

Abstract
Stromelysin-3 is an unusual matrix metalloproteinase, being released in the active rather than zymogen form and having a distinct substrate specificity, targeting serine proteinase inhibitors (serpins), which regulate cellular functions involved in atherosclerosis. We report here that human atherosclerotic plaques (n = 7) express stromelysin-3 in situ, whereas fatty streaks (n = 5) and normal arterial specimens (n = 5) contain little or no stromelysin-3. Stromelysin-3 mRNA and protein colocalized with endothelial cells, smooth muscle cells, and macrophages within the lesion. In vitro, usual inducers of matrix metalloproteinases such as interleukin-1, interferon-γ, or tumor necrosis factor α did not augment stromelysin-3 in vascular wall cells. However, T cell–derived as well as recombinant CD40 ligand (CD40L, CD154), an inflammatory mediator recently localized in atheroma, induced de novo synthesis of stromelysin-3. In addition, stromelysin-3 mRNA and protein colocalized with CD40L and CD40 within atheroma. In accordance with the in situ and in vitro data obtained with human material, interruption of the CD40–CD40L signaling pathway in low density lipoprotein receptor–deficient hyperlipidemic mice substantially decreased expression of the enzyme within atherosclerotic plaques. These observations establish the expression of the unusual matrix metalloproteinase stromelysin-3 in human atherosclerotic lesions and implicate CD40–CD40L signaling in its regulation, thus providing a possible new pathway that triggers complications within atherosclerotic lesions.
Keywords: atherosclerosis, CD40L/CD154, matrix metalloproteinase, serpins, stromelysin-3
The proteinase stromelysin-3 (MMP-11) belongs to the matrix metalloproteinase (MMP)1 family, enzymes which participate in tissue remodeling in a variety of diseases, including tumor metastasis and invasion, arthritis, and atherogenesis (1–8). In contrast to “classical” MMPs, secreted as inactive zymogens, stromelysin-3 is released as an active enzyme (9, 10). The zymogen contains a unique 10– amino acid insert between the pro and catalytic domain that includes a recognition motif for furin, the Golgi-associated pro protein convertase, which processes stromelysin-3 to its enzymatically active form during traversal through the constitutive secretory pathway.
Stromelysin-3 was originally identified by differential screening of cDNA libraries derived from an invasive breast cancer surgical specimen and from benign breast fibroadenoma (11). Stromelysin-3 mRNA and protein expression correlates with the invasiveness of human carcinomas and also occurs in some sarcomas or other nonepithelial tumors (3, 12–15). These, as well as other studies (16–20), indicated a crucial role for the proteinase in processes characterized by extensive extracellular matrix turnover. Stromelysin-3, however, has a restricted substrate specificity distinct from all other matrix metalloproteinases. The enzyme only weakly, if at all, degrades extracellular matrix constituents such as laminin, fibronectin, and collagen (21, 22). Instead, substrates of stromelysin-3 include serine proteinase inhibitors, termed serpins (23, 24).
The serpin superfamily includes more than 60 proteins, which can function as inhibitory mediators by acting as suicide substrates for the respective enzyme, with the vast majority targeted towards the serine proteinases known to be involved in extracellular matrix remodeling, regulation of blood pressure, modulation of inflammatory responses, cell migration and differentiation, fibrinolysis, or blood coagulation (24). This group of serpins consists of single chain proteins, such as α1-proteinase inhibitor (α1-PI), α1-antitrypsin (α1-AT), α2-antiplasmin (α2-AP), α2-macroglobulin (α2-M), plasminogen activator inhibitor, antithrombin III (AT-III), C1 inhibitor, angiotensinogen, etc. Most of these serpins have known functions: α1-PI acts on elastolytic proteases, such as leukocyte elastase, and cathepsin G (25, 26); α1-AT and α2-M are involved in lipoprotein catabolism (27–29); α1-AT also inhibits renin and thus the renin– angiotensinogen interaction (30); α2-AP and plasminogen activator inhibitor are involved in the regulation of fibrinolysis; C1 inhibitor is essential for the regulation of activation of the complement and kinin generating system; and AT-III as well as heparin cofactor II play a central role in the regulation of blood coagulation (31). Interestingly, serpins also inhibit matrix degrading enzymes, such as cathepsins (32). The regulation of matrix degradation may thus not only be promoted by tissue inhibitors of matrix metalloproteinases/MMP imbalance but also accelerated by decreased levels of certain serpins such as α2-AP or α1-PI (33), inhibitors of matrix degrading activity. In this manner, stromelysin-3, despite possessing weak direct matrix degrading capabilities, might yet play a crucial role in matrix turnover.
Serpins play critical roles in maintaining homeostasis. Therefore, any mechanism that reduces the functional level of members of this superfamily, including inactivation by proteinases, may result in substantial pathological problems (23, 24). Several of the functions listed above relate to atherosclerosis, and atherosclerotic patients exhibit changes in the levels of several of these serpins, including α1-AT, α2-M (34, 35), and AT-III (36). In addition, animal studies indicate a potential role of serpins in the inhibition of arterial intimal thickening (37) and atherosclerotic plaque development post injury (38) in vivo. For these reasons, the present study analyzed the expression of stromelysin-3 within human atherosclerotic lesions.
Although the expression of stromelysin-3 at sites of pathologic processes such as Alzheimer's disease or cancer has prompted great interest in its regulation, little is known about the mechanisms involved (11, 39, 40). The stromelysin-3 gene promoter differs markedly from previously described matrix metalloproteinase promoters as it lacks a consensus activator protein 1 binding site and has a functional retinoic acid responsive element. We recently demonstrated the presence of the immune mediators CD40 ligand (CD40L) and its receptor CD40 on endothelial cells (EC), smooth muscle cells (SMC), and macrophages (MØ) within human atherosclerotic lesions (41) and showed that ligation of CD40 induces de novo synthesis of the classical MMP interstitial collagenase (MMP-1) and the gelatinases A and B (MMP-2 and -9, respectively) (42, 43) in these cells. Therefore, this study also tested the hypothesis that CD40 signaling regulates the expression of the constitutively active matrix metalloproteinase stromelysin-3 in human atheroma-associated cells and in mouse atheroma in vivo.
We report here that (a) stromelysin-3 (MMP-11) is present in human atherosclerotic lesions in situ, (b) cultured human vascular EC, SMC, and MØ synthesize stromelysin-3 de novo upon stimulation with CD40L, and (c) interruption of CD40–CD40L signaling in hyperlipidemic mice diminishes stromelysin-3 expression in atherosclerotic lesions in vivo.
Materials and Methods
Materials.
Human recombinant IL-1β, TNFα, and IFNγ were obtained from Endogen. Phorbol-12 myristate 13-acetate (PMA) and polymyxin B were purchased from Sigma Chemical Co. Human recombinant CD40L (rCD40L) was generated as described previously (44) and the mouse anti–human stromelysin-3 antibody 5ST-4A9 was produced in a program sponsored by Bristol Myers Squibb and is subject of an issued U.S. utility patent number 5484726 (45). Experiments employing rCD40L were performed in the presence of polymyxin B. Anti-CD40L, a rat mAb IgG2 antibody raised against mouse CD40L was prepared as described (46) and provided by Immunex Corp. Rat IgG salt-free crystalline powder (Sigma Chemical Co.), reconstituted in pyrogen-free normal saline and sterile filtered, was obtained from Immunex Corp. Both rat anti–mouse CD40L antibody and rat IgG contained <2 pg/μl of endotoxin. Anti–human CD40 as well as control IgG1 mAb (FITC conjugated) used for immunohistochemistry were obtained from PharMingen. Low density lipoprotein receptor–deficient (LDLR−/−) mice (B6/129LDRr-tm1Her) were obtained from Jackson Laboratory.
Cell Isolation and Culture.
Human vascular EC were isolated from saphenous veins by collagenase treatment (1 mg/ml; Worthington Biochemicals) and cultured in dishes coated with fibronectin (1.5 μg/cm2; New York Blood Center Reagents). Cells were maintained in medium 199 (M199; BioWhittaker) supplemented with 1% penicillin/streptomycin (BioWhittaker), 5% fetal bovine serum (FBS) (Atlanta Biologicals), 50 μg/ml heparin (Sigma Chemical Co.), and endothelial cell growth factor (Pel-Freez Biological). SMC were isolated from human saphenous veins by explant outgrowth (47) and cultured in DMEM (BioWhittaker) supplemented with 1% l-glutamine (BioWhittaker), 1% penicillin/streptomycin, and 10% FBS. Both cell types were subcultured following trypsinization (0.5% trypsin [Worthington Biochemicals]/0.2% EDTA [EM Science]) in 75 cm2 culture flasks (Becton Dickinson) and used throughout passages two to four. Culture media and FBS contained <40 pg endotoxin/ml as determined by chromogenic Limulus amoebocyte assay analysis (QLC-1000; BioWhittaker). EC and SMC were characterized by immunostaining with anti von Willebrand factor and anti SMC α-actin antibody (Dako), respectively. Both cell types were cultured 24 h before the experiment in media lacking FBS: vascular EC were cultured in M199 supplemented with 0.1% human serum albumin and vascular SMC cultured in insulin/transferrin medium as described previously (48).
Mononuclear phagocytes were isolated by density gradient centrifugation (49), using lymphocyte separation medium (Organon-Teknika), and subsequent counterflow elutriation from freshly prepared human PBMCs obtained from leukopacs of healthy donors (provided by Steve K. Clinton, Dana-Farber Cancer Institute, Boston, MA). Mononuclear phagocytes were used directly (monocytes) for the experiments or cultured for 1, 3, or 9 d (MØ) in RPMI 1640 containing 2% human serum (Sigma Chemical Co.). The purity of monocytes/MØ was ≥96%, as determined by FACS® analysis (anti–human CD68 mAb FITC; PharMingen). For certain studies, MØ were stimulated in RPMI 1640 lacking serum.
Freshly isolated human CD4+ T cells were a gift from Dr. F.W. Luscinskas (Brigham and Women's Hospital, Boston, MA). The purity of CD4+ T cells was ≥98%, as determined by FACS® analysis (anti–human CD4 mAb FITC; Calbiochem). Human CD4+ T cells were activated with PMA (50 ng/ml, 12 h) and CD40L cell surface expression confirmed by FACS® analysis using FITC-labeled anti–human CD40L mAb (Calbiochem). Finally, activated CD4+ T cell membranes were prepared as described previously (42) and used for stimulation at a ratio of T cells to SMC/EC/MØ of 10:1.
Immunohistochemistry.
Surgical specimens of human carotid atheroma and aorta were obtained by protocols approved by the Human Investigation Review Committee at the Brigham and Women's Hospital. Serial cryostat sections (5 μm) were cut, air dried onto microscope slides (Fisher Scientific), and fixed in acetone at −20°C for 5 min. Sections were preincubated with PBS containing 0.3% hydrogen peroxidase activity. The sections were then incubated (90 min) with primary or control (mouse myeloma protein MOPC-21; Sigma Chemical Co.) antibody diluted in PBS supplemented with 5% appropriate serum. After washing three times in PBS, sections were incubated with the respective biotinylated secondary antibody (45 min; Vector) followed by avidin–biotin–peroxidase complex (Vectastain ABC kit; Vector), and antibody binding was visualized with 3-amino-9-ethyl carbazole (Vector) according to the recommendations provided by the supplier. For colocalization of stromelysin-3 with CD40 or the respective cell type, double immunofluorescence staining was performed. The anti–human stromelysin-3 mAb (1:200) was applied for 60 min followed by biotinylated anti–mouse secondary antibody for 45 min and Texas red–conjugated streptavidin (Amersham). Subsequent to application of the avidin–biotin blocking kit (Vector), rabbit–anti human CD40 antibody (1:250; Santa Cruz), anti-muscle actin mAb for SMC (Enzo Diagnostics), anti-CD31 mAb for EC (1:400, Dako), or anti-CD68 mAb for MØ (1:600; Dako) were added and sections incubated overnight at 4°C. Subsequently, the appropriate secondary antibodies were applied for 30 min followed by Streptavidin–FITC (Amersham). To detect stromelysin-3 in mouse atherosclerotic lesions, the stromelysin-3 mAb and a specific anti– mouse absorbed rat biotinylated secondary antibody (1:500), followed by avidin–biotin–peroxidase complex, were applied.
Biochemical Analysis of Human Atherosclerotic Lesions.
Frozen tissue from five nonatherosclerotic arteries and seven atheromatous carotid plaques were homogenized (IKA-Labortechnik, Ultra-turrax T 25) and lysed (0.3 mg tissue/ml lysis buffer) as described previously (5). The lysates were clarified (16,000 g, 15 min) and the protein concentration for each tissue extract as well as for the cell culture samples was determined using a bicinchoninic acid protein assay according to the instructions of the manufacturer (Pierce). 50 μg total protein were applied to Western blot analysis.
In Situ Hybridization.
In situ hybridization was performed according to the instructions of the manufacturer (Hyb-Probe™; Shandon/Lipshaw). Frozen tissue sections, obtained as described above, were fixed in cold acetone, air-dried, and incubated with a mixture of FITC labeled stromelysin-3–specific (5′-GGTACCGTCAACCAGGTCCTCGTCCACG-3′; 5′-CTCAGAGTCGGGTCTACTGACCGTCC-3′; 5′-CCTACTGGTCCCGTGTCTGGACGACGTCCA-3′; 5′-ACGGTCCGGTGCTTATAGTCC-GATCTCCTGG-3′), CD40L-specific (5′-TTATGGGTGTCAA-GGCGGTTTGGAACGCCCGTT-3′; 5′-TGAAAAACGACA-CATAGAAGTATCTTCCAA-3′; 5′-TACTAGCTTTGTATGTTGGTTTGAAGAGGGGCT-3′; 5′-TGCAGGAAACCGAATGAGTTTGAGACTTGT-3′), or random oligomers in hybridization buffer (30% formamide, 0.6 M NaCl2, 10% dextran sulfate, 50 mM Tris [pH 7.5], 0.1% sodiumpyrophosphate, 0.2% Ficoll, 5 mM EDTA) for 10 min at 65°C and subsequently for 2 h at 37°C in a moist chamber. Thereafter, slides were immersed in TBS/ Triton (50 mM Tris, 150 mM NaCl, pH 7.6/0.1% Triton X-100) to allow coverslips to float off and were washed (TBS/ Triton) three times for 3 min at 37°C. The slides were forwarded to the immunological reaction by incubation with blocking solution (10 min, rt) and subsequent addition of the alkaline phosphatase–conjugated rabbit Fab′ anti FITC (30 min, rt, moist chamber) as the primary detection reagent. Finally, the slides were washed twice in TBS (3 min, rt), covered with alkaline phosphatase substrate buffer (5 min, rt), and developed using the NBT/BCIP (nitro-blue-tetrazolium/5-bromo-4-chloro-3-indolyl phosphate) chromogen solution (1–2 h, rt, moist chamber).
Western Blotting and Radioimmunoprecipitation.
Cell extracts (25 μg total protein/lane) and culture supernatants were separated by standard SDS-PAGE under reducing conditions and blotted to polyvinylidene difluoride membranes (Bio-Rad) using a semidry blotting apparatus (0.8 mA/cm2, 30 min; Bio-Rad). Blots were blocked and first and second monoclonal antibodies were diluted in 5% defatted dry milk/PBS/0.1% Tween 20. After 1 h of incubation with the respective primary antibody, blots were washed three times (PBS/0.1% Tween 20) and the secondary, peroxidase–conjugated, goat anti–mouse antibody (Jackson ImmunoResearch) was added for another hour. Finally, the blots were washed (20 min, PBS/0.1% Tween 20) and immunoreactive proteins were visualized using the Western blot chemiluminescence system (NEN™).
For radioimmunoprecipitation experiments, cells were washed and further incubated with unlabeled medium lacking methionine/cysteine. Subsequently, medium containing 10% FBS and 50 mCi/ml [35S]methionine/cysteine (NEN™) was added to the cells for 24 h. Supernatants were harvested and concentrated (10×) using Centricon 3 devices (Amicon). Immunoprecipitation buffer (50 mM Tris-HCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% NP-40, 150 mM NaCl, 5 mM EDTA, 20 μg/ml soybean trypsin inhibitor, 0.1% mM PMSF, 0.2 U/ml aprotinin, 0.025% sodium azide) was added to the cultures and cells were harvested by scraping. Subsequently, non immune mouse serum (Vector) was added (24 h, 4°C) to preclear the samples. Antigens in supernatants and cell extracts were immunoprecipitated with the specific anti stromelysin-3 antibody 5ST-4A9 (2 h, 4°C) and pelleted by subsequent addition of rabbit anti–mouse IgG (18 h, 4°C) as well as protein A–Sepharose beads (2 h, 4°C). The beads were washed four times in 50 mM Tris-HCl and finally 50 μl SDS-PAGE loading buffer (200 mmol/liter Tris, 5% glycerol, 0.1% SDS, 3% β-mercaptoethanol, 0.1 mg/ml bromophenol blue) was added. After heating the samples 5 min at 95°C, supernatants were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Blots were dried and exposed to x-ray film for detection of the immunoprecipitated antigen.
Isolation of RNA and PCR.
Total RNA from unstimulated or stimulated (24 h) EC, SMC, or MØ was isolated by a one-step preparation according to the method of Chomczynski and Sacchi (50). The cDNA was prepared by reverse transcription (RT) of total RNA (1 μg) with oligo(dT) using superscript reverse transcriptase (GIBCO BRL). The RT products were diluted in 480 μl twice distilled water and 10 μl of these cDNA preparations were mixed on ice with 10 μl primers (20 μM), 80 μl reaction mix (including 10 ml of PCR buffer, 2.5 mM MgCl), 4 μl dNTPs (200 μM; final concentration), and 0.5 μl Taq-polymerase (2.5 U; all GIBCO BRL). PCR was performed for 35 cycles at 95°C (120 s), 62°C (120 s), and 72°C (180 s, 2 s prolongation per cycle) after hot start. The sequences of primers for human stromelysin-3 were 5′-CTGCAGTCATCTGGGCTGAGACTC-3′ and 5′-CCATGGCAGTTGGTGCAGGAGCAG-3′. The primers were obtained from Integrated DNA Technologies. Aliquots of the PCR products were run on 1.3% agarose gels and visualized by UV transillumination.
In Vivo Analysis of CD40L-mediated Stromelysin-3 Expression.
LDLR−/− at a minimum age of 8 wk were fed a high cholesterol diet (1.25% cholesterol, 0% cholate; Research Diets) and treated for a period of 12 wk with either rat IgG (control) or rat anti– mouse CD40L (M158) antibody (both at 250 μg per mouse twice a week intraperitoneal; both provided by Immunex Corp.) (51). Thereafter, the mice (10 per group) were killed, the aortic arches embedded, and tissue sections examined by immunohistochemistry as described below. The extent of lesion formation and the expression of vascular adhesion molecule-1 in these animals has been reported separately (51). No difference in the serum lipid profiles (total cholesterol, very low density lipoprotein, low density lipoprotein, and high density lipoprotein [analyzed by fast protein liquid chromatography] or triglycerides) or in circulating leukocytes, hematocrit, or body weight was observed among the groups.
Results
Expression of Stromelysin-3 in Human Atherosclerotic Plaques.
Immunohistochemical analysis of the expression of stromelysin-3 in normal aortic specimens (n = 5; Fig. 1 A, left) and human atherosclerotic fatty streaks (n = 5; data not shown) revealed little or no expression of the enzyme. In contrast, well-developed human carotid atherosclerotic lesions (n = 7) consistently showed strong stromelysin-3 immunoreactivity, most prominently at the luminal border and in the shoulder region of the plaque (Fig. 1 A, right). Western Blot analysis, performed on protein extracts of the surgical specimens and using the identical antibody used for the immunohistochemistry studies, revealed barely detectable immunoreactive stromelysin-3 in control specimens but markedly increased levels of the proteinase in atherosclerotic tissue (Fig. 1 B). The immunoreactive bands detected had molecular masses of ∼64, 48, 35, and 28 kD, corresponding to the zymogen, intermediate, and active forms of stromelysin-3 (9, 10, 22, 23, 52, and 53). Higher magnifications of the immunohistochemical analysis, as well as immunofluorescent double staining with respective cell-selective antibodies, localized stromelysin-3 within EC, SMC, and MØ of the plaque (Fig. 2). Tissues showed no staining with the respective control IgG1 antibody (data not shown). Because we recently localized CD40 and CD40L in human atherosclerotic plaques and have shown that CD40 ligation induces interstitial collagenases and gelatinases in atheroma-associated cells (41–43), we investigated the possible colocalization of stromelysin-3 with CD40. Indeed, cells expressing stromelysin-3 also bear CD40 (Fig. 3). Furthermore, we analyzed the cellular localization of stromelysin-3 transcripts by in situ hybridization (Fig. 4). Human atheroma (Fig. 4, C–E), but not normal arteries (Fig. 4, A and B), contained stromelysin-3 mRNA. Within the atherosclerotic lesion, stromelysin-3 transcripts localized most prominently at the luminal border and the shoulder region of the plaques, areas described above as positive for the immunoreactive protein. The staining for the transcripts colocalized with smooth muscle cell- and macrophage-like cells (Fig. 4, D and E) as well as the endothelium (Fig. 4 E). Furthermore, transcripts for the immune mediator CD40L showed a similar distribution on adjacent sections (Fig. 4, G and H). In situ hybridization with negative control probes did not yield any signal (Fig. 4 F).
Figure 1.

Expression of stromelysin-3 in human atherosclerotic plaques. (A) Frozen sections of normal human arterial tissue and human atheromatous plaques were stained for stromelysin-3. The tissue was analyzed using horseradish peroxidase–mediated immunohistochemistry on adjacent sections (red reaction product). No immunoreactivity was observed in tissue stained with the respective control IgG1 antibody (data not shown). The lumen of the artery is at the top of each photomicrograph (×100). Analysis of five normal aortic tissue and seven atheroma obtained from different donors showed similar results. (B) Extracts (50 μg/ml) of three nonatherosclerotic tissue (Normal) and atheromatous plaques (Atheromatous) were separated by standard SDS-PAGE under reducing conditions and analyzed by Western blotting for stromelysin-3 expression. The positions of the molecular mass markers are indicated (kD). Analysis of five normal tissues as well as seven atheromatous lesions from different donors showed similar results.
Figure 2.

Colocalization of stromelysin-3 with endothelial cells, smooth muscle cells, and macrophages in human atheroma. High power views (×400) of frozen sections of human carotid lesions showed specific staining for stromelysin-3 (right, red staining) on human vascular endothelial cells (EC), smooth muscle cells (SMC), and macrophages (MØ) within the plaque. Cell types were characterized by immunofluorescence double staining (left, green staining) as described in Materials and Methods. The lumen of the artery is at the top of each photomicrograph. Analysis of seven atheroma from different donors showed similar results.
Figure 3.
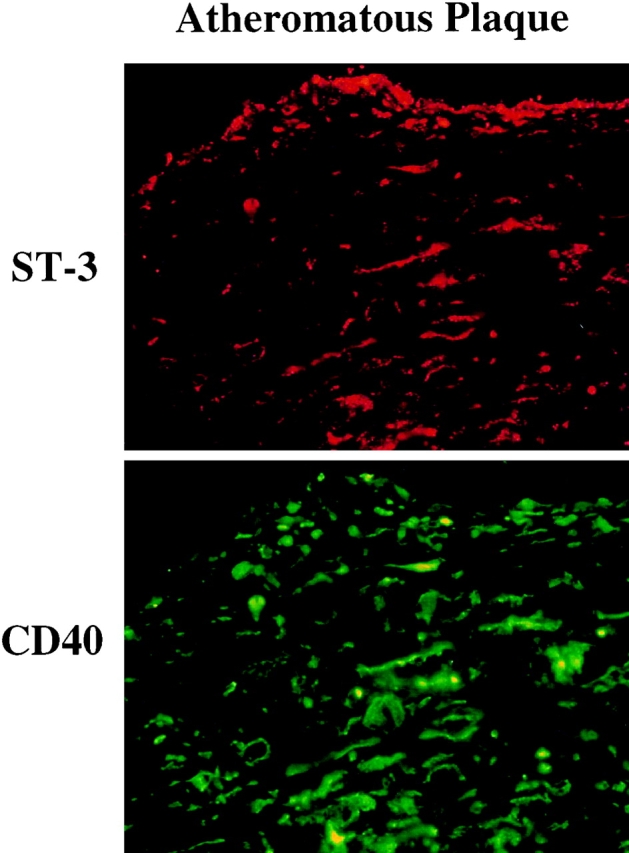
Colocalization of stromelysin-3 with CD40 in human atherosclerotic lesions. Frozen sections of normal human atheromatous plaques were stained for stromelysin-3 and CD40 by immunofluorescence double staining (stromelysin-3 in red, CD40 in green). No immunoreactivity was observed in tissue stained with the respective control IgG1 antibody (data not shown). The lumen of the artery is at the top of each photomicrograph (×100). Analysis of five normal aortic tissue and seven atheroma obtained from different donors showed similar results.
Figure 4.
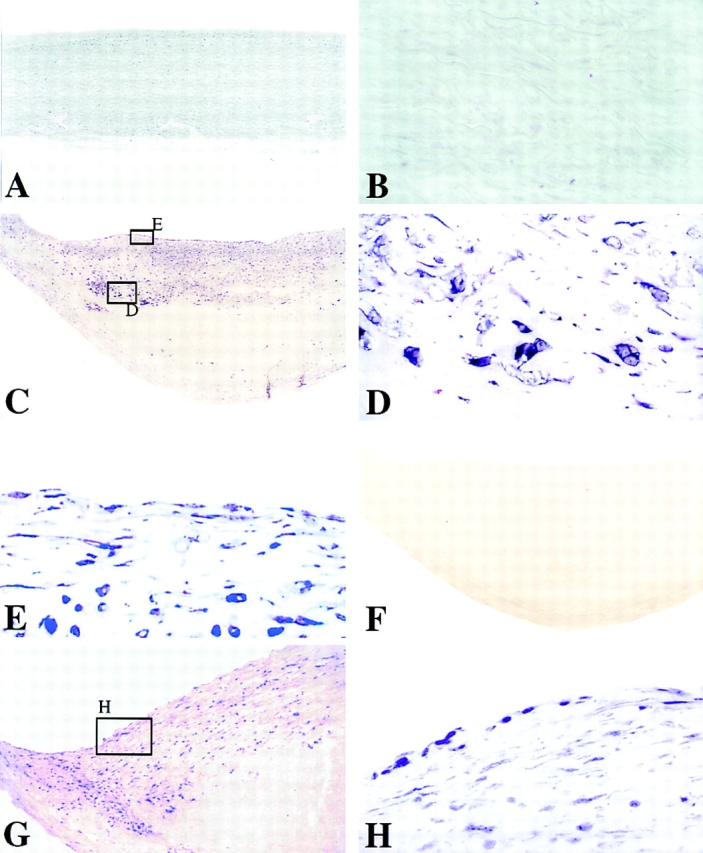
Human stromelysin-3 mRNA colocalizes with EC, SMC, and MØ of the atherosclerotic lesion. In situ hybridization analysis of nonatherosclerotic tissue (A and B) revealed no detectable stromelysin-3 mRNA signal, in contrast to atheromatous plaques (C–E). Higher magnification (×400) of stromelysin-3 mRNA-positive areas revealed colocalization with MØ-like (D), as well as EC- and SMC-like (E) cell types within the lesion. Negative control probes yielded no detectable signal (F). Areas of the plaque and cell types similar to those found positive for stromelysin-3 stained also for CD40L mRNA (G and H). Analysis of three atheroma from different donors showed similar results.
Ligation of CD40 on Human Vascular Endothelial and SMC as well as MØ Induces De Novo Expression of Stromelysin-3.
To determine the mechanisms involved in stromelysin-3 expression in atheroma-associated cells, we analyzed the expression of the enzyme in human vascular EC and SMC as well as in human mononuclear phagocytes in vitro. Human vascular EC, SMC, and MØ released immunoreactive stromelysin-3 constitutively in moderate amounts. Stimulation of the cells with the classical mediators of MMP regulation, IL-1 (1 and 10 ng/ml), TNF-α (5 and 50 ng/ml), or IFNγ (100 and 1,000 U/ml), did not affect the expression of stromelysin-3, as illustrated here for IL-1 and SMC (Fig. 5). However, the immunohistochemical studies presented above indicated possible involvement of the CD40– CD40L signaling pathway. Stimulation with both T cell– derived as well as recombinant human CD40L induced stromelysin-3 expression in all three cell types (Figs. 5 and 6). Besides an increased intensity of the higher (∼64 kD) molecular mass band, which corresponds to the molecular mass of the stromelysin-3 zymogen, CD40 ligation induced the expression of immunoreactive proteins of lower molecular mass, corresponding to the molecular mass of the active cleavage products of the zymogen (Fig. 5). In control experiments, addition of blocking anti-CD40L antibody during stimulation of the cells with native or recombinant ligand inhibited induction of stromelysin-3 expression.
Figure 5.

CD40L induces stromelysin-3 in human vascular smooth muscle cells in vitro. Human vascular SMC, cultured 24 h before the experiment in insulin/transferrin medium, were incubated (24 h) with membranes of 50 ng/ml PMA-stimulated (12 h) CD4+ T lymphocytes (T cell membranes, ratio equivalent to 1 SMC:10 T cells), 1 or 10 μg/ml recombinant human CD40 ligand (rCD40L), or 10 ng/ml IL-1β in the absence or presence of an anti-CD40L antibody (1 μg/ml α-CD40L). Concentrated (10×) supernatants were analyzed by Western blotting for stromelysin-3 protein expression. The positions of the molecular weight markers are indicated (kD). Data shown are representative of three experiments performed with SMC from different donors.
Figure 6.

CD40L induces stromelysin-3 in atheroma-associated cells in a concentration dependent fashion. Human vascular smooth muscle cells (SMC), endothelial cells (EC), monocyte–derived macrophages after 9 d in culture (MØ), and freshly isolated peripheral blood monocytes (PBMC), cultured 24 h before the experiment in the respective serum-free medium, were incubated (24 h) with the respective concentrations of recombinant human CD40 ligand (rCD40L). Concentrated (10×) supernatants were analyzed by Western blotting for stromelysin-3 protein expression. For specificity control, the primary antibody was blocked (1 h, 37°C) with recombinant stromelysin-3 (10 μg/ml) before applied to parallel samples of supernatants of 3 μg/ml rCD40L–stimulated cultures (blocking, right lanes). The positions of the molecular mass markers are indicated (kD). Data shown are representative of three to six experiments performed with cells from different donors.
Among the cell types analyzed, the immunoreactive forms of stromelysin-3 detected after CD40 ligation showed slight differences in the intensity of the respective bands but were identical in molecular mass. In vascular SMC, CD40 ligation concentration and time dependently elevated expression of the 64-kD protein, as well as induced immunoreactive proteins with molecular masses of ∼48 and ∼28 kD. The induction of the lower molecular mass forms of stromelysin-3 in cultures of SMC required stimulation with 3–10 μg/ml rCD40L (Fig. 6 A). An increase in the 64-kD protein, as well as in the ∼48-kD immunoreactive protein, occurred after 1 h of stimulation, whereas detection of the 28-kD band required at least 6–12 h of exposure to CD40L, as determined by Western blot analysis (data not shown) and radioimmunoprecipitation experiments (Fig. 7). The patterns of immunoreactive proteins detected in cultures of vascular smooth muscle cells resembled those found in fibroblasts, an established source of stromelysin-3 (data not shown). The pattern of immunoreactive bands observed with supernatants of CD40L-stimulated EC (Fig. 6 B) resembled that obtained with cultures of SMC, except that the 64-kD form was much less abundant in EC.
Figure 7.

CD40 ligation induces de novo stromelysin-3 expression in human vascular SMC. Human vascular SMC, cultured 24 h before the experiment in DMEM lacking methionine/cysteine, were incubated for the indicated times with 10 μg/ml rCD40L in the presence of 50 μCi/ml [35S]methionine/cysteine. Antigen was immunoprecipitated from concentrated (10×) culture supernatants with the specific antistromelysin-3 antibody and blots were dried and exposed to x-ray film for detection. The positions of the molecular mass markers are indicated (kD). Data shown are representative of three experiments performed with SMC obtained from different donors.
Macrophages, derived from monocytes cultured for 9 d, showed an additional immunoreactive protein at ∼35 kD. Stimulation with ≥0.3 μg/ml rCD40L increased levels of immunoreactive proteins with apparent molecular masses of 64, 48, 35, and 28 kD (Fig. 6 C). Induction of these immunoreactive bands required at least 6 h of stimulation (data not shown). Specificity of the antibody used was confirmed by Western blot analysis performed with the antistromelysin-3 antibody preincubated with recombinant stromelysin-3 (Fig. 6, blocking). In contrast to monocyte-derived MØ cultured for 9 d, freshly isolated peripheral blood monocytes expressed stromelysin-3 neither constitutively nor when stimulated with rCD40L (Fig. 6 D). Responsiveness of monocyte-derived cells to CD40 ligation required a minimum of three days of in vitro culture (data not shown).
The stromelysin-3 expression induced by CD40 ligation in human vascular EC, SMC, and MØ resulted from de novo synthesis of the protein. Metabolic labeling and immunoprecipitation experiments yielded autoradiographic bands resembling the patterns of immunoreactive proteins observed by Western blot analysis as shown here for vascular SMC (Fig. 7). In accordance with our protein analysis, RT-PCR experiments showed increased product corresponding to stromelysin-3 transcript after CD40 ligation in EC and SMC as well as MØ (data not shown).
Interruption of CD40–CD40L Signaling Diminished Stromelysin-3 Expression in Mouse Atherosclerotic Lesions.
The in situ observations of stromelysin-3 expression in human atherosclerotic plaques, in combination with the in vitro findings that CD40 ligation selectively mediates the expression of stromelysin-3, suggested an in vivo evaluation of the importance of CD40–CD40L signaling for the expression of this enzyme within atherosclerotic plaques. For this purpose, an established animal model of arteriosclerosis was used: LDLR−/− mice were fed a high cholesterol (1.25%) diet to develop atherosclerotic lesions (51). Immunohistochemical analysis of lesions within the aortic arch as well as the thoracic portion of the aorta revealed a stromelysin-3 expression pattern similar to that found in human atherosclerotic plaques. All three vascular cell types—EC, SMC and MØ—within the lesions stained for the proteinase. Treatment of the mice with rat IgG (n = 8) did not affect the stromelysin-3 expression (Fig. 8 A) compared to controls. In contrast, mice treated with the anti–mouse CD40L antibody (n = 8) showed substantially reduced immunoreactivity for stromelysin-3 within the atherosclerotic lesions (Fig. 8 B). Since the anti-CD40L antibody treatment also resulted in a decrease of total plaque number and area (51), lesions of similar sizes within the treatment groups were compared. No immunoreactivity was observed in tissues stained with the control IgG1 antibody (data not shown).
Figure 8.
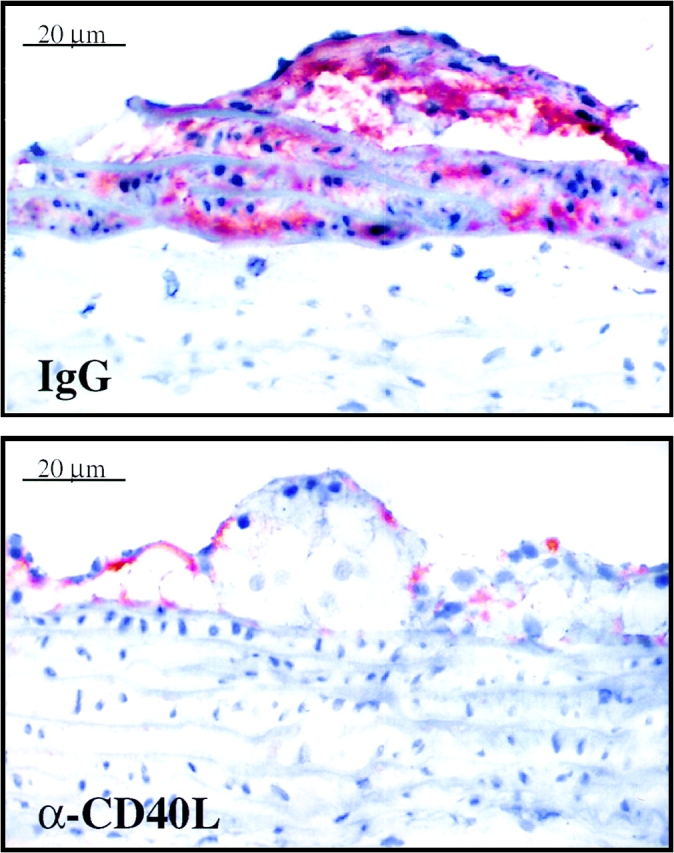
Interruption of CD40–CD40L signaling in hyperlipidemic LDLR−/− mice reduces stromelysin-3 expression in atherosclerotic plaques. High power views of frozen sections of aorta from LDLR−/− mice fed a high cholesterol diet and treated with rat IgG for control (IgG) or rat anti–murine CD40L antibody (α-CD40L) (both at 250 μg/ml, intraperitonally, twice per week) are shown. Plaques of approximately similar size were chosen for comparison. The lumen of the artery is at the top of each photomicrograph. Analysis of aortas obtained from eight different mice of each group showed similar results.
Discussion
This study establishes the expression of stromelysin-3 in advanced human atheroma and furthermore provides evidence for (a) colocalization of this matrix metalloproteinase with CD40 on lesional EC, SMC, and MØ in situ and (b) regulation of de novo expression of stromelysin-3 by CD40 ligation, rather than by the classical soluble mediators of MMPs such as IL-1, TNF-α, or IFN-γ. We also demonstrated in vivo that the interruption of CD40– CD40L interaction markedly reduced the expression of stromelysin-3 in mouse atheroma. The finding that EC, SMC, and MØ express stromelysin-3 establishes atheroma-associated cells as novel sources of stromelysin-3. Thus, this report provides evidence that stromelysin-3, whose expression correlates with the invasiveness of malignancies (3, 11, 12, 16, 17, 54), might also participate in the pathogenesis of another common human disease, atherosclerosis. In view of its unusual substrate specificity, stromelysin-3 might participate in several pathways. The pattern of stromelysin-3 expression within tissues that undergo extensive remodeling in pathological (carcinomas; 11, 12, 16, 17, 54) as well as physiological (placenta and uterus; 11, 18) events, indicates a role for stromelysin-3 in these processes. Although related by sequence to the MMP family, stromelysin-3 does not hydrolyze many of the extracellular matrix components that are substrates for other MMPs such as fibronectin, laminin, elastin, or collagen type I and type IV (23). From this perspective, the enzyme has been considered a nonmatrix–degrading metalloproteinase. Instead, stromelysin-3 appears to act as a predominant regulator of serpin function. Interestingly, a recent report postulated that extracellular matrix degradation might depend not only on an imbalance between matrix metalloproteinases and their inhibitors, but might accelerate in face of decreased levels of certain serpins such as α2-M, α2-AP, or α1-PI (33), preferred substrates of stromelysin-3 (23). Pathways by which this enzyme might regulate matrix degradation include (a) inactivation of matrix-degrading, enzyme-inhibiting serpins as described for cathepsins (32) or for elastolytic activity by α1-PI, shown to simultaneously augment the proliferative and invasive activity of cells (23); (b) activation of other members of the MMP family (17) copiously expressed in atheroma, such as interstitial collagenase, gelatinases A and B, and stromelysin-1 (5) (at sites of chronic inflammation, such as atherosclerosis, constitutively active stromelysin-3 [9] might act proximally to promote conversion of the zymogen forms of these classical MMPs to their active forms); and (c) degradation of matrix molecules not cleaved by the classical MMPs such as extracellular proteins containing amino acids with unusual long side chains, including those generated in vivo by certain posttranslational modifications (55). Thus, matrix degradation, a critical step in the progression from the stable atheroma to one prone to rupture and capable of causing thrombotic complications, might indeed involve the action of stromelysin-3. The serpin-degrading function of stromelysin-3 may thus have significance beyond tumor invasion and metastasis.
Furthermore, the action of stromelysin-3 on serpins also might affect atherosclerosis by pathways other than extracellular matrix remodeling. Serpins regulate multiple functions associated with the disease, including (a) blood pressure (e.g., the serpin α1–antitrypsin inhibits renin and thus renin–angiotensinogen interaction, as well as angiotensinogen itself [30]), (b) fibrinolysis (e.g., the serpin α1–antiplasmin targets plasmin, a key effector of fibrinolysis [23]), (c) blood coagulation (e.g., the serpins anti-thrombin III and heparin cofactor II rapidly interact with thrombin in the presence of heparin [31]), or (d) lipoprotein uptake (cleavage of the serpins α1-antitrypsin and α2-macroglobulin is associated with increased low density lipoprotein uptake into cells, indicating that those cleaved serpins disturb the intracellular cholesterol homeostasis [27–29]). Aside from these effects, stromelysin-3 might further affect atherosclerosis via the insulin-like growth factor–insulin-like growth factor binding protein (IGF–IGFBP) system. Human atheroma contain IGF-1 and IGFBPs (56). These mediators are associated with cardiovascular pathophysiology particularly via their critical role in vascular growth (57). IGF-1 directly accelerates arteriosclerosis in rat aorta allografts via myointimal proliferation and intimal thickening (58). The level of free, biologically active IGF-1 depends on the degree of complex formation with the respective binding proteins. A recent study identified IGFBP-1 as a potential physiological substrate for human stromelysin-3 (59). Finally, IGF also acts as a survival factor for human vascular SMC derived from normal vessels as well as coronary atherosclerotic plaques (60), suggesting stromelysin-3 may prevent apoptosis of vascular SMC by augmenting IGF-1 levels.
The potential relevance of serpin degradation for the pathogenesis of atherosclerosis in humans is supported by reports showing that levels of several serpins, including AT-III (36), α2-M, and α2-AP (35), decrease in this prevalent disease. In addition, recent in vivo studies demonstrated that serpins inhibit coronary restenosis in atherosclerotic swine (37) and atherosclerotic plaque development in rabbits post injury (38). Our findings that (a) differentiated atherosclerotic lesions but not their precursor, the fatty streak, bear stromelysin-3; (b) the immunoreactive bands detected within the atheromatous plaques resembled mostly the pattern obtained with MØ in vitro; and (c) only differentiated MØ, but not peripheral blood monocytes, exhibit stromelysin-3 induction; support a role of this enzyme in the late rather than the early states of atherosclerosis. Interestingly, the Rotterdam study showed increased AT-III levels in moderate peripheral arterial atherosclerosis and decreased levels in more severe atherosclerosis (61).
Future studies will have to establish whether these various functions apply to human atherosclerosis and whether stromelysin-3 is the crucial mediator in reduction of all or only certain atheroma-associated serpin activities. However, based on the recently burgeoning background information on stromelysin-3, our findings suggest functions of this proteinase in regulation of plaque progression and (in)stability by mechanisms distinct from those ascribed to members of the MMP family, including augmented extracellular matrix degradation, promotion of blood coagulation, decreased fibrinolytic activity, and dysregulation of blood pressure as well as lipoprotein catabolism. The colocalization of stromelysin-3 with CD40 and CD40L within the human atherosclerotic lesion, as well as the demonstration that CD40L, rather than the classical mediators of MMPs, induce stromelysin-3 expression in atheroma-associated cells demonstrate CD40–CD40L signaling as a new, and, as indicated by the in vivo studies, probably crucial pathway of stromelysin-3 induction.
Acknowledgments
We thank Maria Muszynski, Eugenia Shvartz, and Elissa Simon-Morrissey (Brigham and Women's Hospital) for their skillful technical assistance.
This work was supported in part by grants from the National Heart, Lung, and Blood Institute to Dr. Peter Libby (HL-43364) and the Swiss National Research Fund to Dr. François Mach and was performed during the tenure of the Paul Dudley White fellowship of the American Heart Association by Dr. Uwe Schönbeck.
Footnotes
Abbreviations used in this paper: α2-AP, α2-antiplasmin; α1-AT, α1-antitrypsin; α2-M, α2-macroglobulin; α1-PI, α1-proteinase inhibitor; AT-III, antithrombin III; EC, endothelial cells; FBS, fetal bovine serum; IGF, insulin-like growth factor; IGFBP, insulin-like growth factor binding protein; LDLR−/−, low density lipoprotein receptor deficient; MMP, matrix metalloproteinase; MØ, macrophages; PMA, Phorbol-12 myristate 13-acetate; RT, reverse transcription; SMC, smooth muscle cells.
References
- 1.Manuel D, Murray C, Risau W, Clauss M. Tumor necrosis: factors and principles. Immunol Today. 1996;17:254–256. doi: 10.1016/0167-5699(96)80539-1. [DOI] [PubMed] [Google Scholar]
- 2.Moscatelli D, Rifkin DB. Membrane and matrix localization of proteinases: A common theme in tumor cell invasion and angiogenesis. Biochim Biophys Acta. 1988;948:67–85. doi: 10.1016/0304-419x(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 3.Rouyer N, Wolf C, Chenard MP, Rio MC, Chambon P, Bellocq JP, Basset P. Stromelysin-3 gene expression in human cancer: an overview. Invasion Metastasis. 1995;14:269–275. [PubMed] [Google Scholar]
- 4.Henney AM, Wakeley PR, Davies MJ, Foster K, Hembry R, Murphy G, Humphries S. Localization of stromelysin gene expression in atherosclerotic plaques by in situhybridization. Proc Natl Acad Sci USA. 1991;88:8154–8158. doi: 10.1073/pnas.88.18.8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown DL, Hibbs MS, Kearney M, Loushin C, Isner JM. Identification of 92 kD gelatinase in human coronary atherosclerotic lesions. Circulation. 1995;91:2125–2131. doi: 10.1161/01.cir.91.8.2125. [DOI] [PubMed] [Google Scholar]
- 7.Lee, R.T., and P. Libby. 1996. Metalloproteinases and atherosclerotic plaque rupture. In The Role of Immune Mechanisms. H.-P. Schultheiss and P. Schwimmbeck, editors. Springer Verlag, New York. 21:238–246.
- 8.Matrisian LM. The matrix-degrading metalloproteinases. Bioessays. 1992;14:455–463. doi: 10.1002/bies.950140705. [DOI] [PubMed] [Google Scholar]
- 9.Pei D, Weiss SJ. Furin-dependent intracellular activation of the human stromelysin-3 zymogen. Nature. 1995;375:244–247. doi: 10.1038/375244a0. [DOI] [PubMed] [Google Scholar]
- 10.Santavicca M, Noel A, Angliker H, Stoll I, Segain J-P, Anglard P, Chretien M, Seidah N, Basset P. Characterization of structural determinants and molecular mechanisms involved in pro-stromelysin-3 activation by 4-aminophenylmercuric acetate and furin-type convertases. Biochem J. 1996;315:953–958. doi: 10.1042/bj3150953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basset P, Bellocq JP, Wolf C, Stoll I, Hutin P, Limacher JM, Podhajcer OL, Chenard MP, Rio MC, Chambon P. A novel metalloproteinase gene specifically expressed in stromal cells of breast carcinomas. Nature. 1990;348:699–704. doi: 10.1038/348699a0. [DOI] [PubMed] [Google Scholar]
- 12.Wagner SN, Ruhri C, Kunth K, Holecek BU, Hoefler H, Atkinson MJ. Expression of stromelysin-3 in the stromal elements of human basal cell carcinoma. Diag Mol Pathol. 1992;1:200–205. [PubMed] [Google Scholar]
- 13.Urbanski SJ, Edwards DR, Maitland A, Leco KJ, Watson A, Kossakowska AE. Expression of matrix metalloproteinase and their inhibitors in primary pulmonary carcinomas. Br J Cancer. 1992;66:1188–1194. doi: 10.1038/bjc.1992.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polette M, Clavel C, Birembaut P, De Clerck YA. Localization by in situ hybridization of mRNAs encoding stromelysin 3 and tissue inhibitors of metalloproteinases TIMP-1 and TIMP-2 in human head and neck carcinomas. Pathol Res Pract. 1993;189:1052–1057. doi: 10.1016/S0344-0338(11)80679-5. [DOI] [PubMed] [Google Scholar]
- 15.Hähnel E, Harvey JM, Joyce R, Robbins PD, Sterrett GF, Hähnel R. Stromelysin-3 expression in breast cancer biopsies: clinico-pathological correlation. Int J Cancer. 1993;55:1–4. doi: 10.1002/ijc.2910550513. [DOI] [PubMed] [Google Scholar]
- 16.Wolf C, Chenard MP, Durand De Grossouvre P, Bell JP, Chambon P, Basset P. Breast-cancer-associated stromelysin-3 gene is expressed in basal cell carcinoma and during cutaneous wound healing. J Investig Dermatol. 1992;99:870–879. doi: 10.1111/1523-1747.ep12614846. [DOI] [PubMed] [Google Scholar]
- 17.Nawrocki B, Polette M, Clavel C, Morrone A, Eschard JP, Etienne JC, Birembaut P. Expression of stromelysin 3 and tissue inhibitors of matrix metalloproteinases, Timp-1 and Timp-2, in rheumatoid arthritis. Pathol Res Pract. 1994;190:690–696. doi: 10.1016/S0344-0338(11)80748-X. [DOI] [PubMed] [Google Scholar]
- 18.Lefebvre O, Wolf C, Limacher JM, Hutin P, Wendling C, LeMeur M, Basset P, Rio MC. The breast cancer–associated stromelysin-3 gene is expressed during mouse mammary gland apoptosis. J Cell Biol. 1992;119:997–1002. doi: 10.1083/jcb.119.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodgers WH, Matrisian LM, Giudice LC, Dsupin B, Cannon P, Svitek C, Gorstein F, Osteen KG. Patterns of matrix metalloproteinase expression in cycling endometrium imply differential functions and regulation by steroid hormones. J Clin Invest. 1994;94:946–953. doi: 10.1172/JCI117461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lefebvre O, Regnier C, Chenard MP, Wendling C, Chambon P, Basset P, Rio MC. Developmental expression of mouse stromelysin-3 mRNA. Development. 1995;121:947–955. doi: 10.1242/dev.121.4.947. [DOI] [PubMed] [Google Scholar]
- 21.Noël A, Santavicca M, Stoll I, L'Hoir C, Staub A, Murphy G, Rio MC, Basset P. Identification of structural determinants controlling human and mouse stromelysin-3 proteolytic activities. J Biol Chem. 1995;270:22866–22872. doi: 10.1074/jbc.270.39.22866. [DOI] [PubMed] [Google Scholar]
- 22.Murphy G, Segain J-P, O'Shea M, Cockett M, Ioannou C, Lefebvre O, Chambon P, Basset P. The 28-kDa N-terminal domain of mouse stromelysin-3 has the general properties of a weak metalloproteinase. J Biol Chem. 1993;268:15435–15441. [PubMed] [Google Scholar]
- 23.Pei D, Majmudar G, Weiss SJ. Hydrolytic inactivation of a breast carcinoma cell-derived serpin by human stromelysin-3. J Biol Chem. 1994;269:25849–25855. [PubMed] [Google Scholar]
- 24.Potempa J, Korzus E, Travis J. The serpin superfamily of proteinase inhibitors: structure, function, and regulation. J Biol Chem. 1994;269:15957–15960. [PubMed] [Google Scholar]
- 25.Beatty K, Bieth J, Travis J. Kinetics of association of serine proteinases with native and oxidized alpha-1-proteinase inhibitor and alpha-1-antichymotrypsin. J Biol Chem. 1990;255:3931–3934. [PubMed] [Google Scholar]
- 26.Duranton J, Adam C, Bieth JG. Kinetic mechanism of the inhibition of cathepsin G by alpha1-antichymotrypsin and alpha1-proteinase inhibitor. Biochemistry. 1998;37:11239–11245. doi: 10.1021/bi980223q. [DOI] [PubMed] [Google Scholar]
- 27.Janciauskiene S, Al O, Rayyes, Floren C-H, Eriksson S. Low density lipoprotein catabolism is enhanced by the cleaved form of alpha-1-antitrypsin. Scand J Clin Lab Investig. 1997;57:325–336. doi: 10.3109/00365519709099406. [DOI] [PubMed] [Google Scholar]
- 28.Kasza A, Petersen HH, Heegaard CW, Oka K, Christensen A, Dubin A, Chan L, Andreasen PA. Specificity of serine proteinase/serpin complex binding to very-low-density lipoprotein receptor and alpha2-macroglobulin receptor/low density-lipoprotein-receptor-related protein. Eur J Biochem. 1997;248:270–281. doi: 10.1111/j.1432-1033.1997.00270.x. [DOI] [PubMed] [Google Scholar]
- 29.Janciauskiene S, Lindgren S, Wright HT. The C-terminal peptide of alpha-1-antitrypsin increases low density lipoprotein binding in HepG2 cells. Eur J Biochem. 1998;254:460–467. doi: 10.1046/j.1432-1327.1998.2540460.x. [DOI] [PubMed] [Google Scholar]
- 30.Scharpe S, Eid M, Cooreman W, Lauwers A. Alpha-1-Anti-trypsin, an inhibitor of renin. Biochem J. 1976;153:505–507. doi: 10.1042/bj1530505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pratt CW, Church FC. General features of the heparin-binding serpins antithrombin, heparin cofactor II, and protein C inhibitor. Blood. 1993;4:479–490. doi: 10.1097/00001721-199306000-00013. [DOI] [PubMed] [Google Scholar]
- 32.Schick C, Kamachi Y, Bartuski AJ, Cataltepe S, Schechter NM, Pemberton PA, Silverman GA. Squamous cell carcinoma-antigen 2 is a novel serpin that inhibits the chymotrypsin-like proteinases cathepsin G and mast cell chymase. J Biol Chem. 1997;272:1849–1855. doi: 10.1074/jbc.272.3.1849. [DOI] [PubMed] [Google Scholar]
- 33.Oleksyszyn J, Augustine AJ. Plasminogen modulation of IL1-stimulated degradation in bovine and human articular cartilage explants. The role of the endogenous inhibitors, PAI-1, alpha-2-antiplasmin, alpha-1-PI, alpha-2-macroglobulin and TIMP. Inflamm Res. 1996;45:464–472. doi: 10.1007/BF02252318. [DOI] [PubMed] [Google Scholar]
- 34.Hornebeck W, Robert L. Elastase-like enzymes in aortas and human breast carcinomas: quantitative variations with age and pathology. Adv Exp Med Biol. 1977;79:145–164. doi: 10.1007/978-1-4684-9093-0_14. [DOI] [PubMed] [Google Scholar]
- 35.Mohacsi A, Fulop T, Kozlovszky B, Hauck M, Kiss I, Leovey A. Sera and leukocyte elastase-type protease and antiprotease activity in healthy and atherosclerotic subjects of various ages. J Gerontol. 1992;47:154–158. doi: 10.1093/geronj/47.5.b154. [DOI] [PubMed] [Google Scholar]
- 36.O'Brien JR, Etherington MD, Jamieson S, Lawford P, Lincoln SV, Alkjaersig NJ. Blood changes in atherosclerosis and long after myocardial infarction and venous thrombosis. Thromb Diath Haemorrh. 1975;34:483–497. [PubMed] [Google Scholar]
- 37.Ali MN, Mazur W, Kleinman NS, Rodgers GP, Abukhalil JM, French BA, Raizner AE. Inhibition of coronary restenosis by antithrombin III in atherosclerotic swine. Coron Artery Dis. 1996;7:851–861. doi: 10.1097/00019501-199611000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Lucas A, Liu L, Macen J, Nash P, Dai E, Stewart M, Graham K, Etches W, Boshkov L, Nation PN, et al. Virus-encoded serine proteinase inhibitor SERP-1 inhibits atherosclerotic plaque development after balloon angioplasty. Circulation. 1996;94:2890–2900. doi: 10.1161/01.cir.94.11.2890. [DOI] [PubMed] [Google Scholar]
- 39.Okada A, Bellocq J-P, Rouyer N, Chenard M-P, Rio MC, Chambon P, Basset P. Membrane-type matrix metalloproteinase (MT-MMP) gene is expressed in stromal cells of human colon, breast, and head and neck carcinomas. Proc Natl Acad Sci USA. 1995;92:2730–2734. doi: 10.1073/pnas.92.7.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anglard P, Melot T, Guerin E, Thomas G, Basset P. Structure and promotor characterization of the human stromelysin-3 gene. J Biol Chem. 1995;270:20337–20344. doi: 10.1074/jbc.270.35.20337. [DOI] [PubMed] [Google Scholar]
- 41.Mach F, Schönbeck U, Sukhova GK, Bourcier T, Bonnefoy J-Y, Pober JS, Libby P. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci USA. 1997;94:1931–1937. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schönbeck U, Mach F, Sukhova GK, Murphy C, Bonnefoy J-Y, Fabumni RP, Libby P. Regulation of matrix-metalloproteinase expression in human vascular smooth muscle cells by T lymphocytes: a role for CD40 signaling in plaque rupture? . Circ Res. 1997;81:448–454. doi: 10.1161/01.res.81.3.448. [DOI] [PubMed] [Google Scholar]
- 43.Mach F, Schönbeck U, Bonnefoy J-Y, Pober JS, Libby P. Activation of monocyte/macrophage function related to acute atheromata complication by ligation of CD40: induction of collagenase, stromelysin, and tissue factor. Circulation. 1997;96:396–399. doi: 10.1161/01.cir.96.2.396. [DOI] [PubMed] [Google Scholar]
- 44.Mazzei GJ, Edgerton MD, Losberger C, Lecoanet-Henchoz S, Graber P, Durandy A, Gauchat J-F, Bernard A, Allet B, Bonnefoy J-Y. Recombinant soluble trimeric CD40 ligand is biologically active. J Biol Chem. 1995;270:7025–7028. doi: 10.1074/jbc.270.13.7025. [DOI] [PubMed] [Google Scholar]
- 45.Santavicca M, Noel A, Chenhard M-P, Lutz Y, Stoll I, Segain J-P, Rouyer N, Rio MC, Wolf C, Belloco J-P, Basset P. Characterization of monoclonal antibodies against stromelysin-3 and their use to evaluate stromelysin-3 levels in breast carcinoma by semi-quantitative immunohistochemistry. Int J Cancer. 1995;64:336–341. doi: 10.1002/ijc.2910640510. [DOI] [PubMed] [Google Scholar]
- 46.Kennedy MK, Picha KS, Fanslow WC, Grabstein KH, Alderson MR, Clifford KN, Chin WA, Mohler KM. CD40/CD40 ligand interactions are required for T cell-dependent production of interleukin-12 by mouse macrophages. Eur J Immunol. 1996;26:370–378. doi: 10.1002/eji.1830260216. [DOI] [PubMed] [Google Scholar]
- 47.Ross, R., and B. Kariya. 1980. Morphogenesis of vascular smooth muscle in atherosclerosis and cell structure. In Handbook of Physiology, The Cardiovascular System. D.F. Bohr, A.P. Somlyo, and H.Y. Sparks, editors. American Physiological Society, Bethesda, MD. 66–91.
- 48.Libby P, O'Brien KV. Culture of quiescent arterial smooth muscle cells in a defined serum-free medium. J Cell Physiol. 1983;115:217–223. doi: 10.1002/jcp.1041150217. [DOI] [PubMed] [Google Scholar]
- 49.Böyum A. Isolation of mononuclear cells and granulocytes from human blood. Scand J Clin Lab Investig. 1968;21:77–89. [PubMed] [Google Scholar]
- 50.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 51.Mach F, Schönbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in a mouse model by interruption of CD40-CD40L signaling. Nature. 1998;394:200–203. doi: 10.1038/28204. [DOI] [PubMed] [Google Scholar]
- 52.Guerin E, Ludwig M-G, Basset P, Anglard P. Stromelysin-3 induction and interstitial collagenase repression by retinoic acid. J Biol Chem. 1997;272:11088–11095. doi: 10.1074/jbc.272.17.11088. [DOI] [PubMed] [Google Scholar]
- 53.Mari BP, Anderson IC, Mari SE, Ning Y, Lutz Y, Kobzik L, Shipp MA. Stromelysin-3 is induced in tumor/stroma cocultures and inactivated via a tumor-specific and basic fibroblast growth factor-dependent mechanism. J Biol Chem. 1998;273:618–626. doi: 10.1074/jbc.273.1.618. [DOI] [PubMed] [Google Scholar]
- 54.Wolf C, Rouyer N, Lutz Y, Adida C, Loriot M, Bell JP, Chambon P, Basset P. Stromelysin 3 belongs to a subgroup of proteinases expressed in breast carcinoma fibroblastic cells and possibly implicated in tumor progression. Proc Natl Acad Sci USA. 1993;90:1843–1847. doi: 10.1073/pnas.90.5.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mucha A, Cuniasse P, Kannan R, Beau F, Yiotakis A, Basset P, Dive V. Membrane type-1 matrix metalloprotease and stromelysin-3 cleave more efficiently substrates containing unusual amino acids in their P1′ positions. J Biol Chem. 1998;273:2763–2768. doi: 10.1074/jbc.273.5.2763. [DOI] [PubMed] [Google Scholar]
- 56.Grant MB, Wargovich TJ, Ellis EA, Tarnuzzer R, Caballero S, Estes K, Rossing M, Spoerri PE, Pepine C. Expression of IGF-I, IGF-I receptor and IGF binding proteins-1, -2, -3, -4, and -5 in human atheroectomy specimens. Regul Pept. 1996;67:137–144. doi: 10.1016/s0167-0115(96)00124-3. [DOI] [PubMed] [Google Scholar]
- 57.Delafontaine P. Insulin-like growth factor I and its binding proteins in the cardiovascular system. Cardiovasc Res. 1995;30:825–834. [PubMed] [Google Scholar]
- 58.Motomura N, Lou H, Maurice P, Foegh ML. Acceleration of arteriosclerosis of the rat aorta allograft by insulin growth factor-I. Transplantation. 1997;63:932–936. doi: 10.1097/00007890-199704150-00004. [DOI] [PubMed] [Google Scholar]
- 59.Manes S, Mira E, Barbacid MM, Cipres A, Fernandez-Resa P, Buesa JM, Merida I, Aracil M, Marquez G, Martinez AC. Identification of insulin-like growth factor-binding protein-1 as a potential physiological substrate for human stromelysin-3. J Biol Chem. 1997;272:25706–25712. doi: 10.1074/jbc.272.41.25706. [DOI] [PubMed] [Google Scholar]
- 60.Bennett MR, Evan GI, Schwartz SM. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaque. J Clin Invest. 1995;95:2266–2274. doi: 10.1172/JCI117917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van der Bom JG, Bots ML, van Vliet HH, Pols HA, Hofman A, Grobbee DE. Antithrombin and atherosclerosis in the Rotterdam Study. Arterioscler Thromb Vasc Biol. 1996;16:864–867. doi: 10.1161/01.atv.16.7.864. [DOI] [PubMed] [Google Scholar]