

Abstract
Polymeric immunoglobulins provide immunological protection at mucosal surfaces to which they are specifically transported by the polymeric immunoglobulin receptor (pIgR). Using a panel of human IgA1/IgG1 constant region “domain swap” mutants, the binding site for the pIgR on dimeric IgA (dIgA) was localized to the Cα3 domain. Selection of random peptides for pIgR binding and comparison with the IgA sequence suggested amino acids 402–410 (QEPSQGTTT), in a predicted exposed loop of the Cα3 domain, as a potential binding site. Alanine substitution of two groups of amino acids in this area abrogated the binding of dIgA to pIgR, whereas adjacent substitutions in a β-strand immediately NH2-terminal to this loop had no effect. All pIgR binding IgA sequences contain a conserved three amino acid insertion, not present in IgG, at this position. These data localize the pIgR binding site on dimeric human IgA to this loop structure in the Cα3 domain, which directs mucosal secretion of polymeric antibodies. We propose that it may be possible to use a pIgR binding motif to deliver antigen-specific dIgA and small-molecule drugs to mucosal epithelia for therapy.
Keywords: immunoglobulin A, secretory immunoglobulin A, polymeric immunoglobulin receptor, J chain, mucosal immunity
Secretory IgA (sIgA) mediates humoral immunological defense at mucosal surfaces (1), the largest surface area of the body normally exposed to pathogens (2). Mucosal secretion of antibody is mediated by the polymeric Ig receptor (pIgR) via a unique cellular transport process, termed transcytosis (3). The precursor of sIgA is dIgA, secreted by plasma cells with bound J chain (4). Compared with IgG, IgA heavy chains have an additional COOH-terminal 18-amino acid tailpiece with a penultimate cysteine residue (5). J chain disulfide bonds to two tailpiece cysteine residues, one on each monomeric IgA subunit, and the other cysteines form a direct tailpiece–tailpiece disulfide bond (6– 9). The pIgR is a type I transmembrane protein with five immunoglobulin superfamily homology domains (I–V) constituting the extracellular region (10). Polymeric Igs IgA and IgM are bound by pIgR at the basolateral surface of mucosal epithelial cells, transported through these cells, and secreted at the mucosae (3). sIgA retains the extracellular region of the pIgR, termed secretory component, covalently bound to dIgA (11–13), although the initial interaction with dIgA is the high-affinity noncovalent binding of pIgR domain I (14, 15).
Materials and Methods
Baculovirus Expression.
Arsonate hapten–specific chimeric IgA1 and IgA1/IgG1 domain swap mutants were expressed as previously described (16, 17). Dimeric IgA was generated by coexpression of IgA with J chain. Affinity purification was carried out on arsonate-sepharose. Antibodies were eluted with 200 mM arsanillic acid (Sigma Chemical Co.) in 200 mM Tris-HCl, pH 8.0, which was removed by extensive dialysis against PBS. Mono- and dimeric IgA were detected by 4% nonreducing SDS-PAGE analysis. The hexahistidine-tagged human pIgR extracellular domain was expressed in a similar manner and purified on a Ni-NTA Agarose (Qiagen) column (18).
Construction of Mutant IgA Antibodies for Baculovirus Expression.
The Cα3 loop mutants L1, L2, and L3 were constructed by PCR SOEing (splicing by overlap extension; reference 19) using the following complementary pairs of sense (S) and antisense (AS) primers: L1S 5′-GAGCCCAGCGCGGGCGCCGCCGCCTTCGCTGTG-3′, L1AS 5′-CTCGGGTCGCGCCCGCGGCGGC-GGAAGCGACAC-3′; L2S 5′-TACCTGACTGCGGCAGCCGCGCAGGAGCCC-3′, L2AS 5′-ATGGACTGACGCCGTC-GGCGCGTCCTCGGG-3′; and L3S 5′-CGGCAGGAGGCCGCCGCGGCCACCACCACC-3′, L3AS 5′-GCCGTCCTCCGGCGGCGCCGGTGGTGGTGG-3′. The outer primers B1-2 (5′-CCTATAACCATGGGATGGAGCTTCATC-3′), specific for the 5′ leader of the VH region of this chimeric IgA heavy chain, and Cα3-3′ (5′-CCCTCTAGATTAGTAGCAGGTGCCGTCCAC-3′), specific for the 3′ tailpiece–encoding sequence of the IgA1 gene, were used with the above primer pairs L1–L3 to generate pairs of 5′ and 3′ fragments with complementary overlaps. These fragments were gel purified then spliced in a further PCR reaction using the outer primers B1-2 and Cα3-3′. Modified IgA1 genes were cloned into the baculovirus transfer vector using XbaI and NcoI digestion and the insert sequences were verified. Recombinant baculovirus were produced using the BacPAK system (Clontech).
FACS® Analysis.
Madin-Derby canine kidney (MDCK) cells were placed in serum-free MEM plus Earle's salts (MediaTech, Inc.) 16 h before the experiment. Cells were harvested in 10 mM EDTA in PBS and washed in PBS 0.1% BSA. pIgR binding of human IgA1 antibodies and mutants was assessed by incubation of 100 μl antibody in PBS/BSA with ∼106 cells for 1 h. Cells were washed three times in PBS/BSA and bound antibody was detected with 100 μl of an anti–human κ FITC conjugate (Sigma Chemical Co.) diluted 1:100. Cells were washed as above and resuspended in 1 ml PBS/BSA. FACS® analysis was carried out on a Becton Dickinson FACScan® instrument. Data collection and analysis were performed with the LYSYSII (Becton Dickinson) and WINMIDI (http://facs.scripps.edu) or with the Cellquest programs (Becton Dickinson).
Phage Display Peptide Library Selection.
The random 40-mer peptide library was constructed in the pCANTAB5e vector and has an actual total diversity of 1.55 × 1010 (20). The random 40-mer is flanked by two peptide tag sequences, preceded by a leader peptide and fused to the membrane-proximal domain of the M13 phage coat protein III. 1–2 × 106 MDCK cells were harvested in 5 ml PBS plus 10 mM EDTA at 37°C, washed twice in 15 ml PBS, and resuspended in 1.8 ml PBS at 4°C. 100 μl phagemid library stock (4.5 × 1012 CFU) was added and incubated for 1 h at 37 or 4°C. The cells were then washed five times with 15 ml PBS at 4°C. Bound phage were eluted with 2 ml of 0.1 M glycine/ HCl, pH 2.2, containing 0.1% BSA for 10 min and neutralized immediately with 400 μl of 2 M Tris base. Phage rescue and amplification were carried out in Escherichia coli strain TG1 (Pharmacia) according to standard procedures (21).
DNA Sequencing and Analysis.
DNA sequencing was carried out on double-stranded plasmid or phagemid DNA using an ABI 377 Prism (Applied Biosystems, Inc.) automated sequencer. Alignments of deduced peptide sequences and Ig-constant regions were carried out using the MAP (22) and PIMA (23) software.
Results and Discussion
Chimeric human IgA1 (16) and a panel of IgA1/IgG1 constant region domain swap mutants (24) with murine-encoded arsonate specificity were expressed in baculovirus as both monomer and dimer, affinity purified, and used to define the pIgR binding site. dIgA was operationally defined as an IgA preparation generated by coexpression of IgA with J chain. MDCK cells, transfected with rabbit pIgR (25), were used to measure binding of recombinant IgA1 mutants to the receptor by FACS® analysis (Fig. 1 a). Specific binding was observed with dIgA and not with monomeric IgA (Fig. 1 b), a medium control (Fig. 1 b) or IgG (data not shown). Mutant VGAA, in which the Cα1 domain was substituted with the Cγ1 domain, bound to the pIgR in a manner similar to wild-type IgA1 (Fig. 1 c). The dimeric molecule (Fig. 1 c, heavy line) bound to the receptor, whereas the monomer (light line) did not. Similarly, the VGGA mutant, in which both Cα1 and Cα2 including the hinge of IgA were replaced with the analogous domains from IgG, bound as a dimer but not as a monomer (Fig. 1 d). Thus, the Cα1 and Cα2 domains of dIgA are not necessary for pIgR binding, suggesting that the presence of the Cα3 domain is required.
Figure 1.

Binding of monomeric and dimeric IgA/IgG domain swap mutant antibodies to pIgR expressed on MDCK cells. (a) Staining of MDCK cells with sheep anti-pIgR (heavy line) antiserum or normal sheep serum (broken line) followed by anti-sheep IgG FITC conjugate. (b) Binding of wild-type IgA monomer (thin line) or dimer (heavy line) to pIgR on MDCK cells. (c) Binding of VGAA mutant expressed as monomer (thin line) or dimer (heavy line) to pIgR on MDCK cells. (d) Binding of VGGA mutant expressed as monomer (thin line) or dimer (heavy line) to pIgR on MDCK cells. Bound IgA or IgA/G chimeric antibodies were detected by rabbit anti–human κ chain-FITC conjugate. Negative controls are shown as broken lines.
dIgA contains four Cα3 domains and the covalently bound J chain which, together with the IgA tailpiece, are responsible for IgA polymerization. To reduce the complexity of this problem, a library of random 40-mer peptides, expressed as a phage display library (20), was selected against pIgR-expressing MDCK cells. The goal was to identify putative pIgR binding sites within IgA by reducing them to a minimum peptide binding unit, a proven approach for several receptor–ligand interactions (26–28). Selection was carried out on live pIgR-expressing MDCK cells in suspension with negative selection on nonreceptor– expressing cells. Bound phage were eluted with acid or by cell lysis. Recovery of both acid-eluted and cell-associated phage increased gradually from ∼6 × 104 to 5 × 107 CFU over 4–6 successive rounds, indicating enrichment for specific binding clones. Individual clones were randomly selected from the final panning from the acid-eluted and membrane-associated fractions and sequenced. Binding of the enriched phage populations to recombinant human pIgR, as measured by ELISA, increased with successive rounds of panning and was inhibited by polymeric IgM (data not shown). Sequencing of phagemid DNA showed that 20 out of 32 acid-eluted clones and 12 out of 32 cell-associated clones had open reading frames (Fig. 2). There is little clonality among these two groups of sequences, although the A22 peptide was recovered three times. These peptides were aligned for maximum homology with the human IgA1 Cα3 region amino acid sequence (Fig. 2) using the PIMA program (23). Many of the peptides, particularly A12 (9 out of 30 identical amino acids) (Fig. 3 a), show homology with human IgA1 Cα3 domain, prompting a further examination of the amino acid sequence and structure in this area.
Figure 2.
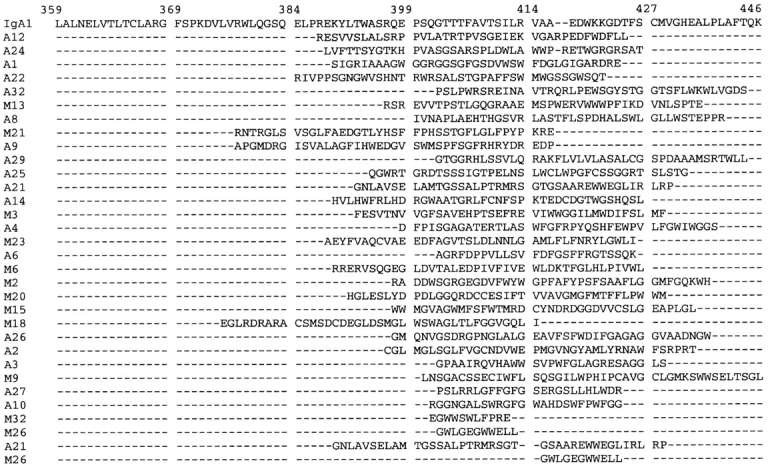
Alignment of deduced peptide sequences from selection of phage display peptide library against pIgR receptor–expressing cells with the human Cα3 domain amino acid sequence. Peptides designated A or M are from the acid-eluted and cell-associated fractions, respectively. Numbering of IgA1 is according to reference 5.
Figure 3.

Comparison of IgG1 and IgA1 CH3 sequences and IgG1 structure in the area homologous to several phage-derived peptides. (a) The A12 peptide alignment with human IgA1 and IgG1. IgGSTR indicates structural features of IgG1 where < denotes a β-strand running in a descending orientation (i.e., hinge to CH3 direction), > denotes a β-strand running in an ascending direction (i.e., CH3 to hinge direction), and – denotes a loop or open structure (29). (b) Comparison of several mammalian IgA sequences with the four human IgG subclasses showing the additional IgA-specific amino acids present in the loop at positions 402–410 in the IgA sequence. hu, human; gr, gorilla; mur, murine; rab, rabbit. (c) IgA1 Cα3 mutants L1, L2, and L3 aligned with the Cα3 and Cγ3 wild-type sequences and Cγ3 structure (IgGSTR). = denotes sequence identity in the mutants, – denotes a space introduced in the IgG sequence to maximize homology, and IgGSTR is labeled according to panel a. Numbering of IgA1 and IgG1 is according to references 5 and 29, respectively.
The human Cα3 domain is 40% identical and 62% homologous to the corresponding region of human IgG1 at the amino acid level. In addition, all the sequence hallmarks of the immunoglobulin superfamily fold are conserved. Accordingly, the human IgG1 crystal structure (29) was used to predict the likely positions of the major structural motifs (β-strands and loops) within the IgA1 sequence, an approach used previously to map the FcαR (CD89) binding site on IgA1 (24). Fig. 3 a shows the alignment of the peptide A12 with the IgA1 sequence and the corresponding IgG1 sequence with its secondary structural features. The A12 peptide is homologous to a region that in the IgG structure forms an exposed 6-amino acid loop between two β-strands. However, in IgA1, this area contains a 3-amino acid insertion to expand the loop to 9-amino acids. The flanking β-strand sequences and part of the loop are conserved between IgA and IgG, which suggests that gross structural features are also conserved. Fig. 3 b shows alignment of this region in the CH3 domain of five mammalian IgA molecules aligned with the four human IgG subclasses. Despite sequence differences in the loop, all IgA sequences have the three additional amino acids, whereas the IgG sequences do not. Similar to IgA, the sequence of IgM contains a 2-amino acid insertion at this site (data not shown). On the basis of these observations, three mutant IgA1 molecules were constructed and expressed in baculovirus to examine the effect of amino acid changes in this area on pIgR binding (Fig. 3 c). Mutations were made in the loop itself (L1 and L3) and in the β-strand NH2-terminal to the loop (L2) as a negative control. Binding was then measured to the physiologically relevant human receptor by ELISA using the purified recombinant extracellular domain of human pIgR expressed in baculovirus as previously described (18). Fig. 4 shows the binding of IgA1 monomer, IgA1 dimer, and IgG compared with the monomeric and dimeric forms of the L1, L2, and L3 mutants to purified human pIgR. Only dimeric wild-type IgA1 and dimeric L2 mutant, in which the mutations are in the β-strand NH2-terminal to the loop, show binding. Mutations within the loop itself, namely L1 and L3, abrogate the binding of the dimeric IgA1 mutant molecules to the pIgR. Similar binding patterns were obtained with the loop mutants and rabbit pIgR-expressing cells as measured by FACS® (data not shown). These results indicate that this Cα3 loop is the major binding motif for the pIgR on dIgA.
Figure 4.

Binding of IgA mutants L1, L2, and L3 to purified human pIgR by ELISA. The extracellular domain of human pIgR was purified after expression in baculovirus and coated onto ELISA plates at 10 μg/ml. Chimeric IgA1 and IgA1 Cα3 mutants L1, L2, and L3 were expressed as both monomeric (m) and dimeric (d) forms along with chimeric IgG1, purified and incubated on the pIgR-coated plates to compare their abilities to bind to pIgR. Bound antibodies were detected with anti–human κ light chain alkaline phosphatase conjugate.
IgA is, in functional terms, closely related to IgM, sharing its ability to polymerize and be secreted. However, the overall IgA domain organization resembles that of IgG. The presence of amino acid sequence insertions in all the polymeric Igs that are ligands for this receptor and the absence of insertions from non-pIgR binding Igs (Fig. 3 b) supports its role in Ig secretion. The variation in the insertion size and the actual IgA and IgM sequences may reflect differences in fine structure of these polymeric antibodies or in their affinity for pIgR binding.
The fact that monomeric IgA is not secreted suggests that either a conformational change induced by polymerization is required for dIgA binding to the receptor or that the binding requires a polyvalent interaction of these Cα3 sites with the receptor. The presence of J chain is required for optimal IgA (or IgM) polymerization but its precise role in Ig secretion remains to be elucidated. The increase in binding observed with dimeric L3 when compared with monomeric L3 (and to a lesser extent with the L1 mutants) suggests that J chain and/or polymerization may play a role in binding (Fig. 4). Although amino acids 402–410 in the Cα3 domain of dIgA define a major pIgR binding site, other dIgA structures may be involved. J chain–deficient mice express lower levels of polymeric IgA and have impaired hepatic transport of IgA (which humans lack) but normal levels of IgA at mucosal epithelial sites, compared with wild-type mice (30, 31). J chain thus may not be necessary for secretion of IgA but still required for stable binding to the secretory component in the mucosal environment; however, alternative secretory mechanisms may also be involved. Further studies are underway with peptides and additional mutations to examine the nature of the interaction between IgA and the pIgR as well as the role of J chain. The ability of a peptide sequence to confer mucosal secretion upon a molecule may prove a powerful means of delivery of therapeutic molecules to mucosal areas where they may prevent the entry of pathogens.
Acknowledgments
This study was supported in part by National Institutes of Health grant AI12127. J. Hexham was supported in part by the Pediatric AIDS Foundation. K. White was a recipient of a Leukemia Research Foundation fellowship.
Footnotes
Rabbit pIgR-expressing MDCK cells, untransfected MDCK cells and sheep anti-pIgR polyclonal antiserum were kind gifts from Keith Mostov, University of California at San Francisco, San Francisco, CA. The vector for baculovirus expression of the human pIgR was a kind gift from Jean-Pierre Kraehenbuhl, University of Lausanne, Lausanne, Switzerland. We also thank Shirley Hall for performing the DNA sequencing.
Present addresses are as follows: J.M. Hexham, Transplantation Research, Novartis Pharmaceuticals Corporation, 556 Morris Ave., Summit, NJ 07901; L.N. Carayannopoulos, Washington University Medical School, St. Louis, MO 87978; W. Mandecki and R. Brisette, DGI Biotechnologies LLC, PO Box 424, Edison, NJ 08818; and Y.-S. Yang, Department of Internal Medicine, UT Southwestern Medical Center, 6000 Harry Hines Blvd., Dallas, TX 75235.
References
- 1.Underdown BJ, Schiff JM. Immunoglobulin A: strategic defense initiative at the mucosal surface. Annu Rev Immunol. 1986;4:389–417. doi: 10.1146/annurev.iy.04.040186.002133. [DOI] [PubMed] [Google Scholar]
- 2.Childers NK, Bruce MG, McGhee JR. Molecular mechanisms of immunoglobulin A defense. Annu Rev Microbiol. 1989;43:503–536. doi: 10.1146/annurev.mi.43.100189.002443. [DOI] [PubMed] [Google Scholar]
- 3.Mostov KE, Blobel G. A transmembrane precursor of secretory component. The receptor for transcellular transport of polymeric immunoglobulins. J Biol Chem. 1982;257:11816–11821. [PubMed] [Google Scholar]
- 4.Koshland ME. The coming of age of the immunoglobulin J chain. Annu Rev Immunol. 1985;3:425–453. doi: 10.1146/annurev.iy.03.040185.002233. [DOI] [PubMed] [Google Scholar]
- 5.Putnam FW, Liu YSV, Low TLK. Primary structure of a human IgA1 immunoglobulin. J Biol Chem. 1979;254:2865–2874. [PubMed] [Google Scholar]
- 6.Garcia-Pardo A, Lamm ME, Plaut AG, Frangione B. J chain is covalently bound to both monomer subunits in human secretory IgA. J Biol Chem. 1981;256:11734–11738. [PubMed] [Google Scholar]
- 7.Bastian A, Kratzin H, Eckart K, Hilschmann N. Intra- and interchain disulfide bridges of the human J chain in secretory immunoglobulin A. Biol Chem Hoppe Seyler. 1992;373:1255–1263. doi: 10.1515/bchm3.1992.373.2.1255. [DOI] [PubMed] [Google Scholar]
- 8.Atkin JD, Pleass RJ, Owens RJ, Woof JM. Mutagenesis of the IgA1 heavy chain tailpiece that prevents dimer assembly. J Immunol. 1996;157:156–159. [PubMed] [Google Scholar]
- 9.Krugman S, Pleass RJ, Atkin JD, Woof JM. Structural requirements for assembly of dimeric IgA probed by site-directed mutagenesis of J chain and a cysteine residue of the alpha-chain CH2 domain. J Immunol. 1997;159:144–249. [PubMed] [Google Scholar]
- 10.Mostov KE, Freidlander M, Blobel G. The receptor for transepithelial transport of IgA and IgM contains multiple immunoglobulin-like domains. Nature. 1984;308:37–43. doi: 10.1038/308037a0. [DOI] [PubMed] [Google Scholar]
- 11.Lindh E, Bjork I. Binding of secretory component to dimers of immunoglobulin A in vitro. Eur J Biochem. 1974;45:261–268. doi: 10.1111/j.1432-1033.1974.tb03550.x. [DOI] [PubMed] [Google Scholar]
- 12.Fallgreen-Gebauer E, Gebauer W, Bastian A, Kratzin HD, Eiffert H, Zimmermann B, Karas M, Hilschmann N. The covalent linkage of secretory component to IgA. Biol Chem Hoppe Seyler. 1993;374:1023–1028. doi: 10.1515/bchm3.1993.374.7-12.1023. [DOI] [PubMed] [Google Scholar]
- 13.Underdown BJ, De Rose J, Plaut A. Disulfide bonding of secretory component to a single monomer subunit in human secretory IgA. J Immunol. 1977;118:1816–1821. [PubMed] [Google Scholar]
- 14.Frutiger S, Hughes GJ, Hanly WC, Kingzette M, Jaton JC. The amino terminal domain of rabbit secretory component is responsible for noncovalent binding to immunoglobulin A dimers. J Biol Chem. 1986;261:16673–16681. [PubMed] [Google Scholar]
- 15.Bakos M, Kurowsky A, Goldblum RM. Characterization of a critical binding site for human polymeric Ig on secretory component. J Immunol. 1991;147:3419–3426. [PubMed] [Google Scholar]
- 16.Carayannopoulos LN, Max EE, Capra JD. Recombinant human IgA expressed in insect cells. Proc Natl Acad Sci USA. 1994;91:8348–8352. doi: 10.1073/pnas.91.18.8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Reilly, D.R., L.K. Miller, and V.A. Luckow. 1992. Baculovirus Expression Vectors: A Laboratory Manual. W.H. Freeman and Co., New York. 347 pp.
- 18.Rindisbacher L, Cottet S, Witteek R, Kraehenbuhl J-P. Production of human secretory component with dimeric IgA binding capacity using viral expression systems. J Biol Chem. 1995;270:14220–14228. doi: 10.1074/jbc.270.23.14220. [DOI] [PubMed] [Google Scholar]
- 19.Horton RM, Cai Z, Ho SN, Pease LR. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques. 1990;8:528–535. [PubMed] [Google Scholar]
- 20.Ravera MW, Carcamo J, Brissette R, Alam-Moghe A, Dedova O, Cheng W, Hsiao KC, Klebanov D, Shen H, Tang P, et al. Identification of an allosteric binding site on the transcription factor p53 using a phage-displayed peptide library. Oncogene. 1998;16:1993–1999. doi: 10.1038/sj.onc.1201717. [DOI] [PubMed] [Google Scholar]
- 21.Hexham JM. Production of human Fab antibody fragments from phage display libraries. Methods Mol Biol. 1998;80:461–474. doi: 10.1007/978-1-59259-257-9_46. [DOI] [PubMed] [Google Scholar]
- 22.Huang X. On global sequence alignment. Comput Appl Biosci. 1994;10:227–235. doi: 10.1093/bioinformatics/10.3.227. [DOI] [PubMed] [Google Scholar]
- 23.Smith RF, Smith TF. Pattern-induced multi-sequence alignment (PIMA) algorithm employing secondary structure-dependent gap penalties for use in comparative protein modelling. Protein Eng. 1992;5:35–41. doi: 10.1093/protein/5.1.35. [DOI] [PubMed] [Google Scholar]
- 24.Carayannopoulos LN, Hexham JM, Capra JD. Localization of the binding site for the monocyte IgA-Fc receptor (CD89) to the domain boundary between Ca2 and Ca3 in human IgA1. J Exp Med. 1996;183:1579–1586. doi: 10.1084/jem.183.4.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mostov KE, Deitcher DL. Polymeric immunoglobulin receptor expressed in MDCK cells transcytoses IgA. Cell. 1986;46:613–621. doi: 10.1016/0092-8674(86)90887-1. [DOI] [PubMed] [Google Scholar]
- 26.Cwirla S, Peters E, Barrett R, Dower W. Peptides on phage: a vast library of peptides for identifying ligands. Proc Natl Acad Sci USA. 1990;87:6378–6382. doi: 10.1073/pnas.87.16.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parmley S, Smith G. Antibody-selectable filamentous fd phage vectors: affinity purification of target genes. Gene. 1988;73:305–318. doi: 10.1016/0378-1119(88)90495-7. [DOI] [PubMed] [Google Scholar]
- 28.Devlin J, Panganiban L, Devlin P. Random peptide libraries: a source for specific protein binding molecules. Science. 1990;249:404–406. doi: 10.1126/science.2143033. [DOI] [PubMed] [Google Scholar]
- 29.Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureusat 2.9 and 2.8 Angstroms resolution. Biochemistry. 1981;20:2361–2370. [PubMed] [Google Scholar]
- 30.Hendrickson BA, Conner DA, Ladd DJ, Kendall D, Casanova JE, Corthesy B, Max EE, Neutra MR, Seidman CE, Seidman JG. Altered hepatic transport of immunoglobulin A in mice lacking the J chain. J Exp Med. 1995;182:1905–1911. doi: 10.1084/jem.182.6.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hendrickson B, Rindisbacher L, Corthesy B, Kendall D, Waltz D, Neutra M, Seidman J. Lack of association of secretory component with IgA in J chain-deficient mice. J Immunol. 1996;157:750–754. [PubMed] [Google Scholar]