

Abstract
We have shown previously that the inactivation of macrophages in nonobese diabetic (NOD) mice results in the prevention of diabetes; however, the mechanisms involved remain unknown. In this study, we found that T cells in a macrophage-depleted environment lost their ability to differentiate into β cell–cytotoxic T cells, resulting in the prevention of autoimmune diabetes, but these T cells regained their β cell–cytotoxic potential when returned to a macrophage-containing environment. To learn why T cells in a macrophage-depleted environment lose their ability to kill β cells, we examined the islet antigen–specific immune response and T cell activation in macrophage-depleted NOD mice. There was a shift in the immune balance, a decrease in the T helper cell type 1 (Th1) immune response, and an increase in the Th2 immune response, due to the reduced expression of the macrophage-derived cytokine IL-12. As well, there was a deficit in T cell activation, evidenced by significant decreases in the expression of Fas ligand and perforin. The administration of IL-12 substantially reversed the prevention of diabetes in NOD mice conferred by macrophage depletion. We conclude that macrophages play an essential role in the development and activation of β cell–cytotoxic T cells that cause β cell destruction, resulting in autoimmune diabetes in NOD mice.
Keywords: macrophages, T cells, T cell activation, nonobese diabetic mice, autoimmune diabetes
Insulin-dependent diabetes mellitus, also known as type I diabetes, is a serious chronic childhood disorder caused by the progressive loss of insulin-producing pancreatic β cells, culminating in a state of hypoinsulinemia and hyperglycemia. Animal models used in the study of type I diabetes, such as the BioBreeding (BB)1 rat and the nonobese diabetic (NOD) mouse, have enhanced our understanding of the pathogenic mechanisms of this disease. The diabetic syndrome in these animals results from the destruction of pancreatic β cells by cell-mediated immune responses. Many studies have demonstrated the role of CD4+ and CD8+ T cells in the development of autoimmune type I diabetes in NOD mice (1–12). However, the role of macrophages in the pathogenesis of autoimmune diabetes in these animals has not been well investigated. We and others have shown that the major populations of cells infiltrating the islets during the early stage of insulitis in BB rats and NOD mice are macrophages and dendritic cells (13–17). This infiltration precedes invasion of the islets by T lymphocytes, NK cells, and B lymphocytes (18). In addition, electron microscopy has revealed that most of the single cells present at an early stage of insulitis in BB rats are macrophages (13). Intraperitoneal injections of silica, a substance known to be toxic to macrophages, into cyclophosphamide (CY)-treated NOD mice or BB rats almost completely prevents the development of diabetes and insulitis (19–21). This result suggests that macrophages play an important role in the development of insulitis and diabetes in NOD mice. However, the mechanisms involved in the prevention of β cell destruction by the inactivation of macrophages in NOD mice remain unknown.
The purpose of this study is to determine the mechanisms involved in the prevention of T cell–mediated autoimmune diabetes in macrophage-depleted NOD mice. We report that T cells in a macrophage-depleted environment lose their ability to differentiate into β cell–cytotoxic T cells that can kill β cells. However, if these T cells are returned to an environment containing macrophages and/or the macrophage-derived cytokine IL-12 for a sufficient period of time, they regain the ability to differentiate into β cell–cytotoxic cells. We conclude that macrophages are primary contributors to the creation of the immune environment for the development and activation of β cell–specific Th1-type CD4+ T cells and CD8+ cytotoxic T cells that cause autoimmune diabetes in NOD mice.
Materials and Methods
Animals.
NOD mice were originally obtained from Taconic Farms; NOD.scid mice were obtained from The Jackson Laboratory. The animals were bred and maintained under specific pathogen-free conditions and provided with sterile food and water ad libitum at the Animal Resource Centre, Faculty of Medicine, University of Calgary. Female NOD mice were used throughout the experiments, with the exception of the male animals used for the preparation of islets for transplantation. The use and care of the animals used in this study were approved by the Animal Care Committee, Faculty of Medicine, University of Calgary.
Preparation and Administration of Liposomal Dichloromethylene Diphosphonate.
Phosphatidyl choline (86 mg; Lipoid GmbH, Ludwigshafen, Germany) and cholesterol (8 mg; Sigma Chemical Co., St. Louis, MO) were dissolved in chloroform as described previously (22). This solution was evaporated by rotation in a vacuum at 37°C and dispersed by mixing with 10 ml of PBS containing 2.5 g of Cl2MDP (a gift from Boehringer Mannheim GmBH). This mixture was sonicated for 3 min in a water bath sonicator at room temperature and then set aside for 2 h at room temperature. The resulting liposomes were centrifuged at 100,000 g for 30 min to remove the nonencapsulated Cl2MDP. The pellet was washed two to three times with PBS by centrifugation at 25,000 g for 30 min. The final liposomal dichloromethylene diphosphonate (lip-Cl2MDP) was suspended in 4 ml of PBS before use (22, 23).
Treatment of NOD Mice with lip-Cl2MDP.
Female NOD mice were treated with lip-Cl2MDP (200 μl/mouse) by intraperitoneal administration once a week from 3 to 20 wk of age to deplete macrophages. As a control group, NOD mice were treated with PBS (200 μl/mouse). The incidence of diabetes was determined by measurement of urine glucose using Diastix (Miles, Etobicoke, ON, Canada) twice a week from 10 to 35 wk of age. Individual mice were classified as diabetic on the basis of positive glycosuria (>2) and hyperglycemia (blood glucose >16.7 mM) as described elsewhere (24–26).
Flow Cytometric Analysis.
Splenocytes were isolated from lip-Cl2MDP– or PBS-treated NOD mice (2–3 mice/group) at 15 wk of age, as described previously (24). Splenocytes (1 × 106 cells) were incubated with Abs against B220, CD4, and CD8 (PharMingen) for 30 min at 4°C in staining buffer composed of PBS containing 1% FCS and 0.1% sodium azide. After washing, the cells were incubated with FITC-labeled secondary Ab (anti– rat IgG) for 30 min at 4°C. For Mac1+ and CD11c+ cell staining, FITC-labeled anti-Mac1 and PE-labeled anti-CD11c Ab were used. The cells were then washed twice with staining buffer and analyzed using a FACS®.
For analysis of Fas ligand (FasL) and perforin expression, splenic T cells (106 cells), purified using a T cell column (R&D Systems) were stimulated with irradiated islets (40 islets) for 3 d in complete RPMI 1640 medium, containing 10% FCS, 10 mM Hepes buffer, 1 mM sodium pyruvate, 2 mM l-glutamine, 100 U/penicillin, 0.1 mg/ml streptomycin, and 0.05 mM β-mercaptoethanol. Live cells were collected on a Ficoll gradient and washed twice. For FasL staining, T cells were incubated with biotinylated anti-FasL Abs (PharMingen) followed by incubation with PE-conjugated streptavidin. For perforin staining, CD8+ T cells were fixed with 4% paraformaldehyde, rendered permeable using 1% saponin in PBS containing 1% FCS and 0.1% sodium azide, and incubated with anti-perforin Abs (Kamiya Biomedical Co.), followed by incubation with FITC-labeled anti–rat IgG.
In Vitro T Cell Proliferation Assay.
Single cell splenocytes were prepared from 8–10-wk-old female NOD mice, and T cells were purified using a T cell column (R&D Systems). The T cells (5 × 105 cells) were cultured with irradiated splenocytes (5 × 105 cells) from PBS- or lip-Cl2MDP–treated NOD mice, as APCs, in the absence or presence of irradiated islets (4 × 104 cells) or glutamic acid carboxylase (GAD; 20 μg/ml) in 200 μl of complete RPMI medium in a 96-well microplate for 96 h. The cells were pulsed with [3H]thymidine (1 μCi/well) for the last 16–18 h of the incubation and then harvested. The incorporation of [3H]thymidine was measured by liquid scintillation counting.
Adoptive Transfer of Splenocytes into NOD.scid Mice.
Splenocytes were isolated from lip-Cl2MDP– and PBS-treated, nondiabetic female NOD mice at 20 wk of age, as previously described (24). The splenocytes from each donor were injected intravenously (2 × 107 cells/recipient) into 6–8-wk-old NOD.scid mice. The development of diabetes was determined by measurement of urine and blood glucose from day 3 to day 90 after the transfer of splenocytes, as described above. As a positive control, splenocytes from acutely diabetic mice were injected into age-matched NOD.scid mice. For long-term observations, recipient mice were kept up to 20 wk after the transfer of splenocytes and the incidence of diabetes was determined by the measurement of urine and blood glucose.
To determine whether long-term treatment with lip-Cl2MDP affects established β cell–cytotoxic T cells, 16-wk-old female nondiabetic NOD mice were treated intraperitoneally with lip-Cl2MDP (200 μl/mouse per week) or PBS, as a control, once a week up to wk 25 of age, and splenic T cells from nondiabetic NOD mice were isolated and purified using a T cell column (R&D Systems) 1 d after the last treatment (∼65% of the lip-Cl2MDP–treated NOD mice had become diabetic by the age of 25 wk). T cells were injected intravenously (107 cells/mouse) into 6–8-wk-old NOD.scid mice. In a parallel experiment, female NOD mice were treated with PBS or lip-Cl2MDP weekly from 19 to 21 wk of age. Splenic T cells (107 cells/mouse) were transfused into NOD.scid mice as described above. The development of diabetes was monitored by 10 wk after the transfer of T cells as described above, and the incidence of diabetes was compared between the two experimental groups.
Transplantation of NOD Mouse Islets into lip-Cl2MDP–treated NOD Mice.
Intact pancreatic islets were isolated from 4-wk-old male NOD mice as previously described (27). To determine whether cytotoxic T cells that can destroy β cells are generated in the lip-Cl2MDP–treated NOD mice, 400 syngeneic islets were transplanted under the renal capsule of each lip-Cl2MDP– and PBS-treated control animal (5 mice/group) at 20 wk of age, as previously described (27). The recipient mice were killed 3 wk after transplantation, and serial sections of the kidney bearing the transplanted islets were examined after hematoxylin and eosin staining (27) to check for lymphocytic infiltration.
Reverse Transcriptase PCR Analysis of Cytokine Gene Expression.
Total RNA was extracted from the total splenocytes or purified splenic T cells of lip-Cl2MDP– and PBS-treated NOD mice with a T cell column (R&D Systems) using Trizol (GIBCO BRL) according to the manufacturer's protocol. For analysis of mRNA of FasL and perforin, purified T cells were activated with irradiated NOD islets for 72 h before the extraction of RNA. 3 μg of total RNA was used to synthesize cDNA using Superscript II reverse transcriptase (GIBCO BRL) and oligo(dT)12–18. PCR was performed using specific primers for various cytokine genes, as previously described (28). The upstream and downstream primers used were as follows: IL-2: sense, CTTGCCCAAGCAGGCCACAG, antisense, GAGCCTTATGTGTTGTAAGC; IFN-γ: sense, AGCTCTGAGACAATGAACGC, antisense, GGACAATCTCTTCCCCACCC; IL-4: sense, TCTTTCTCGAATGTACCAGG, antisense, CATGGTGGCTCAGTACTACG; IL-10: sense, CAAACAAAGGACCAGCTGGAC, antisense, GAGTCCAGCAGACTCAATAC; TNF-α: sense, CCTGTAGCCCACGTCGTAGC, antisense, TTGACCTCAGCGCTGAGTTG; IL-1β: sense, GAATGACCTGTTCTTTGAAGTT, antisense, TTTTGTTGTTCATCTCGGAGCC; IL-12: sense, AGAGGTGGACTGGACTCCCGA, antisense, TTTGGTGCTTCACACTTCAGC; IL-12Rβ1: sense, GTACTCCACGACACGTGTCGG, antisense, GTCCGCTCGCACCTGAACCTT; IL-12Rβ2: sense, AGTGGACCAAACAATCTGAC, antisense, ATCTTCCCACTGCAGTGTAC; perforin: sense, AACTTCCTAATGAGAGACGCC, antisense, AAGTCAAGGTGGAGTGGAGG; FasL: sense, CAGCTCTTCCACCTGCAG, antisense, TTAAAGCTTATACAAGCC. Hypoxanthine phosphoribosyl transferase (HPRT) was used as an internal standard. The primers used for HPRT were: sense, GTAATGATCAGTCAACGGGGGAC, antisense, CCAGCAAGCTTGCAACCTTAACCA. The PCR condition was optimized for each set of primers. PCR was performed using different numbers of cycles to ensure that amplification occurred in a linear range. The PCR mixture (50 μl) contained 0.2 mM of each deoxynucleotide triphosphate, 1 μM of each specific primer, 1.5 mM or 2 mM MgCl2, 50 mM KCl, 10 mM Tris-Cl, pH 9.0, and 2.5 U of Taq polymerase (Pharmacia Biotech). After amplification, the products were subjected to electrophoresis on a 1.5% agarose gel and detected by ethidium bromide staining.
Quantitative ELISA of Cytokine Production.
Splenic T cells (106 cells) were prepared from 15-wk-old lip-Cl2MDP– or PBS-treated mice (3 mice/group). The cells were washed with serum-free RPMI 1640 medium and incubated for 48 h in anti-CD3 Ab (10 μg/ml)-coated wells or for 72 h with irradiated NOD islets in complete RPMI 1640 medium. The supernatant was collected and cytokine release was measured using a Quantikine ELISA kit (R&D Systems) according to the manufacturer's protocol (28).
Administration of rIL-12.
Recombinant mouse IL-12 (R&D Systems) (0.4 μg/200 μl of PBS, containing 1% syngeneic NOD mouse serum, per mouse for 1 wk followed by 0.2 μg/200 μl of PBS per mouse for 3 wk) was administered intraperitoneally each day commencing at 9 wk of age, to NOD mice which had been treated with lip-Cl2MDP or PBS from 3 wk of age. Control groups of lip-Cl2MDP or PBS treated NOD mice received 200 μl of PBS, containing 1% syngeneic mouse serum, in place of IL-12 each day. The development of diabetes was monitored by measuring glycosuria (>2) every second day (24) and confirmed by the measurement of blood glucose (>16.7 mM) (25, 26).
Histology.
The pancreata were removed from lip-Cl2MDP– and PBS-treated NOD mice killed at 25 wk of age. Pieces of each pancreas were fixed with 10% buffered formalin, embedded in paraffin, sectioned at 4.5 μM, and stained with hematoxylin and eosin.
Statistical Analysis.
The statistical significance of differences between groups was analyzed by Student's t test. A level of P < 0.05 was accepted as significant. Data are expressed as means ± SD.
Results
Macrophages Are Required for the Development of β Cell– cytotoxic T Cells in NOD Mice.
Previously, we found that the depletion of macrophages by treatment with silica prevents the development of diabetes in CY-treated NOD mice (19). First, to confirm that the depletion of macrophages prevents the development of diabetes and to determine whether lip-Cl2MDP has the same effect as silica on the prevention of diabetes in NOD mice, we treated NOD mice from 3 to 20 wk of age with lip-Cl2MDP, which specifically depletes macrophages (22, 29, 30). We then examined the cumulative incidence of diabetes by 35 wk of age. We found that none of the lip-Cl2MDP–treated NOD mice developed diabetes. In contrast, 80% (8 out of 10) of the PBS-treated control NOD mice developed diabetes by the same age (Fig. 1). We also examined the development of insulitis in the lip-Cl2MDP– and PBS-treated NOD mice at 25 wk of age. 91% of the examined islets from the lip-Cl2MDP–treated mice were intact (Table I, Fig. 2 A). In contrast, 88% of the examined islets from the PBS-treated control mice showed moderate to severe insulitis (Table I, Fig. 2 B). These results indicate that macrophages are an absolute requirement for the development of autoimmune diabetes in NOD mice.
Figure 1.

Depletion of macrophages in NOD mice by treatment with lip-Cl2MDP results in the prevention of diabetes. 12 3-wk-old female NOD mice were treated weekly with 200 μl of lip-Cl2MDP (•) until 20 wk of age; 10 female NOD mice were treated similarly with PBS (○). The cumulative incidence of diabetes was measured.
Table I.
Depletion of Macrophages Results in the Prevention of Insulitis in NOD Mice
200 μl of lip-Cl2MDP per mouse was injected intraperitoneally into female NOD mice weekly from 3 to 20 wk of age.
At 25 wk of age, five randomly selected nondiabetic mice were killed and at least 20 islets per mouse were examined. Grade: 0, normal islets; 1, mononuclear infiltration, largely in the periphery, in <25% of the islets; 2, 25–50% of islets infiltrated; 3, >50% of islets infiltrated; 4, small, retracted islets with few mononuclear cells.
Figure 2.
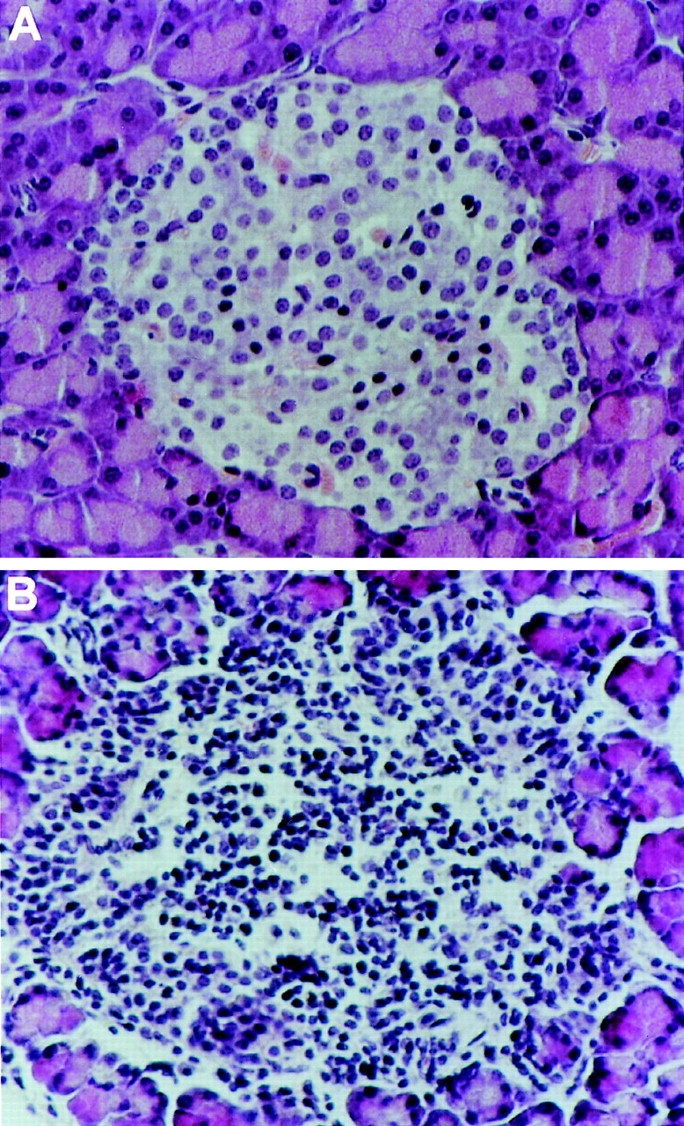
Photomicrographs of the pancreatic islets of NOD mice. Pancreatic sections from 25-wk-old female NOD mice treated with lip-Cl2MDP (A) or PBS (B) were stained with hematoxylin and eosin. Islets from the lip-Cl2MDP–treated mice are intact (A). In contrast, islets from the PBS-treated animals show severe insulitis (B). Original magnification: ×250.
It is known that the development of autoimmune diabetes in NOD mice is T cell mediated. To determine whether macrophages are required for the development of β cell–cytotoxic T cells, we transfused splenocytes from either lip-Cl2MDP– or PBS-treated control NOD mice into 6–8-wk-old NOD.scid mice. None (0 out of 6) of the recipients of lip-Cl2MDP–treated NOD splenocytes developed diabetes, whereas 83% (5 out of 6) of the recipients of splenocytes from PBS-treated control NOD mice became diabetic within 8 wk of transfusion. Similarly, 100% (7 out of 7) of the recipients of splenocytes from newly diabetic NOD mice became diabetic by 9 wk after transfer of the splenocytes (Fig. 3).
Figure 3.

Depletion of macrophages in NOD mice results in the failure of adoptive transfer of diabetes to NOD.scid mice by splenocytes. NOD.scid mice (6–8-wk-old) received splenocytes (2 × 107 cells) isolated from 20-wk-old lip-Cl2MDP–treated (n = 6, •) or PBS-treated (n = 6, ○) NOD mice or from newly diabetic (n = 7, ▴) NOD mice.
To determine whether T lymphocytes in lip-Cl2MDP– treated NOD mice can destroy pancreatic β cells, we transplanted intact, syngeneic NOD mouse pancreatic islets into the renal capsule of lip-Cl2MDP–treated NOD mice (20 wk of age). A majority of the grafted islets remained intact >3 wk after transplantation (Table II, Fig. 4 A). In contrast, most of the syngeneic islets transplanted into age-matched, PBS-treated NOD mice were destroyed within 3 wk of transplantation (Table II, Fig. 4 B). These results indicate that macrophages are required for the development of cytotoxic T cells that can cause β cell destruction in NOD mice.
Table II.
Prevention of Insulitis in Islets Grafted into lip-Cl2MDP–treated NOD Mice
| Islet recipient* | Insulitis grade in islet grafts‡ | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Strain | Treatment | 0 | 1 | 2 | 3 | 4 | ||||||
| % | ||||||||||||
| NOD | PBS | 0 | 2 | 12 | 38 | 48 | ||||||
| lip-Cl2MDP | 85 | 11 | 3 | 1 | 0 | |||||||
Pancreatic islets were prepared from 4-wk-old male NOD mice and transplanted under the renal capsule of NOD mice at 20 wk of age that had been treated with lip-Cl2MDP from 3 to 20 wk of age (5 mice/ group). Insulitis was examined 3 wk after transplantation.
Insulitis in the islet grafts was graded as in Table I.
Figure 4.
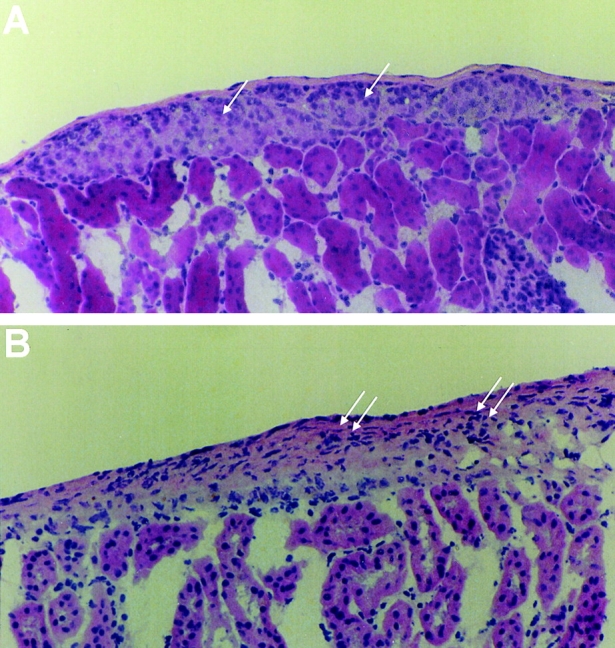
Histology of islet grafts in the renal capsule of lip-Cl2MDP– and PBS-treated NOD recipients. Syngeneic islets were transplanted under the renal capsule of lip-Cl2MDP– and PBS-treated NOD mice at 20 wk of age and the animals were killed 3 wk later. Sections of the kidney bearing the islet grafts were stained with hematoxylin and eosin. (A) Section from lip-Cl2MDP–treated NOD mouse recipient shows intact islet graft (single arrow). (B) Section from PBS-treated mouse recipient shows necrosis and massive infiltration by lymphocytes (double arrows). Original magnification: ×200.
Splenic T Cells from Macrophage-depleted NOD Mice Are Able to Recover Their Ability to Differentiate into β Cell–cytotoxic T Cells in the Presence of Macrophages.
To determine whether splenic T cells from macrophage-depleted NOD mice are able to recover their ability to differentiate into β cell–cytotoxic T cells in the presence of macrophages, we transfused splenocytes isolated from macrophage-depleted NOD mice into NOD.scid mice and made observations for a prolonged period of time (13–20 wk). None of the recipients developed diabetes earlier than 13 wk after splenocyte transfer; however 71% (5 out of 7) developed diabetes between 14 and 17 wk after the transfusion (Fig. 5). In contrast, 86% (6 out of 7) of the recipients of splenocytes from PBS-treated NOD mice became diabetic within 8 wk of transfusion and 100% (5 out of 5) of the recipients of splenocytes from newly diabetic NOD mice became diabetic within 7 wk of the transfusion (Fig. 5). This suggests that prolonged exposure to the endogenous macrophages in NOD.scid mice can provide the necessary immune environment required to differentiate the splenic T cells from macrophage-depleted mice into β cell–cytotoxic T cells.
Figure 5.

Splenic T cells from lip-Cl2MDP–treated mice can recover the capability to differentiate into cytotoxic effector T cells in the presence of macrophages. NOD.scid mice (7 wk old) received splenocytes (2 × 107 cells) isolated from 20-wk-old lip-Cl2MDP–treated (n = 7, •) or PBS-treated (n = 7, ○) NOD mice or from newly diabetic NOD mice (n = 5, ▵).
Long-Term Treatment with lip-Cl2MDP Does Not Alter the Function of Established β Cell–cytotoxic T Cells in NOD Mice.
To determine whether treatment with lip-Cl2MDP alters the function of established β cell–cytotoxic T cells in NOD mice, we treated NOD mice with lip-Cl2MDP or PBS, as a control, weekly for a short period (3 wk) and a longer period (9 wk) at the age of ∼20 wk (19–21 and 16–25 wk of age, respectively). These particular age ranges were chosen because β cell–cytotoxic T cells have developed in NOD mice by around these ages. We isolated splenic T cells from short-term or long-term PBS- or lip-Cl2MDP–treated NOD mice, transfused them into 6–8-wk-old NOD.scid mice, and examined the incidence of diabetes in the NOD.scid mice. We found no difference in the incidence of diabetes between the NOD.scid recipients of T cells from PBS-treated NOD mice and the recipients of T cells from lip-Cl2MDP–treated NOD mice (Table III). In addition, no statistically significant difference was found in the incidence of diabetes between NOD.scid recipients of T cells from short-term lip-Cl2MDP–treated (3 wk) and long-term lip-Cl2MDP–treated (9 wk) NOD mice. This result indicates that neither short- nor long-term treatment with lip-Cl2MDP alters the function of established β cell– cytotoxic T cells in NOD mice, since these T cells are able to induce diabetes.
Table III.
Long-Term Treatment with lip-Cl2MDP Does Not Alter the Function of Established β Cell–cytotoxic T Cells in NOD Mice
| Treatment of donors (NOD mice) | Incidence of diabetes in recipients (NOD.scid mice)* | |||
|---|---|---|---|---|
| Treatment | Duration | |||
| wk | ||||
| PBS | 3 | 80% (4/5) | ||
| 9 | 83% (5/6) | |||
| lip-Cl2MDP | 3 | 75% (3/4) | ||
| 9 | 86% (6/7) | |||
Female NOD mice were treated with PBS or lip-Cl2MDP once a week from 19 to 21 wk of age (3-wk duration of treatment) or from 16 to 25 wk (9-wk duration of treatment). Splenic T cells (107 T cells/ mouse) were transfused intravenously into 6–8-wk-old NOD.scid mice.
Cumulative incidence of diabetes by 10 wk after transfer of T cells. Parentheses indicate number of animals which developed diabetes over total number of animals tested.
Depletion of Macrophages Results in a Decrease in the Th1 Immune Response and an Increase in the Th2 Immune Response in NOD Mice.
As Th1 development may be mediated by macrophage-derived cytokines, particularly IL-12, we determined whether the depletion of macrophages would result in a change in the Th1 and Th2 immune responses by examining the expression of cytokine mRNAs (IL-2 and IFN-γ for the Th1 response and IL-4 and IL-10 for the Th2 response) in the splenic T cells from lip-Cl2MDP– or PBS-treated NOD mice. The expression of both IL-2 and IFN-γ mRNA was decreased in splenic T cells from lip-Cl2MDP–treated NOD mice as compared with that in PBS-treated control NOD mice. In contrast, the expression of both IL-4 and IL-10 mRNA was increased in the splenic T cells from lip-Cl2MDP–treated NOD mice as compared with the expression in PBS-treated controls (Fig. 6 A).
Figure 6.

Depletion of macrophages results in a decrease in the Th1 immune response and an increase in the Th2 immune response in NOD mice. Splenic T cells were isolated from three mice (15 wk of age) from each group. Reverse transcriptase PCR analysis for cytokine gene expression was performed using primers specific for IL-2, IFN-γ, IL-4, and IL-10 (A) and IL-12Rβ1 and IL-12Rβ2 (B). Primers for HPRT were used as standards. M, 100-bp ladder; 1, amplified product from splenic T cells of PBS-treated NOD mice; 2, amplified product from splenic T cells of lip-Cl2MDP– treated NOD mice. Representative data for three independent experiments are shown. For the measurement of cytokine secretion from splenic T cell cultures, splenic T cells were isolated from lip-Cl2MDP– and PBS-treated NOD mice (n = 3 per group, 15 wk of age), and activated with anti-CD3 Ab (10 μg/ml) for 2 d (C) or with irradiated NOD islets for 3 d (D). The production of cytokine levels was determined by ELISA. Values are shown as mean ± SD of three independent experiments. *P < 0.05 compared with PBS-treated control group.
To ascertain the islet antigen-specific cytokine secretion, we activated splenic T cells isolated from lip-Cl2MDP– or PBS-treated NOD mice using anti-CD3 Ab or NOD mouse pancreatic islets, and measured the IFN-γ and IL-4 cytokine secretion. A low level of IFN-γ secretion, but a high level of IL-4 secretion, was found in lip-Cl2MDP– treated NOD mice as compared with the levels in PBS-treated control NOD mice when the splenic T cells were activated with anti-CD3 Ab (Fig. 6 C) or NOD mouse pancreatic islets (Fig. 6 D). These results indicate that the inactivation of macrophages in NOD mice results in a decrease in the Th1 immune response along with an increase in a Th2-type immune response after both antigen-specific and nonspecific stimulation.
It has been found that the IL-12Rβ2 subunit, but not the IL-12Rβ1 subunit, is preferentially expressed in Th1-type CD4+ T cells (31). To determine whether there is any change in the expression of the IL-12Rβ2 subunit in splenic T cells from lip-Cl2MDP–treated NOD mice, we examined the expression of the IL-12R in splenic T cells from these mice and PBS-treated control NOD mice. The expression of the IL-12Rβ2 subunit was significantly decreased in lip-Cl2MDP–treated NOD mice as compared with that in the PBS-treated control animals (Fig. 6 B). This result supports the finding that the depletion of macrophages results in a decrease in the Th1 immune response in NOD mice.
Depletion of Macrophages Does Not Alter the Lymphocyte Subset Population.
To determine whether there is any change in the number of immunocytes in the spleens from lip-Cl2MDP–treated NOD mice, FACS® analysis of immunocytes was carried out. Mac1+ cells were clearly decreased in lip-Cl2MDP–treated NOD mice. There was no significant difference in the number of B220-positive B cells or CD4+ T cells between lip-Cl2MDP– and PBS-treated NOD mice. The number of CD8+ T cells appears to have declined in the lip-Cl2MDP–treated NOD mice as compared with the PBS-treated control mice (15% in PBS-treated control mice versus 11% in lip-Cl2MDP–treated mice). However, there was no statistically significant difference (P > 0.05) between the macrophage-depleted and control animals (Table IV). Similarly, CD11c+ cells, a marker for dendritic cells, appear to have declined in lip-Cl2MDP–treated NOD mice (2.7%) as compared with PBS-treated control mice (3.6%). However, there was no statistically significant difference (P = 0.067) between the lip-Cl2MDP– and PBS-treated groups (Table IV).
Table IV.
Treatment with lip-Cl2MDP Does Not Alter T and B Lymphocyte Populations in NOD Mice
| Antibody against* | Treatment | |||
|---|---|---|---|---|
| PBS‡ | lip-Cl2MDP‡ | |||
| % | ||||
| Mac1 | 8.3 ± 1.2 | 1.9 ± 0.3§ | ||
| CD4 | 35.4 ± 7.0 | 31.1 ± 4.6 | ||
| CD8 | 14.5 ± 2.6 | 11.4 ± 2.2‖ | ||
| B220 | 36.7 ± 4.0 | 38.2 ± 5.2 | ||
| CD11c | 3.6 ± 0.4 | 2.7 ± 0.3‖ | ||
Splenocytes were isolated from PBS- and lip-Cl2MDP–treated NOD mice at 15 wk of age, stained with the designated antibody, and analyzed by FACScan®.
Data shown represent mean ± SD from three different experiments.
P < 0.05 compared with PBS-treated control mice.
P > 0.05 compared with PBS-treated control mice.
Treatment of NOD Mice with lip-Cl2MDP Results in a Decrease in the Gene Expression of the Macrophage-derived Cytokines.
To determine whether a shift in the immune response of macrophage-depleted mice, specifically a decrease in the Th1 immune response and an increase in the Th2 immune response, is due to changes in the macrophage-derived cytokine IL-12, we examined the expression of IL-12 in splenic macrophages from lip-Cl2MDP– and PBS-treated NOD mice using reverse transcriptase PCR analysis. We found that the gene expression of IL-12 was significantly decreased in the lip-Cl2MDP–treated NOD mice as compared with that in the PBS-treated controls (Fig. 7). In addition, the expression of other macrophage-derived cytokines, IL-1β and TNF-α, also were decreased in the lip-Cl2MDP–treated NOD mice (Fig. 7). This result suggests that a shift from a Th1 immune response to Th2 immune response may be due to the decreased expression of macrophage-derived cytokines, particularly IL-12.
Figure 7.

Treatment of NOD mice with lip-Cl2MDP results in a decrease in the gene expression of macrophage-derived cytokines. Splenocytes were pooled from three mice (15 wk of age) in each group. RNA was extracted from the splenocytes and reverse transcriptase PCR analysis for cytokine gene expression was performed using primers specific for IL-1β, TNF-α, and IL-12. M, 100-bp ladder; 1, amplified product from splenocytes of PBS-treated NOD mice; 2, amplified product from splenocytes of lip-Cl2MDP–treated NOD mice. Representative data from three independent experiments are shown.
Administration of IL-12 Substantially Reverses the Preventive Effect of lip-Cl2MDP in NOD Mice.
The treatment of NOD mice with lip-Cl2MDP results in a significant decrease in the expression of the macrophage-derived cytokine IL-12 and in the decrease of Th1-type immune responses. To determine whether IL-12, which is known to play an important role in the induction of Th1-type immune responses, reverses the preventive effect of lip-Cl2MDP in NOD mice, we treated NOD mice with lip-Cl2MDP with or without IL-12. 43% (3 out of 7) of the IL-12 co-injected NOD mice developed diabetes, whereas none (0 out of 9) of the mice treated with lip-Cl2MDP alone developed the disease (Fig. 8). In contrast, 83% (5 out of 6) of the age-matched PBS-treated control mice (no lip-Cl2MDP treatment) that were injected with IL-12 developed diabetes by 8 wk after IL-12 injection (17 wk of age at the time of onset of diabetes). 60% (6 out of 10) of the mice that were treated with PBS alone developed diabetes by 22 wk of age (Fig. 8). These results indicate that IL-12 substantially reverses the preventive effect of lip-Cl2MDP in NOD mice, probably due to the induction of Th1-type immune responses.
Figure 8.

Administration of IL-12 substantially reverses the preventive effect of lip-Cl2MDP in NOD mice. Female NOD mice were treated with lip-Cl2MDP once a week from 3 wk of age. Recombinant mouse IL-12 (0.4 μg/200 μl PBS/mouse) was administered intraperitoneally each day for the first week beginning at 9 wk of age; and then 0.2 μg of IL-12 was injected daily for the next 3 wk (○). As a control, IL-12 was administered as described above to NOD mice that had not received lip-Cl2MDP treatment (▵). As an additional control, to determine the incidence of spontaneously developed diabetes in NOD mice, NOD female mice received PBS (200 μl/mouse) daily (▴). To determine the effect of lip-Cl2MDP treatment on the prevention of diabetes, NOD female mice were treated with lip-Cl2MDP only (•). The incidence of diabetes was assessed by the measurement of urine glucose every other day.
The Function of Antigen Presentation Is Reduced in Macrophage-depleted NOD Mice.
One role of macrophages is the processing and presentation of antigens to CD4+ helper T cells in association with MHC class II molecules. If these antigen-processing and presenting functions are decreased, then CD4+ T cell activation also would be reduced. Thus, we determined the T cell proliferative response to islet antigens and GAD, a well known β cell autoantigen, in the presence of irradiated splenocytes from either lip-Cl2MDP– or PBS-treated NOD mice. The T cell proliferative response against islets and GAD was significantly decreased in the presence of lip-Cl2MDP–treated splenocytes, as compared with the response in the presence of PBS-treated splenocytes (Fig. 9). These results indicate that the function of antigen presentation in macrophage-depleted NOD mice is significantly impaired.
Figure 9.

The antigen-presenting function is significantly decreased in macrophage-depleted NOD mice. Splenic T cells (5 × 105 cells) from 8–10- wk-old female NOD mice (n = 4) were cultured with irradiated splenocytes (5 × 105 cells) from PBS- or lip-Cl2MDP–treated NOD mice in the presence or absence of the antigens indicated, in 200 μl of complete RPMI 1640 medium. [3H]thymidine incorporation was measured. The bar represents SD value from 4 different mice. *P < 0.05 compared with PBS-treated controls.
Inactivation of Macrophages Results in a Decrease in the Expression of FasL and Perforin.
FasL and perforin are known to show increased expression in activated T cells (32). To determine whether there is any change in the expression level of FasL and perforin in the splenic T cells in macrophage-depleted NOD mice, we measured the mRNA expression of FasL and perforin in splenic T cells from lip-Cl2MDP–treated NOD mice after stimulation with NOD islets. We found a significant decrease in expression as compared with that in PBS-treated control NOD mice (Fig. 10 A). To confirm this result at the protein level, we analyzed the expression of FasL and perforin after activation with syngeneic NOD islets, using flow cytometry. We found that the expression of FasL and perforin in T cells were also significantly downregulated in lip-Cl2MDP– treated NOD mice as compared with PBS-treated control NOD mice (Fig. 10, B and C).
Figure 10.

Inactivation of macrophages results in a decrease in the expression of FasL and perforin. In each group, splenic T cells were pooled from three mice (15 wk of age) and activated with irradiated NOD islets for 72 h. Reverse transcriptase PCR was performed using primers specific for FasL and perforin (A). Primers for HPRT were used as standards. M, 100-bp ladder; 1, amplified product from splenic T cells of PBS-treated NOD mice; 2, amplified product from splenic T cells of lip-Cl2MDP– treated NOD mice. Representative data from three independent experiments. For FACS® analysis of FasL (B) and perforin (C) expression, splenic T cells (106 cells) were stimulated with irradiated islets (40 islets) for 3 d and stained with biotinylated anti-FasL or anti-perforin Abs followed by incubation with PE-streptavidin and FITC-labeled anti–rat IgG, respectively. Solid-line histograms represent cells incubated with PE-streptavidin or FITC-labeled anti–rat IgG only. Dashed line histograms represent cells stained with anti-FasL or anti-perforin Abs. Upper panel shows the results for PBS-treated control mice; lower panel shows the results for lip-Cl2MDP–treated mice.
Discussion
Type I diabetes is a T cell–mediated autoimmune disease found both in humans and in animal models. The role of T cells in the pathogenesis of autoimmune type I diabetes in NOD mice has been studied extensively (1–12). Activated Th1-type CD4+ and CD8+ T cells are believed to kill β cells by apoptosis induced by TNF-α–TNF-α receptor interactions (33) and Fas–FasL and/or perforin–granzyme pathways (1, 3). However, the mechanisms by which these cytotoxic T cells develop are not well understood. The depletion of macrophages results in the complete prevention of T cell–mediated autoimmune type I diabetes in CY-treated NOD mice (19). Thus, it has been suggested that macrophages play an important role in the pathogenesis of autoimmune type I diabetes (3). However, the role of macrophages in the T cell–mediated autoimmune process has not been elucidated.
The first question posed in this study was whether macrophages are required for the development of cytotoxic T cells that can destroy β cells. We transfused splenocytes from macrophage-depleted NOD mice into NOD.scid mice, and found that none of the recipients developed diabetes, whereas the majority of the NOD.scid recipients of splenocytes from age-matched, PBS-treated control NOD mice became diabetic within 8 wk of transfer. These results clearly indicate that macrophages are an absolute requirement for the development of β cell–cytotoxic T cells in NOD mice. Next, to determine whether T lymphocytes from macrophage-depleted NOD mice can destroy pancreatic β cells, we transplanted insulitis-free islets into the renal capsule of macrophage-depleted NOD mice at 20 wk of age. The T cells in the macrophage-depleted NOD mice recipients did not destroy the transplanted islets, whereas the T cells in age-matched control NOD mice recipients destroyed the transplanted islet cells within 3 wk after transplantation. These results confirm that T cells in a macrophage-depleted environment lose their ability to differentiate into cytotoxic T cells that can destroy pancreatic β cells.
The second question posed in this study was whether long-term treatment of NOD mice with lip-Cl2MDP affects the established β cell–cytotoxic T cells, resulting in a loss of their ability to destroy β cells. To answer this question, we treated NOD mice with lip-Cl2MDP for a prolonged period (9 wk) after the development of β cell–cytotoxic T cells in these animals, and transfused T cells from these mice into NOD.scid mice. We found that the transfused T cells were able to induce diabetes as did the T cells from PBS-treated control NOD mice. This result indicates that long-term treatment with lip-Cl2MDP does not alter the function of the established β cell–cytotoxic T cells in NOD mice.
Several reports have shown that lip-Cl2MDP selectively depletes macrophages without affecting nonphagocytic cells (22, 29, 30, 34–39). The large liposomes (>0.8 μm) used in this study are selectively phagocytosed by macrophages, whereas small liposomes (<0.2 μm) may be internalized by nonphagocytic cells. In fact, macrophages phagocytose lip-Cl2MDP, and the phospholipid bilayers of the liposomes are disrupted by lysosomal phospholipase. Cl2MDP is then released intracellularly, and kills macrophages by apoptosis. Therefore, only cell populations that possess both phagocytic activity and phospholipase can be affected. Thus, we cannot exclude the possibility that lip-Cl2MDP treatment may have some effect on a certain dendritic cell population, which possesses both a phagocytic function and lysosomal phospholipase. However, CD4+ and CD8+ T and B cells, which have neither a phagocytic function nor lysosomal phospholipase, are not affected by treatment with lip-Cl2MDP. It had been thought that free Cl2MDP released from dying macrophages may be toxic to other immune cells. However, free Cl2MDP is rapidly excreted by the kidneys (<10 min) (van Rooijen, N., unpublished observation). Furthermore, lip-Cl2MDP has a very short half-life in circulation and within body fluids, explaining why it does not affect nonphagocytic cells (35). Finally, the specific depletion of macrophages by lip-Cl2MDP treatment has been confirmed by immunocytochemical and electron microscopic methods and by functional assays (36, 37).
The third question posed in this study was whether T cells from macrophage-depleted NOD mice would recover their ability to differentiate into cytotoxic T cells when transferred into a macrophage-containing environment. To answer this question, we transfused splenocytes from macrophage-depleted NOD mice into NOD.scid mice (which contain endogenous macrophages but lack T cells) and monitored urine glucose for a prolonged time period. In agreement with the previous experiment, none of the mice became diabetic up to 13 wk after transfusion, but ∼70% became diabetic between 14 and 17 wk after the transfer of splenocytes. This supports our findings that macrophages are a major contributor to the creation of the immune environment required for the development of β cell–cytotoxic T cells.
The fourth question posed in this study was why T cells from a macrophage-depleted environment lose their ability to destroy β cells. One possible cause for the impaired capability of T cells to kill β cells in macrophage-depleted NOD mice is a change in the finely tuned balance of immunoregulatory T cells, involving a decrease in the Th1 immune response along with an increase in the Th2 immune response. As macrophage-derived IL-12 is a dominant factor in directing the development of Th1 cells, the depletion of macrophages would be expected to impair the production of IL-12 and suppress the Th1 immune response. It has been suggested that Th1-type T cells play a pathogenic role in the autoimmune destruction of β cells in autoimmune diabetes (12, 40–42), whereas Th2-type T cells play a nondestructive or preventive role in the disease (12, 42–44). Thus, it is thought that the destruction of β cells may depend on which way the finely tuned balance of immunoregulatory T cells (i.e., Th1- and Th2-type T cells) and/or immunoregulatory cytokines (i.e., IFN-γ and IL-2 secreted from Th1-type T cells or IL-4 secreted from Th2-type T cells) is tipped. If this balance is tipped in favor of Th1-type effector T cells and/or their immune-activating cytokines, the autoimmune process leading to β cell destruction may be enhanced. In contrast, Th2-type T cells and/or their immunosuppressive cytokines may protect the β cells against immune-activating effector T cell–mediated damage when the immune balance is tipped in their favor, blocking the autoimmune destructive process. We found that the level of IL-4 secreted from Th2-type T cells increased, whereas IFN-γ secreted from Th1-type T cells decreased in macrophage-depleted NOD mice.
In addition, a recent study shows that the IL-12Rβ2 subunit is preferentially expressed in Th1-type T cells, whereas the IL-12Rβ1 subunit is expressed in both Th1- and Th2-type T cells (31). Therefore, we examined the expression of IL-12Rβ2 in the splenic T cells to confirm our finding of a decrease in the Th1 immune response in T cells from macrophage-depleted NOD mice. We found that the expression of IL-12Rβ2 was significantly decreased in these T cells, as compared with the expression in the T cells of control NOD mice that were not depleted of macrophages. Thus, the decrease in the Th1 immune response along with an increase in the Th2 immune response in macrophage-depleted mice may be one major factor contributing to the impairment of the capability of T cells to kill β cells.
The IL-12 expressed in macrophages is known to play an important role in the development of Th1-type CD4+ T cells, leading to a T cell–mediated immune response (45, 46). Therefore, we examined whether the decrease of the Th1 immune response is due to the decrease of IL-12 expression in lip-Cl2MDP–treated NOD mice. We found that the expression of IL-12 in macrophage-depleted NOD mice was significantly decreased as compared with age-matched, control NOD mice. The fifth question posed in this study was whether the administration of IL-12 would negate the protective effect of macrophage depletion in NOD mice. We found that the administration of IL-12 substantially, but not completely, reversed the prevention of diabetes in macrophage-depleted NOD mice. These results suggest that the downregulation of IL-12 indirectly protects β cells from destruction in macrophage-depleted NOD mice by changing the finely tuned balance of the Th1/Th2 immune response.
We also considered other factors that might protect β cells from destruction in macrophage-depleted mice. These include macrophage-derived soluble mediators such as oxygen free radicals and other cytokines including IL-1β, TNF-α, and IFN-γ. We found that the expression of the cytokines IL-1β, TNF-α, and IFN-γ was significantly decreased in macrophage-depleted NOD mice as compared with PBS-treated control NOD mice. These cytokines, which are released from activated macrophages, are believed to be toxic to β cells (47–50). The toxic effect produced by activated macrophages on β cells is thought to be mediated by the superoxide anion and hydrogen peroxide. The β cell is very sensitive to the production of free radicals because islet cells exhibit very low free radical scavenging activity (51, 52). Cytokines produced by islet-infiltrating macrophages may contribute to β cell damage by inducing the production of oxygen free radicals in the islets (53, 54). Our recent studies have shown that the viral infection of β cells results in the recruitment of macrophages into the pancreatic islets, and that the soluble mediators IL-1β, TNF-α, and inducible nitric oxide synthase, known to be major factors produced by activated macrophages, contribute to the destruction of β cells (55). Therefore, we cannot exclude the possibility that the downregulation of these macrophage-derived soluble mediators also may play a role in the protection of β cells in macrophage-depleted NOD mice.
A role of macrophages is the processing and presentation of antigens to CD4+ helper T cells in association with MHC class II molecules (56, 57). Activation of CD4+ T cells can be through the processing and presentation of antigens by APCs. Therefore, the sixth question posed in this study was whether depletion of macrophages would affect the antigen-presenting function in NOD mice. We measured the T cell proliferation response to islet antigens and GAD (a well-known β cell autoantigen) in the presence of splenocytes, as APCs, from macrophage-depleted or control NOD mice. We found that the T cell proliferation response was significantly decreased when we used splenocytes from macrophage-depleted NOD mice as APCs. This result suggests that the depletion of macrophages results in the downregulation of antigen-specific CD4+ T cell activation.
Recent studies have shown that activated CD8+ T cells are capable of performing cytotoxic functions through the use of perforin and granzymes as well as by the induction of apoptosis through Fas–FasL interactions. Perforin-deficient NOD mice showed a significantly decreased incidence of diabetes and a delayed onset of the disease (58). In addition, NODlpr/lpr mice, in which Fas is not expressed, do not develop diabetes or insulitis (59, 60). Thus, the final question put forth in this study was whether the depletion of macrophages would influence the level of T cell activation. We determined the level of expression of FasL and perforin in splenic T cells from macrophage-depleted NOD mice. We found that there was a significant decrease in the expression of FasL and perforin in splenic T cells from lip-Cl2MDP– treated NOD mice as compared with PBS-treated controls. These results suggest that macrophages are required for the activation of the cytotoxic T cells that can destroy pancreatic β cells. The precise mechanism for the involvement of macrophages in T cell activation is not known. The IL-12 secreted by macrophages could activate Th1-type CD4+ T cells, and subsequently, the IL-2 and IFN-γ produced by these activated CD4+ T cells may help with the maximal activation of CD8+ T cells. The downregulation of islet cell–specific T cell activation may be another major factor contributing to the impairment of the capability of T cells to kill β cells in macrophage-depleted NOD mice.
In conclusion, macrophages are primary contributors to the creation of the immune environment for the development and activation of cytotoxic T cells that destroy pancreatic β cells, resulting in the development of autoimmune diabetes in NOD mice.
Acknowledgments
This work was supported by grants MA9584 and MTI3224 from the Medical Research Council of Canada to J.W. Yoon. J.W. Yoon is a Heritage Medical Scientist awardee of the Alberta Heritage Foundation for Medical Research.
Abbreviations used in this paper
- BB
BioBreeding
- CY
cyclophosphamide
- FasL
Fas ligand
- GAD
glutamic acid decarboxylase
- HPRT
hypoxanthine phosphoribosyl transferase
- lip-Cl2MDP
liposomal dichloromethylene diphosphonate
- NOD
nonobese diabetic
Footnotes
The authors gratefully acknowledge the excellent technical assistance of Ms. L. Bryant for FACS® analyses and the editorial assistance of Ms. Karen Clarke and Dr. A.L. Kyle.
References
- 1.Wong, F.S., and C.A. Janeway. 1997. The role of CD4+ and CD8+ T cells in type I diabetes in the NOD mouse. 7th Forum in Immunology. 327–332. [DOI] [PubMed]
- 2.Delovitch TL, Bhagirath S. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 1997;7:727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- 3.Yoon JW, Jun HS, Santamaria PS. Cellular and molecular mechanisms for the initiation and progression of β-cell destruction resulting from the collaboration between macrophages and T cells. Autoimmunity. 1998;127:109–122. doi: 10.3109/08916939809008041. [DOI] [PubMed] [Google Scholar]
- 4.Nagata M, Yoon JW. Studies on autoimmunity for T-cell-mediated β cell destruction. Distinct difference in β cell destruction between CD4+ and CD8+T-cell clones derived from lymphocytes infiltrating the islets of NOD mice. Diabetes. 1992;41:998–1008. doi: 10.2337/diab.41.8.998. [DOI] [PubMed] [Google Scholar]
- 5.Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+islet-specific T cell clone. Science. 1990;249:1433–1436. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 6.Haskins K, Portas M, Bradley B, Wegmann D, Lafferty K. T-lymphocyte clone specific for pancreatic islet antigen. Diabetes. 1988;37:1444–1448. doi: 10.2337/diab.37.10.1444. [DOI] [PubMed] [Google Scholar]
- 7.Wong S, Wen L, Visintin I, Flavell RA, Janeway CA., Jr CD8 T cell clones from young nonobese diabetic (NOD) islets can transfer rapid onset of diabetes in the absence of CD4 T cells. J Exp Med. 1996;183:67–76. doi: 10.1084/jem.183.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagata M, Santamaria P, Kawamura T, Utsugi T, Yoon JW. Evidence for the role of CD8+ cytotoxic T cells in the destruction of pancreatic beta cells in NOD mice. J Immunol. 1994;152:2042–2050. [PubMed] [Google Scholar]
- 9.Utsugi T, Yoon JW, Park BJ, Imamura M, Averil N, Kawazu S, Santamaria P. Major histocompatibility complex class I-restricted infiltration and destruction of pancreatic islets by NOD mouse-derived beta cell cytotoxic CD8+ T cell clones in vivo. Diabetes. 1996;45:1121–1131. doi: 10.2337/diab.45.8.1121. [DOI] [PubMed] [Google Scholar]
- 10.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 11.Verdaguer J, Yoon JW, Averil N, Utsugi T, Park BJ, Santamaria P. Acceleration of diabetes in NOD mice expressing beta cell-cytotoxic T cell-derived TCRβ transgene. J Immunol. 1996;157:4726–4735. [PubMed] [Google Scholar]
- 12.Katz JD, Benoist C, Mathis D. T helper cell subsets in insulin-dependent diabetes. Science. 1995;268:1185–1190. doi: 10.1126/science.7761837. [DOI] [PubMed] [Google Scholar]
- 13.Kolb, H., G. Kantwerk, U. Treichel, T. Kurner, U. Kiesel, T. Hoppe, and V. Kolb-Bachofen. 1986. Prospective analysis of islet lesions in BB rats. Diabetologia. 29(Suppl. 1):A559.
- 14.Lee KU, Kim MK, Amano K, Pak CY, Jaworski MA, Mehta JG, Yoon JW. Preferential infiltration of macrophages during early stages of insulitis in diabetes-prone BB rats. Diabetes. 1989;37:1053–1058. doi: 10.2337/diab.37.8.1053. [DOI] [PubMed] [Google Scholar]
- 15.Voorbij HAM, Jeucken PHM, Kabel PJ, DeHaan M, Drexhage HA. Dendritic cells and scavenger macrophages in pancreatic islets of prediabetic BB rats. Diabetes. 1989;38:1623–1629. doi: 10.2337/diab.38.12.1623. [DOI] [PubMed] [Google Scholar]
- 16.Walker R, Bone AJ, Cooke A, Baird JD. Distinct macrophage subpopulations in pancreas of prediabetic BB/E rats: possible role for macrophages in pathogenesis of IDDM. Diabetes. 1988;37:1301–1304. doi: 10.2337/diab.37.9.1301. [DOI] [PubMed] [Google Scholar]
- 17.Jasen A, Homo-Delarche R, Hooijkaas H, Leenen PJ, Dardenne M, Drexhage HA. Immunohistochemical characterization of monocyte-macrophages and dendritic cells involved in the initiation of insulitis and beta-cell destruction in NOD mice. Diabetes. 1994;43:667–675. doi: 10.2337/diab.43.5.667. [DOI] [PubMed] [Google Scholar]
- 18.Amano K, Yoon JW. Studies on autoimmunity for initiation of beta-cell destruction. V. Decrease of macrophage dependent T-effector cells and natural killer cytotoxicity in silica-treated BB rats. Diabetes. 1990;39:590–596. doi: 10.2337/diab.39.5.590. [DOI] [PubMed] [Google Scholar]
- 19.Lee KU, Amano K, Yoon JW. Evidence for initial involvement of macrophage in development of insulitis in NOD mice. Diabetes. 1988;37:989–991. doi: 10.2337/diab.37.7.989. [DOI] [PubMed] [Google Scholar]
- 20.Oschilewski U, Kiesel U, Kolb H. Administration of silica prevents diabetes in BB rats. Diabetes. 1985;34:197–199. doi: 10.2337/diab.34.2.197. [DOI] [PubMed] [Google Scholar]
- 21.Lee KU, Pak CY, Amano K, Yoon JW. Prevention of lymphocytic thyroiditis and insulitis in diabetes-prone BB rats by the depletion of macrophages. Diabetologia. 1988;31:400–402. doi: 10.1007/BF02341511. [DOI] [PubMed] [Google Scholar]
- 22.van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 23.Chung YH, Jun HS, Kang Y, Hirasawa K, Lee BR, Van Rooijen N, Yoon JW. Role of macrophages and macrophage-derived cytokines in the pathogenesis of Kilham rat virus-induced diabetes in diabetes-resistant BB rats. J Immunol. 1997;159:466–471. [PubMed] [Google Scholar]
- 24.Kawamura T, Nagata M, Utsugi T, Yoon JW. Prevention of autoimmune type I diabetes by CD4+ suppressor T cells in superantigen-treated non-obese diabetic mice. J Immunol. 1993;151:4362–4370. [PubMed] [Google Scholar]
- 25.Yoon JW, McClintock PR, Onodera T, Notkins AL. Virus-induced diabetes mellitus. XVIII. Inhibition by a nondiabetogenic variant of encephalomyocarditis virus. J Exp Med. 1980;152:878–892. doi: 10.1084/jem.152.4.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoon JW, Rodriguez MM, Currier C, Notkins A. Long-term complications of virus-induced diabetes mellitus in mice. Nature. 1982;296:566–569. doi: 10.1038/296566a0. [DOI] [PubMed] [Google Scholar]
- 27.Utsugi T, Nagata M, Kawamura T, Yoon JW. Prevention of recurrent diabetes in syngeneic islet-transplanted NOD mice by transfusion of autoreactive T lymphocytes. Transplantation. 1994;57:1799–1804. [PubMed] [Google Scholar]
- 28.Han HS, Jun HS, Utsugi T, Yoon JW. A new type of CD4+ suppressor T cell completely prevents spontaneous autoimmune diabetes and recurrent diabetes in syngeneic islet-transplanted NOD mice. J Autoimmunity. 1996;9:331–339. doi: 10.1006/jaut.1996.0045. [DOI] [PubMed] [Google Scholar]
- 29.Naito M, Hirotaka N, Kawano S, Umezu H, Zhu H, Moriyama H, Yamomoto T, Takatsuka H, Takei Y. Liposome-encapsulated dichloro-methylene diphosphonate induces macrophage apoptosis in vivo and in vitro. J Leukocyte Biol. 1996;60:337–344. doi: 10.1002/jlb.60.3.337. [DOI] [PubMed] [Google Scholar]
- 30.van Rooijen N, Sanders AM, van den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J Immunol Methods. 1996;193:93–99. doi: 10.1016/0022-1759(96)00056-7. [DOI] [PubMed] [Google Scholar]
- 31.Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the IL-12R β2 subunit expression in developing Th1 and Th2 cells. J Exp Med. 1997;185:817–824. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bretscher P. The two-signal model of lymphocyte activation twenty-one years later. Immunol Today. 1992;13:74–76. doi: 10.1016/0167-5699(92)90138-W. [DOI] [PubMed] [Google Scholar]
- 33.Kurrer MO, Pakala SV, Hanson HL, Katz JD. β cell apoptosis in T cell-mediated autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:213–218. doi: 10.1073/pnas.94.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Rooijen N, Bakker J, Sanders AM. Transient suppression of macrophage functions by liposome- encapsulated drugs. Trends Biotechnol. 1997;15:178–185. doi: 10.1016/s0167-7799(97)01019-6. [DOI] [PubMed] [Google Scholar]
- 35.van Rooijen N, Sanders AM. Elimination, blocking, and activation of macrophages: three of a kind? . J Leukocyte Biol. 1997;62:702–709. doi: 10.1002/jlb.62.6.702. [DOI] [PubMed] [Google Scholar]
- 36.van Rooijen N, van Nieuwmegen R. Elimination of phagocytic cells in the spleen after intravenous injection of liposome encapsulated dichloromethylene diphosphonate. An enzyme histochemical study. Cell Tissue Res. 1984;238:355–358. doi: 10.1007/BF00217308. [DOI] [PubMed] [Google Scholar]
- 37.van Rooijen N, van Nieuwmegen R, Kamperdijk EWA. Elimination of phagocytic cells in the spleen after intravenous injection of liposome encapsulated dichloromethylene diphosphonate. Ultra-structural aspects of elimination of marginal zone macrophages. Virchows Arch B Cell Pathol. 1985;49:375–383. doi: 10.1007/BF02912114. [DOI] [PubMed] [Google Scholar]
- 38.Fraser CC, Chen BP, Webb S, van Rooijen N, Kraal G. Circulation of human hematopoietic cells in severe combined immunodeficient mice after Cl2MDP-liposome-mediated macrophage depletion. J Immunol Methods. 1990;134:153–161. [PubMed] [Google Scholar]
- 39.Claassen I, van Rooijen N, Claassen E. A new method for removal of mononuclear phagocytes from heterogeneous cell populations in vitro, using the liposome-mediated ‘suicide' technique. Blood. 1995;86:183–192. doi: 10.1016/0022-1759(90)90376-7. [DOI] [PubMed] [Google Scholar]
- 40.Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today. 1995;16:34–38. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 41.Zipris D, Greiner DL, Malkani S, Whalen B, Mordes JP, Rossini AA. Cytokine gene expression in islets and thyroids of BB rats: IFN-γ and IL-12p40 mRNA increase with age in both diabetic and insulin-treated nondiabetic BB rats. J Immunol. 1996;156:1315–1321. [PubMed] [Google Scholar]
- 42.Rabinovitch A. Immunoregulatory and cytokine imbalances in the pathogenesis of IDDM: therapeutic intervention by immunostimulation? . Diabetes. 1994;43:613–621. doi: 10.2337/diab.43.5.613. [DOI] [PubMed] [Google Scholar]
- 43.Fox CJ, Danska JS. IL-4 expression at the onset of islet inflammation predicts nondestructive insulitis in nonobese diabetic mice. J Immunol. 1997;158:2414–2424. [PubMed] [Google Scholar]
- 44.Cameron MJ, Arreaza GA, Zucker P, Chensue SW, Strieter RM, Chakrabart S, Delovitch TL. IL-4 prevents insulitis and insulin-dependent diabetes mellitus in nonobese diabetic mice by potentiation of regulatory T helper-2 cell function. J Immunol. 1997;159:4686–4692. [PubMed] [Google Scholar]
- 45.Trinchieri G. Interleukin-12 and its role in the generation of Th1 cells. Immunol Today. 1993;14:335–337. doi: 10.1016/0167-5699(93)90230-I. [DOI] [PubMed] [Google Scholar]
- 46.Kennedy MK, Picha KS, Shanebeck KD, Anderson DM, Grabstein KH. Interleukin-12 regulates the proliferation of Th1, but not Th2 or Th0 clones. Eur J Immunol. 1994;24:2271–2278. doi: 10.1002/eji.1830241002. [DOI] [PubMed] [Google Scholar]
- 47.Pankewycz OG, Guan JX, Benedict JF. Cytokines as mediators of autoimmune diabetes and diabetic complications. Endocrinol Rev. 1995;16:164–176. doi: 10.1210/edrv-16-2-164. [DOI] [PubMed] [Google Scholar]
- 48.Mandrup-Poulsen T, Bendtzen K, Dinarello C, Nerup J. Human tumor-necrosis factor potentiates human interleukin 1-mediated rat pancreatic beta cell-cytotoxicity. J Immunol. 1987;139:4077–4082. [PubMed] [Google Scholar]
- 49.Appels B, Burkart V, Kantwerk-Funke M, Funda J, Kolb-Bachofen V, Kolb H. Spontaneous cytotoxicity of macrophages against pancreatic islet cells. J Immunol. 1989;142:3803–3808. [PubMed] [Google Scholar]
- 50.Pukel C, Baquerizo H, Rabinovitch A. Destruction of rat islet cell monolayers by cytokines. Synergistic interactions of interferon-gamma, tumor necrosis factor, lymphotoxin, and interleukin-1. Diabetes. 1988;37:133–136. doi: 10.2337/diab.37.1.133. [DOI] [PubMed] [Google Scholar]
- 51.Faust, A., R. Kleemann, H. Rothe, and H. Kolb. 1996. Role of macrophages and cytokines in β-cell death. In Lessons from Animal Diabetes VI. E. Shafrir, editor. Birkhäuser, Cambridge, Massachusetts. 47–56.
- 52.Asayama K, Kooy NW, Burr IM. Effect of vitamin E deficiency and selenium deficiency on insulin secretory reserve and free radical scavenging systems in islets; decrease of islet manganosuperoxide dismutase. J Lab Clin Med. 1986;107:4559–4564. [PubMed] [Google Scholar]
- 53.Malaisse WJ, Malaisse-Lagae F, Sener A, Pipeleers DG. Determinants of the selective toxicity of alloxan to the pancreatic β cell. Proc Natl Acad Sci USA. 1982;79:927–930. doi: 10.1073/pnas.79.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corbett JA, McDaniel ML. Does nitric oxide mediate autoimmune destruction of β cells? Possible therapeutic interventions in IDDM. Diabetes. 1992;41:897–903. doi: 10.2337/diab.41.8.897. [DOI] [PubMed] [Google Scholar]
- 55.Hirasawa K, Jun HS, Maeda K, Kawaguchi Y, Itagaki S, Mikami T, Baek HS, Doi K, Yoon JW. Possible role of macrophage-derived soluble mediators in the pathogenesis of EMC virus-induced diabetes in mice. J Virol. 1997;71:4024–4031. doi: 10.1128/jvi.71.5.4024-4031.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Unuaue ER. Antigen-presenting function of the macrophage. Annu Rev Immunol. 1984;2:395–428. doi: 10.1146/annurev.iy.02.040184.002143. [DOI] [PubMed] [Google Scholar]
- 57.Unuaue ER, Allen PM. The basis for the immunoregulatory role of macrophages and other accessory cells. Science. 1987;236:551–557. doi: 10.1126/science.2437650. [DOI] [PubMed] [Google Scholar]
- 58.Kagi D, Odermatt B, Seiler P, Zinkernagel RM, Mak TW, Hengartner H. Reduced incidence and delayed onset of diabetes in perforin-deficient nonobese diabetic mice. J Exp Med. 1997;186:989–997. doi: 10.1084/jem.186.7.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Itoh N, Imagawa A, Hanafusa T, Waguri M, Yamamoto K, Iwahashi H, Moriwaki M, Nakajima H, Miyagawa J, Namba M, et al. Requirement of Fas for the development of autoimmune diabetes in nonobese diabetic mice. J Exp Med. 1997;186:613–618. doi: 10.1084/jem.186.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chervonsky AV, Wang Y, Wong FS, Visintin I, Flavell RA, Janeway CA, Jr, Matis L. The role of Fas in autoimmune diabetes. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]