of which bind endogenous chemoattractants. Thus, thioglycollate challenge in FPR knockout mice does not reveal evidence for the widely held speculation that an endogenous ligand for FPR exists.
Increased Susceptibility of FPR−/− Mice to Challenge with L. monocytogenes.
To test the role of FPR in host defense, we compared the susceptibility of FPR−/− and FPR+/+ mice to infection with L. monocytogenes. This organism was chosen because it produces an acute infection in mice with well-characterized immunopathogenesis (30), involving both innate and acquired arms of the immune system, and because, like other bacteria, it produces N-formylpeptides (31). It is not known whether Listeria-derived N-formylpeptides are released in vivo in amounts required to activate FPR; however, this is also unknown for other bacteria. FPR−/− mice exhibited a markedly higher rate and extent of mortality compared with FPR+/+ littermates (Fig. 3 a). 73% of FPR−/− mice injected with 2 × 104 CFU died by day 4 in contrast to only 6% in FPR+/+ mice. Only 6% of FPR−/− mice survived beyond 6 d, whereas 50% of FPR+/+ mice remained healthy. The survival curve of +/− mice fell between those of −/− and +/+ mice, suggesting a gene dosage effect for this phenotype. Consistent with this, we observed intermediate responsiveness of neutrophils from +/− mice versus −/− and +/+ mice to fMLF in the calcium flux assay (data not shown).
Figure 3.
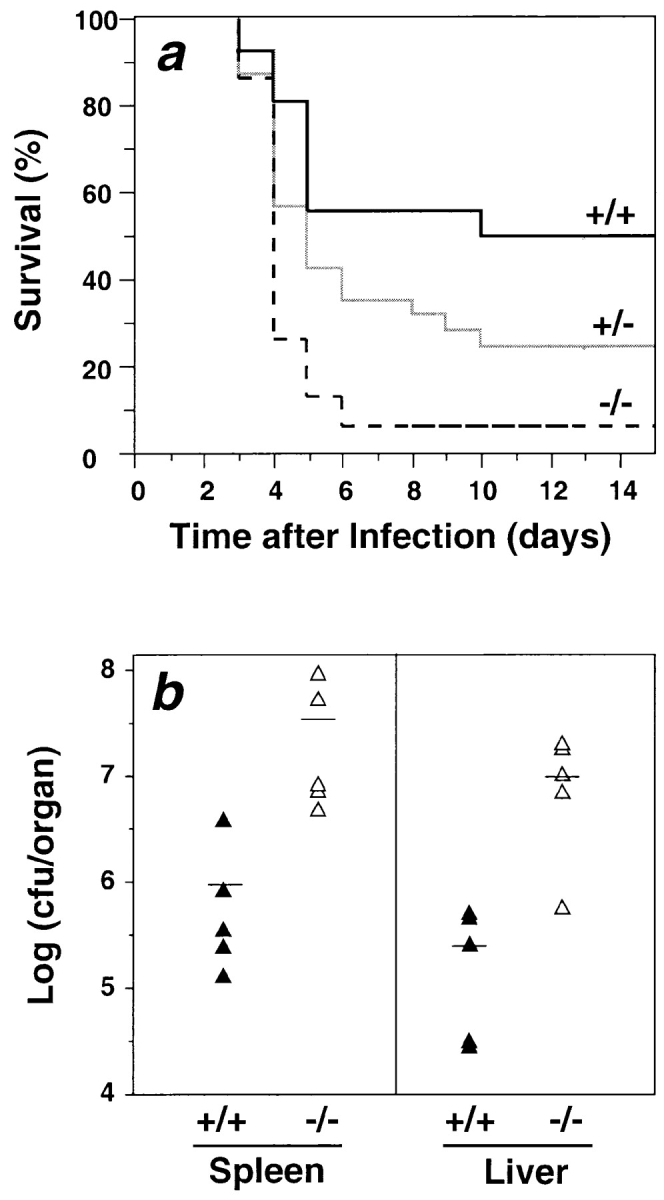
Anti-listerial host defense in mice lacking FPR. (a) Accelerated lethality in FPR−/− mice challenged with L. monocytogenes. Mice were injected with 2 × 104 CFU in the tail vein. Results are from a single experiment with −/− and +/+ sex-matched littermates (n = 15 in each group), and are representative of three separate experiments with a consistent pattern. P = 0.003 for +/+ versus −/−; P = 0.038 for +/− versus −/− (log-rank test). (b) Listeria clearance. Under the same conditions as in panel a, the bacterial burden from whole spleen and liver was determined 2 d after infection. The mean values are indicated as solid bars. +/+, wild-type mice; −/−, knockout mice.
Early mortality suggests a defect in innate immunity. To test this further, we infected mice with 2 × 104 CFU and measured bacterial burden 2 d later, a time when nonspecific immune responses control infection (30). Consistent with a defect in innate immunity, FPR−/− mice showed 32- and 45-fold more bacteria in spleen and liver, respectively, at this time, relative to +/+ control mice (Fig. 3 b).
Although our results show that FPR deficiency causes increased susceptibility to Listeria infection, they do not clearly identify its mechanism of action. The histopathologic appearance of the liver and spleen was indistinguishable in infected FPR+/+ and FPR−/− mice killed on days 1, 2, and 3 after infection, and in infected animals with discordant genotypes that were allowed to die naturally. In both cases, neutrophilic abscesses were observed, suggesting that FPR deficiency does not affect normal neutrophil trafficking to these organs in this model. Additional work will be needed to identify the precise mechanism of action of FPR in host defense in this model.
It has been demonstrated previously that, as in FPR-deficient mice, neutrophil-depleted mice rapidly succumb to overwhelming listeriosis after experimental challenge, with marked increases in bacterial burden in both liver and spleen (32, 33). Disruption of other genes that regulate phagocyte function, including the genes encoding gp91phox (a component of the phagocyte NADPH oxidase; reference 34), the IFN-γ receptor (35), and the chemokine receptors CCR2 (24) and CCR5 (25), has also been linked to increased susceptibility to Listeria. Since not all FPR−/− mice died from Listeria challenge in our experiments, these and other genes may compensate, at least partially, when FPR is absent.
In summary, we have found that FPR−/− mice have no obvious developmental defects and do not develop spontaneous infection when derived in specific pathogen-free conditions. This suggest that, under these conditions, FPR is dispensable. However, when challenged with L. monocytogenes, FPR-deficient mice have accelerated mortality and increased bacterial burden in liver and spleen early after infection, which suggests a role for FPR in host defense, specifically through regulation of innate immunity. This is consistent with expression of FPR on phagocytes. Additional work will be needed to establish whether FPR is a host defense factor versus other types of microorganisms, and a factor in inflammation induced by noninfectious agents, as well as to establish whether its mechanism of action in listeriosis involves binding of N-formylpeptides and trafficking of phagocytes.
Acknowledgments
We thank Karen Elkins for Listeria strains, helpful advice, and comments on the manuscript, and Heiner Westphal for helpful advice.
Abbreviations used in this paper
- fMLF
formyl-methionyl-leucyl-phenylalanine
- FPR
high affinity N-formylpeptide receptor
- FPRL1
low affinity N-formylpeptide receptor or lipoxin A4 receptor
- MIP
macrophage inflammatory protein
- ORF
open reading frame
- TP neutrophils
thioglycollate-elicited peritoneal neutrophils
References
- 1.Murphy, P.M. 1996. The N-formylpeptide chemotactic receptor. In Chemoattractant Ligands and Their Receptors. R. Horuk, editor. CRC Press, Inc., Boca Raton, FL. 269–299.
- 2.Schiffmann E, Corcoran BA, Wahl SM. N-formylmethionyl peptides as chemoattractants for leukocytes. Proc Natl Acad Sci USA. 1975;72:1059–1063. doi: 10.1073/pnas.72.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schiffmann E, Showell HV, Corcoran BA, Ward PA, Smith E, Becker EL. The isolation and partial characterization of neutrophil chemotactic factors from Escherichia coli. . J Immunol. 1975;114:1831–1837. [PubMed] [Google Scholar]
- 4.Marasco WA, Phan SH, Krutzsch H, Showell HJ, Feltner DC, Nairn R, Becker EL, Ward PA. Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. . J Biol Chem. 1984;259:5430–5439. [PubMed] [Google Scholar]
- 5.Boulay F, Tardif M, Brouchon L, Vignais P. Synthesis and use of a novel N-formyl peptide derivative to isolate a human N-formyl peptide receptor cDNA. Biochem Biophys Res Commun. 1990;168:1103–1109. doi: 10.1016/0006-291x(90)91143-g. [DOI] [PubMed] [Google Scholar]
- 6.Murphy PM, McDermott D. Functional expression of the human formyl peptide receptor in Xenopusoocytes requires a complementary human factor. J Biol Chem. 1991;266:12560–12567. [PubMed] [Google Scholar]
- 7.Murphy PM, Ozcelik T, Kenney RT, Tiffany HL, McDermott D, Francke U. A structural homologue of the N-formyl peptide receptor. Characterization and chromosome mapping of a peptide chemoattractant receptor family. J Biol Chem. 1992;267:7637–7643. [PubMed] [Google Scholar]
- 8.Ye RD, Cavanagh SL, Quehenberger O, Prossnitz ER, Cochrane CG. Isolation of a cDNA that encodes a novel granulocyte N-formyl peptide receptor. Biochem Biophys Res Commun. 1992;184:582–589. doi: 10.1016/0006-291x(92)90629-y. [DOI] [PubMed] [Google Scholar]
- 9.Bao L, Gerard NP, Eddy RL, Jr, Shows TB, Gerard C. Mapping of genes for the human C5a receptor (C5AR), human FMLP receptor (FPR), and two FMLP receptor homologue orphan receptors (FPRH1, FPRH2) to chromosome 19. Genomics. 1992;13:437–440. doi: 10.1016/0888-7543(92)90265-t. [DOI] [PubMed] [Google Scholar]
- 10.Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 11.Durstin M, Gao J-L, Tiffany HL, McDermott D, Murphy PM. Differential expression of the members of the N-formylpeptide receptor gene cluster in human phagocytes. Biochem Biophys Res Commun. 1994;201:174–179. doi: 10.1006/bbrc.1994.1685. [DOI] [PubMed] [Google Scholar]
- 12.Laudanna C, Campbell JJ, Butcher EC. Role of Rho in chemoattractant-activated leukocyte adhesion through integrins. Science. 1996;271:981–983. doi: 10.1126/science.271.5251.981. [DOI] [PubMed] [Google Scholar]
- 13.Fiore S, Maddox JF, Perez HD, Serhan CN. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J Exp Med. 1994;180:253–260. doi: 10.1084/jem.180.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerard NP, Bao L, He X-P, Eddy RL, Jr, Shows TB, Gerard C. Human chemotactic receptor genes cluster at 19q13.3/13.4. Characterization of the human C5a receptor gene. Biochemistry. 1993;32:1243–1250. doi: 10.1021/bi00056a007. [DOI] [PubMed] [Google Scholar]
- 15.Gao J-L, Murphy PM. Species and subtype variants of the N-formyl peptide chemotactic receptor reveal multiple important functional domains. J Biol Chem. 1993;268:25395–25401. [PubMed] [Google Scholar]
- 16.Gao J-L, Chen H, Filie JD, Kozak CA, Murphy PM. Differential expansion of the N-formylpeptide receptor gene cluster in human and mouse. Genomics. 1998;51:270–276. doi: 10.1006/geno.1998.5376. [DOI] [PubMed] [Google Scholar]
- 17.Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J Exp Med. 1997;185:1693–1704. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wenzel-Seifert K, Seifert R. Cyclosporin H is a potent and selective formyl peptide receptor antagonist. Comparison with N-t-butoxycarbonyl-L-phenylalanyl-L-leucyl-L-phenylalanyl-L-leucyl-L-phenylalanine and cyclosporins A, B, C, D, and E. J Immunol. 1993;150:4591–4599. [PubMed] [Google Scholar]
- 19.Tybulewicz VLJ, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 20.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 21.Gao J-L, Becker E, Freer RJ, Muthukumaraswamy N, Murphy PM. A high potency non-formylated peptide agonist for the phagocyte N-formylpeptide chemotactic receptor. J Exp Med. 1994;180:2191–2198. doi: 10.1084/jem.180.6.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elkins KL, MacIntyre AT, Rhinehart-Jones TR. Nonspecific early protective immunity in Francisella and Listeriainfections can be dependent on lymphocytes. Infect Immun. 1998;66:3467–3469. doi: 10.1128/iai.66.7.3467-3469.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J-L, Wynn TA, Chang Y, Lee EJ, Broxmeyer HE, Cooper S, Tiffany HL, Westphal H, Kwon-Chung J, Murphy PM. Impaired host defense, hematopoiesis, granulomatous inflammation and type 1–type 2 cytokine balance in mice lacking CC chemokine receptor 1. J Exp Med. 1997;185:1959–1968. doi: 10.1084/jem.185.11.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Y, Kurihara T, Ryseck RP, Yang Y, Ryan C, Loy J, Warr G, Bravo R. Impaired macrophage function and enhanced T cell-dependent immune response in mice lacking CCR5, the mouse homologue of the major HIV-1 coreceptor. J Immunol. 1998;160:4018–4025. [PubMed] [Google Scholar]
- 26.Cacalano, G., J. Lee, K. Kikly, A.M. Ryan, S. Pitts-Meek, B. Hultgren, W.I. Wood, and M.W. Moore. 1994. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 265:682–684. (See published erratum. 270:365.) [DOI] [PubMed]
- 27.Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Jr, Broxmeyer HE, Charo IF. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. 1997;100:2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hopken UE, Lu B, Gerard NP, Gerard C. The C5a chemoattractant receptor mediates mucosal defence to infection. Nature. 1996;383:86–89. doi: 10.1038/383086a0. [DOI] [PubMed] [Google Scholar]
- 30.Unanue ER. Studies in listeriosis show the strong symbiosis between the innate cellular system and the T-cell response. Immunol Rev. 1997;158:11–25. doi: 10.1111/j.1600-065x.1997.tb00988.x. [DOI] [PubMed] [Google Scholar]
- 31.Nataraj C, Huffman GR, Kurlander RJ. H2M3 (wt)-restricted, Listeria monocytogenes-immune CD8 T cells respond to multiple formylated peptides and to a variety of Gram-positive and Gram-negative bacteria. Int Immunol. 1998;10:7–15. doi: 10.1093/intimm/10.1.7. [DOI] [PubMed] [Google Scholar]
- 32.Conlan JW, North RJ. Neutrophil-mediated dissolution of infected host cells as a defense strategy against a facultative intracellular bacterium. J Exp Med. 1991;3:741–744. doi: 10.1084/jem.174.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogers HW, Unanue ER. Neutrophils are involved in acute, nonspecific resistance to Listeria monocytogenesin mice. Infect Immun. 1993;12:5090–5096. doi: 10.1128/iai.61.12.5090-5096.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dinauer MC, Deck MB, Unanue ER. Mice lacking reduced nicotinamide adenine dinucleotide phosphate oxidase activity show increased susceptibility to early infection with Listeria monocytogenes. . J Immunol. 1997;12:5581–5583. [PubMed] [Google Scholar]
- 35.Dai WJ, Bartens W, Kohler G, Hufnagel M, Kopf M, Brombacher F. Impaired macrophage listericidal and cytokine activities are responsible for the rapid death of Listeria monocytogenes-infected IFN-gamma receptor-deficient mice. J Immunol. 1997;158:5297–5304. [PubMed] [Google Scholar]