

Abstract
Activation-induced cell death (AICD) is a process that regulates the size and the duration of the primary immune T cell response. In this report, we investigated the mechanisms involved in the regulation of AICD by transforming growth factor β1 (TGF-β1). We found that TGF-β1 decreased apoptosis of human T cells or T cell hybridomas after activation by anti-CD3. This decrease was associated with inhibition of Fas (Apo-1/CD95) ligand (FasL) expression, whereas Fas signaling was not affected by TGF-β1. In parallel, TGF-β1 inhibited c-Myc expression in T cell hybridomas, and ectopic expression of a chimeric molecule composed of c-Myc and the steroid binding domain of the estrogen receptor (Myc-ER) blocked both the inhibition of FasL and the decrease of AICD induced by TGF-β1, providing that 4-hydroxytamoxifen was present. These results identify one mechanism by which TGF-β1 blocks AICD to allow the clonal expansion of effector T cells and the generation of memory T cells during immune responses.
Keywords: apoptosis, T cells, transforming growth factor β, Fas ligand, c-myc
Transforming growth factor (TGF)-β1 is a multifunctional cytokine that regulates cell growth, adhesion, and differentiation in a wide variety of cell types (1). In the immune system, TGF-β1 controls the growth and differentiation of T cells, B cells, NK cells, and dendritic cells (2–7). TGF-β1 has also been shown to be effective in treating experimental allergic encephalomyelitis and collagen-induced arthritis, two T cell–mediated experimental autoimmune diseases (8–10). In contrast, the expansion of T lymphocytes expressing a naive surface phenotype can be enhanced rather than suppressed by TGF-β1 (7, 11, 12).
TGF-β1 may be one of the candidate molecules involved in memory and effector T cell generation. Indeed, TGF-β1 has recently been implicated in the generation of a large population of effector CD4+ T cells in response to antigen stimulation (13, 14). Furthermore, in this system TGF-β1 did not affect proliferation or influence cytokine secretion but partially blocked activation-induced apoptosis (13, 14). The protective effect of TGF-β1 on activation-induced cell death (AICD)1 has also been reported for CD8+ T cells (12).
AICD in T cells in vivo has been proposed to limit the expansion of an immune response by eliminating effector cells that are no longer needed (15). Indeed, after antigen or pathogens have been eliminated from the organism, these T cells are potentially dangerous because of their potent effector functions and low activation requirements. We and others recently reported that activation of T cell clones, T cell lines, or T cell hybridomas induces FasL expression and that interaction between Fas and its ligand is the major mechanism involved in AICD (16–20).
The expression of FasL during AICD requires the activation of transcription factors. Previous studies using c-myc antisense oligonucleotides (21) or dominant negative reciprocal exchange mutants of Myc or Max (22), which antagonize the functional Myc/Max heterodimer, demonstrated that c-Myc function is required for AICD in T cells. More recently, Hueber et al. (23) reported that c-myc–induced apoptosis of serum-starved fibroblasts requires the expression of Fas and FasL. Therefore, it appears that c-Myc may be essential for the function of FasL and subsequent apoptosis in some systems.
TGF-β1 has been reported to exert its growth inhibitory effects on various cell types through the downregulation of the expression of genes involved in cellular proliferation, such as cyclin-dependent kinases (CDKs [24–26]), and the c-myc protooncogene (27, 28). Furthermore TGF-β1 suppresses constitutive and inducible c-Myc expression in two constitutively activated murine T clones (29).
In this report, we investigated the mechanisms involved in the regulation of AICD by TGF-β1. We determined that TGF-β1 inhibits FasL expression at the level of mRNA expression. TGF-β1 also inhibits the constitutive c-Myc expression in A1.1 T cell hybridomas, and since c-Myc has been demonstrated to regulate AICD, we prepared stable transfectants constitutively expressing a chimeric molecule composed of c-Myc and the steroid binding domain of the estrogen receptor (Myc-ER). In these cells, TGF-β1 did not inhibit FasL expression and subsequent AICD after anti-CD3 antibody treatment, providing that 4-hydroxytamoxifen (4-OHT) was present. These results demonstrate that TGF-β1 inhibits FasL expression and subsequent AICD via downregulation of c-Myc expression.
Materials and Methods
Cell Cultures and Reagents.
The T cell hybridomas A1.1 and 2B4.11 have been described previously (17, 30). PBMCs were isolated from healthy donors by density gradient centrifugation of heparinized blood on a layer of histopaque (Sigma). All cells were grown in RPMI 1640 medium containing 10% FCS, 5 × 10−5 M β-mercaptoethanol, 2 mM l-glutamine, and 100 U/ml each of penicillin and streptomycin (complete medium). PMA, ionomycin, cyclosporine A (CsA), 4-OHT, and M2 anti-Flag antibody were purchased from Sigma. The mouse anti–human CD3 (OKT3) and the hamster anti–mouse CD3ε (145-2C11) antibodies were purified from the culture supernatant by protein A affinity chromatography. Phosphorothioate antisense c-myc (ASc-myc) 5′-CACGTTGAGGGGCAT-3′ and nonsense c-myc (NSc-myc) 5′-AGTGGCGGAGACTCT-3′ oligonucleotides were purchased from Quality Controlled Biochemicals, Inc. Soluble recombinant human FasL was obtained from Dr. Jurg Tschopp (University of Lausanne, Epalinges, Switzerland [31]).
Induction and Analysis of Apoptosis.
For the induction of apoptosis, T cell hybridomas (0.5 × 106/ml) were cultured 16 h in triplicate in 96-well plates precoated with anti-CD3 antibody (2C11). PBMCs (106/ml) were activated for 6 d with 100 ng/ml OKT3, and after elimination of dead cells, were restimulated with PMA (50 ng/ml) and ionomycin (1 μg/ml) for 16 h. Viability was assessed by addition of 5 μg/ml propidium iodide and immediate analysis using a FACScan® (Becton Dickinson). Dead cells were identified as those taking up the dye. Apoptosis was confirmed by morphological inspection by fluorescent microscope after staining with 10 μg/ml Hoechst 33342 (Sigma).
Activation-induced FasL expression on the A1.1 murine T cell hybridoma was assessed by determining the ability of these cells to cause DNA fragmentation in Fas+ target cells as described previously (17). In brief, A1.1 cells were activated with plate-bound anti-CD3 antibodies in the presence or absence of different concentrations of TGF-β1 and cultured for 6 h to allow FasL expression. The cells were washed twice and incubated for another 8 h with [3H]TdR-labeled L1210 or L1210-Fas target cells. In other experiments, TGF-β1 was added at 6 h to determine its effect on the induction of Fas-mediated death in the target cells. [3H]TdR-labeled unfragmented DNA was harvested on glass fiber filters and assessed in a liquid scintillation counter. DNA fragmentation was calculated as follows: % DNA fragmentation = 100 × (cpm control group − cpm experimental group/cpm control group) ± SD. No DNA fragmentation was observed when target cells were cultured with anti-CD3 in the absence of T cell hybridomas or with T cell hybridomas in the absence of anti-CD3.
DNA Gel Electrophoresis.
DNA preparations were obtained following a procedure described previously (32). In brief, 2 × 106 2B4.11 cells were lysed in buffer containing 10 mM EDTA, 100 mM NaCl, 0.5% (wt/vol) SDS, 100 mM Tris-HCl, pH 7.4, and 0.1 mg/ml proteinase K (Boehringer Mannheim). The DNA was extracted twice with phenol and twice with chloroform/isoamyl alcohol. The aqueous phase was precipitated with 2 vol of ethanol. The pellet was discarded, and one tenth of the volume of 3 M sodium acetate was added to the supernatant which was left at −20°C overnight. The precipitate was centrifuged and the pellet was dried under vacuum before resuspension in 100 μl RNase buffer containing 10 mM Tris and 1 mM EDTA, pH 7.5. The samples were diluted in loading buffer and loaded onto a 2% agarose gel containing 0.1 μg/ml ethidium bromide (Sigma). After electrophoresis, gels were examined under UV light.
RT-PCR for fas, fasL, and c-myc Expression.
The expression of fas, fasL, and c-myc was determined by reverse transcription (RT) of total RNA followed by PCR amplification (RT-PCR). Approximately 3 × 106 cells were homogenized with 1 ml Trizol reagent (GIBCO BRL), and total RNA was isolated according to the manufacturer's protocol. cDNAs were synthesized by extension of (dT) primers with 200 U of SuperScript II reverse transcriptase (GIBCO BRL) in a mixture containing 1 μg of total RNA digested by RNase-free DNase (2 U/μg of RNA) for 15 min at 37°C. PCR of the cDNA was performed in a final volume of 50 μl containing all four dNTPs, 2 mM MgCl2, 2.5 U of AmpliTaq (GIBCO BRL), and each primer at 0.2 μM using the geneAmp 2400 PCR system (Perkin Elmer Corp.). The amplification cycles were 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min. The PCR products were separated by electrophoresis on a 1.5% agarose gel after 27–33 cycles for mouse fas, fasL, and c-myc or 22–28 cycles for mouse β-actin and visualized by ethidium bromide staining. Amplification of β-actin served as a control for sample loading and integrity. The following primers were designed to discriminate between the amplification of cDNA (low size PCR products) and contaminating genomic cDNA (high size PCR products): mouse fasL sense, 5′-CAG-CAG-TGC-CAC-TTC-ATC-TTG-G-3′; mouse fasL antisense, 5′-TTC-ACT-CCA-GAG-ATC-AGA-GCG-G-3′; mouse fas sense, 5′-GAG-GAC-TGC-AAA-ATG-AAT-GGG-G-3′; mouse fas antisense, 5′-ACA-ACC-ATA-GGC-GAT-TTC-TGG-G-3′; mouse c-myc sense, 5′-ACA-GAG-GGA-GTG-AGC-GGA-CG-3′; mouse c-myc antisense, 5′-TTC-ACG-TTG-AGG-GGC-ATC-G-3′; mouse β-actin sense, 5′-TGG-AAT-CCT-GTG-GCA-TCC-ATG-AAA-C-3′; and mouse β-actin antisense: 5′-TAA-AAC-GCA-GCT-CAG-TAA-CAG-TCC-G-3′.
Plasmids and Transfections.
Moloney retroviral virions were produced as described previously (33). In brief, amphotropic packaging cell line was plated at 2.5 × 106 cells/10-cm2 culture dish for 18–24 h before transfection as described (34). Cells were transfected with 7.5 μg of pBABE puroMyc-ER G525R construct (35) using a standard calcium phosphate protocol except for chloroquine (25 μM final), which was added to the cells 5 min before addition of calcium phosphate DNA precipitate. After 24 h the cells were gently washed, and fresh medium was added. Virus-containing supernatant was harvested at 24 and 48 h after transfection, filtered, and stored at 4°C. For virus infection, A1.1 cells (0.5 × 106/ml) were resuspended in 3 ml of viral supernatant containing 5 μg/ml polybrene for 12 h. Cells were then washed and resuspended in RPMI medium without phenol red (GIBCO BRL) containing 10% steroid-free FCS, 5 × 10−5 M β-mercaptoethanol, 2 mM l-glutamine, and 100 U/ml each of penicillin and streptomycin. 48 h after transfection, cells were selected with puromycin (1.5 μg/ml) for 7 d.
Assessment of c-Myc and Myc-ER Proteins.
2 × 106 A1.1 or A1.1 Myc-ER cells were incubated with or without 10 ng/ml TGF-β1 for 8 h. Cells were then harvested, washed once in PBS, and lysed in 100 μl 1× SDS-PAGE sample buffer (50 mM Tris, pH 6.8, 100 mM dithiothreitol, 2% SDS, 10% glycerol). Equal volumes of samples (30 μl) were separated on an 8% gel and transferred to a nitrocellulose membrane (Amersham Pharmacia Biotech). Membranes were blocked with 5% nonfat dry milk in TBS, 0.02% sodium azide for 2 h and then incubated with mouse anti–human c-Myc 9E10 (1:1,000; Santa Cruz Biotechnology, Inc.) for 4 h at room temperature. Membranes were then washed three times in TBS, 0.1% Tween 20 for 15 min and incubated with a rabbit anti–mouse horseradish peroxidase conjugate (1:2,000; Amersham Pharmacia Biotech) for 1 h. After three washes, membranes were incubated with ECL chemiluminescence detection solutions (Amersham Pharmacia Biotech) and exposed to x-ray films for 1 min.
Cell Cycle Analysis.
Cell cycle analysis was performed as described previously (36), with slight modifications. 106 A1.1 cells were washed twice in PBS and resuspended in 0.5 ml of hypotonic fluorochrome solution (50 μg/ml propidium iodide and 0.1% Triton X-100 in 0.1% sodium citrate). After 4 h at 4°C in the dark, the fluorescence of gated live cells was analyzed using a FACScan® flow cytometer (Becton Dickinson).
Results
TGF-β1 Decreases AICD in Human T Cells as well as Mouse T Cell Hybridomas.
TGF-β1 has been reported to increase long-term cell expansion of Th2 cells by decreasing activation-induced apoptosis in human T cells (14). To confirm these data, we activated PBMCs for 6 d with anti-CD3, before restimulation with PMA plus ionomycin for 16 h in the presence or absence of increasing concentrations of TGF-β1. CsA, which has been shown to effectively inhibit AICD (37), was used as a positive control. As illustrated in Fig. 1 A, activated PBMCs restimulated by addition of PMA plus ionomycin showed ∼50% death. In contrast, if the cells were stimulated in the presence of TGF-β1, the percentage of apoptotic cells was dramatically decreased.
Figure 1.

TGF-β1 decreases AICD in human peripheral blood T cells and murine T cell hybridomas. (A) PBMCs were activated for 6 d with OKT3 (100 ng/ml) and then restimulated with PMA (50 ng/ml) and ionomycin (1 μg/ml) (P+I) for 16 h with the indicated concentration of TGF-β1 or CsA (100 ng/ml). Viability was assessed by propidium iodide uptake and analyzed using a FACScan®. Apoptosis was confirmed by morphological assessment after staining with Hoechst 33342 at 10 μg/ml (not shown). (B) A1.1 or 2B4.11 T hybridoma cells were left unactivated (open symbols) or were activated (filled symbols) for 16 h with anti-CD3 antibody (2C11) in the presence of the indicated concentration of TGF-β1. Viability was assessed as in A. (C) A1.1 T hybridoma cells were cultured with medium alone or activated for 12 h with anti-CD3 antibody in the presence of the indicated concentrations of TGF-β1 (ng/ml) or CsA (100 ng/ml). DNA fragmentation associated with apoptosis was assessed by agarose gel electrophoresis.
To study the mechanism by which TGF-β1 inhibits AICD, we used the T cell hybridomas A1.1 and 2B4.11, two cell lines used extensively to examine AICD (17, 30). We first investigated the effect of TGF-β1 on AICD in these T cell hybridomas. As expected, TGF-β1 decreased anti-CD3–induced apoptosis in both cell types, as measured by propidium iodide uptake (Fig. 1 B) or DNA laddering (Fig. 1 C).
TGF-β1 Inhibits Activation-induced FasL Expression.
We demonstrated previously that AICD in both A1.1 and 2B4.11 T cell hybridomas proceeds via expression of FasL and subsequent Fas/FasL interaction (17). Therefore, we envisioned three nonexclusive possibilities for the mechanism of inhibition by TGF-β1: inhibition of FasL expression, inhibition of Fas expression, and/or inhibition of the Fas signaling pathway. To assess whether the inhibitory activity of TGF-β1 alters Fas or FasL gene expression, RNA was isolated and analyzed by semiquantitative RT-PCR. A1.1 T cell hybridomas constitutively expressed a low level of Fas mRNA, which increased approximately two- to threefold after activation, and TGF-β1 only slightly decreased Fas expression at high doses (10–100 ng/ml; Fig. 2 A). In contrast, activation-induced expression of FasL in A1.1 cells was greatly reduced by TGF-β1 at 0.1 ng/ml and completely prevented at higher concentrations (Fig. 2 A). This decrease in FasL mRNA was reflected in the biological activity of this molecule, since A1.1 cells that were stimulated with anti-CD3 antibody in the presence of TGF-β1 did not induce apoptosis in Fas+ target cells (Fig. 2 B). Addition of TGF-β1 to the cocultures of L1210-Fas target cells and activated A1.1 cells (6 h after activation), rather than to the A1.1 cells at the onset of the activation, failed to inhibit cytotoxicity (Fig. 2 B). These data suggest that TGF-β1 inhibits AICD by preventing FasL expression and not via a blockade in the Fas signaling pathway. To confirm the absence of effect of TGF-β1 on Fas signaling, we treated A1.1 cells with recombinant soluble FasL cross-linked with anti-Flag antibody (31) in the absence or presence of TGF-β1. As shown in Fig. 3, addition of TGF-β1 did not prevent soluble FasL–induced apoptosis in A1.1 cells.
Figure 2.

TGF-β1 inhibits activation-induced apoptosis by blocking FasL mRNA expression and functional activity. (A) TGF-β1 inhibits activation-induced FasL mRNA expression. RT-PCR analysis of total mRNA obtained from A1.1 cells incubated in medium alone or activated for 4 h by anti-CD3 antibodies in the presence or absence of increasing concentrations of TGF-β1 or CsA (100 ng/ml). mRNA was reverse transcribed by using oligo(dT) primers, and PCR amplification was performed using different numbers of cycles with the primers pairs indicated. The products were electrophoresed on 1.5% agarose gel and stained with ethidium bromide. (B) TGF-β1 inhibits AICD by modulating the expression of FasL functional activity but not by blocking the signaling through Fas receptor. A1.1 cells were activated at time 0 with coated anti-CD3 antibodies in the presence or absence of different concentrations of TGF-β1. The L1210 or L1210-Fas target cells were not added at this time. The T cell hybridomas were then cultured for 6 h to allow FasL expression, harvested, washed twice, and incubated for an additional 8 h with [3H]TdR-labeled L1210 or L1210-Fas target cells. In a parallel experiment, A1.1 cells were activated with anti-CD3 alone and TGF-β1 was added when A1.1 and target cells were mixed (T = +6 h). Percentage of DNA fragmentation was calculated as described in Materials and Methods.
Figure 3.

TGF-β1 does not block recombinant soluble FasL– induced apoptosis. A1.1 T cell hybridomas were preincubated for 1 h in the presence of TGF-β1 (10 ng/ml) and then treated with different doses of soluble recombinant human FasL (sFasL). 15 min after addition of soluble FasL, anti-Flag M2 antibody (1 μg/ml) was added to cross-link soluble FasL. After 12 h incubation, percentage of apoptosis was assessed by propidium iodide uptake and analyzed using a FACScan®.
TGF-β1 Inhibits c-myc Expression in T Cell Hybridomas.
TGF-β1 has been reported to downregulate constitutive and inducible c-Myc expression in two constitutively activated murine T clones (29). To determine whether TGF-β1 regulates c-myc expression in T cell hybridomas, we analyzed the mRNA expression of the c-myc gene by RT-PCR. TGF-β1 induced a dose-dependent inhibition of constitutive c-myc expression (Fig. 4 A), whereas CsA, which also completely blocked FasL mRNA expression (Fig. 2 A), did not inhibit c-myc expression. This effect was confirmed by analysis of c-Myc protein expression after treatment of A1.1 cells for 8 h in the presence of 1 ng/ml TGF-β1 (Fig. 4 B, lanes 1 and 2).
Figure 4.

TGF-β1 inhibits endogenous c-myc mRNA and protein expression but not ectopic expression of the chimeric Myc-ER protein. (A) TGF-β1 inhibits c-myc mRNA expression. RT-PCR analysis of total mRNA obtained from A1.1 cells incubated in medium alone or activated for 4 h by anti-CD3 antibody with the indicated concentration of TGF-β1 or CsA (100 ng/ml). mRNA was reverse transcribed by using oligo(dT) primers, and PCR amplification was performed using different numbers of cycles with the primers pairs indicated. The products were resolved by agarose gel electrophoresis. (B) TGF-β1 inhibits constitutive endogenous c-Myc protein expression but not ectopic expression of the chimeric Myc-ER protein. Total cell extracts were prepared from A1.1 or A1.1 Myc-ER cells treated for 8 h in the presence or absence of TGF-β1 (1 ng/ml). Samples were analyzed by immunoblot analysis using anti–human c-Myc antibody.
Regulation of FasL Expression and AICD by TGF-β1 Is Mediated by c-Myc.
We next addressed whether the inhibition of FasL expression by TGF-β1 was a consequence of c-myc downregulation. To test the role of c-Myc on FasL inhibition induced by TGF-β1, we prepared stable transfectants constitutively expressing a chimeric molecule composed of c-Myc and the estrogen-binding domain of the estrogen receptor (38; Myc-ER). In these A1.1 Myc-ER cells, the activity of the chimeric Myc protein is dependent on the availability of exogenous 4-OHT (35). The expression of Myc-ER and endogenous c-Myc protein before and after treatment of A1.1 Myc-ER cells with TGF-β1 was then assessed by immunoblotting with anti–c-Myc antibody. As shown in Fig. 4 B (lanes 3 and 4), A1.1 Myc-ER cells expressed both Myc-ER and endogenous c-Myc protein, and after TGF-β1 treatment the endogenous c-Myc protein completely disappeared while the chimeric Myc-ER protein remained.
A1.1 or A1.1 Myc-ER cells were then preincubated in the presence or absence of 4-OHT (50 nM) for 4 h to induce Myc-ER function (35), and activation-induced FasL mRNA expression was analyzed by RT-PCR. Results are shown in Fig. 5 A. In all cases, treatment with anti-CD3 induced the expression of FasL (lanes 2) versus unstimulated cells (lanes 1). In the absence of Myc-ER function (A1.1 cells with or without 4-OHT, or A1.1 Myc-ER without 4-OHT), TGF-β1 treatment inhibited expression of FasL in the anti-CD3–stimulated cells (lanes 3). Strikingly, however, functional Myc-ER (A1.1 Myc-ER cells with 4-OHT) permitted expression of FasL in anti-CD3– stimulated cells even in the presence of TGF-β1. Thus, the ability of TGF-β1 to inhibit c-Myc expression is directly responsible for its ability to block FasL expression in these cells. In contrast, CsA, which inhibits FasL expression by inhibition of nuclear factor of activated T cells (NF-AT) activity (39, 40), was effective in inhibiting A1.1 Myc-ER cells even in the presence of 4-OHT (Fig. 5 A).
Figure 5.

Ectopic expression of chimeric Myc-ER protein prevents TGF-β1–mediated inhibition of FasL mRNA and subsequent AICD. (A) Functional Myc-ER interferes with the inhibitory effect of TGF-β1 on activation-induced FasL expression in A1.1 cells. A1.1 or A1.1 Myc-ER cells exposed to 4-OHT (50 nM) were first preincubated for 4 h with the drug, which was then also present during subsequent culture. Cells were then incubated in medium alone (lane 1) or activated for 4 h with anti-CD3 antibodies (lanes 2, 3, and 4). 1 ng/ml TGF-β1 (lane 3) or 100 ng/ ml CsA (lane 4) was added to some cultures. FasL expression was then assessed by RT-PCR. (B) Ectopic expression of a chimeric Myc-ER protein prevents AICD after TGF-β1 treatment. A1.1 or A1.1 Myc-ER T hybridoma cells were first preincubated for 4 h in the presence or absence of 4-OHT (50 nM) and then activated for 16 h with anti-CD3 antibodies with the indicated concentrations of TGF-β1 (1 ng/ml) or CsA (100 ng/ml). Cell death was assessed by propidium iodide uptake using a FACScan®.
The ability of c-Myc function to reverse the inhibitory activity of TGF-β1 on FasL expression also affected the regulation of AICD in A1.1 Myc-ER cells. In the absence of 4-OHT, TGF-β1 inhibited AICD in A1.1 Myc-ER cells. However, after addition of 50 nM 4-OHT, TGF-β1 was ineffective in inhibiting AICD in A1.1 Myc-ER cells (Fig. 5 B). No effect was seen of 4-OHT in the parental A1.1 cells. Thus, active Myc-ER reversed the inhibition of AICD by TGF-β1, most likely via activation-induced FasL expression. To further confirm these data, we cloned A1.1 Myc-ER cells and selected three clones which express the Myc-ER fusion protein (Fig. 6 A). TGF-β1 prevented AICD in the absence of 4-OHT. As with the “bulk” transfectants, addition of 4-OHT almost completely reversed the protective effect of TGF-β1 on AICD for all three Myc-ER clones but not for A1.1 parental cells (Fig. 6 B).
Figure 6.
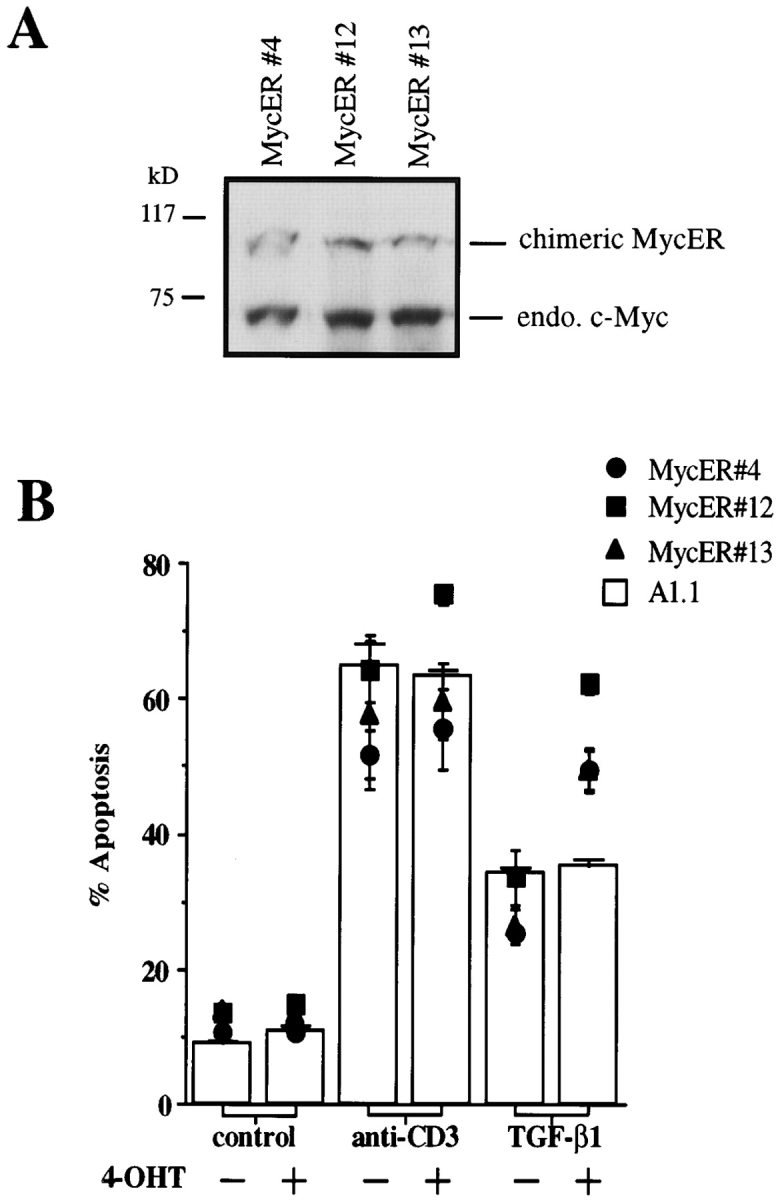
Ectopic expression of chimeric Myc-ER protein prevents TGF-β1–mediated inhibition of AICD in A1.1 T cell hybridomas. (A) Expression of Myc-ER fusion protein in A1.1 Myc-ER clones. Total cell extracts were prepared from three different A1.1 Myc-ER clones. Samples were analyzed by immunoblot analysis using anti–human c-Myc antibody. (B) Ectopic expression of a chimeric Myc-ER protein prevents AICD after TGF-β1 treatment. A1.1 or A1.1 Myc-ER clones were activated for 16 h with anti-CD3 antibodies with the indicated concentrations of TGF-β1 (1 ng/ml) in the presence or absence of 4-OHT (50 nM) added at time 0 of the activation. Cell death was assessed by propidium iodide uptake using a FACScan®.
Thus, when c-Myc activity is maintained, no inhibitory effect of TGF-β1 on either FasL expression or AICD in T cell hybridomas is observed. These results formally demonstrate that the ability of TGF-β1 to inhibit FasL expression and subsequent AICD is dependent on its ability to downregulate c-Myc.
TGF-β1–mediated Inhibition of c-Myc Does Not Block Activation-induced FasL Expression via Perturbation of the Cell Cycle.
In some models, TGF-β1 has been shown to cause cell cycle arrest in G1 by upregulation of CDK inhibitors (25) or by repression of Cdc25A, a CDK tyrosine phosphatase which activates CDK (26). Since Cdc25A is transcriptionally induced by c-Myc (41), we addressed whether TGF-β1–induced inhibition of c-Myc decreased FasL expression via cell cycle arrest. As shown in Fig. 7, addition of TGF-β1, ASc-myc, or NSc-myc oligonucleotides to A1.1 cells cultured in medium alone did not modify the proportion of cells in G0/G1, S, or G2/M phase of the cell cycle. When A1.1 cells were activated with anti-CD3 alone, live cells accumulated in G0/G1 phase with a decreased proportion of cells in S and G2/M phase at 24 h as described (30). Addition of TGF-β1, ASc-myc, or NSc-myc oligonucleotides did not significantly alter this profile (Fig. 7). Although TGF-β1 and ASc-myc oligonucleotides did not modify the proportion of cycling cells, they decreased AICD in A1.1 cells from 60 to 20% and 60 to 29%, respectively, after 12 h of culture (21; data not shown). DNA synthesis studies using [3H]TdR incorporation also demonstrated that TGF-β1 did not inhibit proliferation of unactivated A1.1 cells (26,678 ± 2,343 cpm in control cells vs. 24,310 ± 4,121 cpm in TGF-β1–treated cells), whereas [3H]TdR incorporation of activated A1.1 cells was almost completely inhibited with or without addition of TGF-β1 (1,625 ± 243 cpm in anti-CD3–treated cells vs. 2,111 ± 328 cpm in anti-CD3 plus TGF-β1–treated cells).
Figure 7.

TGF-β1 as well as ASc-myc oligonucleotides do not induce perturbation of the cell cycle in A1.1 T cell hybridomas. 106 A1.1 cells were untreated or activated with anti-CD3 in the presence of 5 μM ASc-myc or NSc-myc oligonucleotides added 4 h before activation or with TGF-β1 (10 ng/ml) added 1 h before activation. Cells were harvested at the indicated times, and cell cycle analysis was performed by propidium iodide staining after permeabilization with Triton X-100. The percentage of cells in G0/G1 (A), S (B), and G2/M (C) phase of the cell cycle was determined for each condition (data shown are means ± SD, n = 3).
Discussion
AICD in vivo has been proposed first to limit the expansion of an immune response by eliminating effector cells that are no longer needed, and second to eliminate potentially autoreactive T cells that have escaped thymic selection (15). Evidence from lpr or gld mice, deficient in functional Fas and FasL, respectively, suggested that Fas/FasL interactions are crucial for the regulation of these physiological processes. Experiments using antagonists of Fas/FasL interaction such as Fas–Fc fusion protein or a neutralizing anti-Fas mAb further demonstrated the direct involvement of these molecules in AICD of mature T cells (16–20). In this study, we have shown that TGF-β1, which is known to block the process of AICD in T cells (13, 14), does so by the selective inhibition of activation-induced FasL expression. This was demonstrated by analysis of FasL mRNA expression by RT-PCR and by functional assays. Indeed, many tumors, such as malignant astrocytoma, produce a high level of TGF-β1 (42) which might inhibit FasL expression on CD8+ T cells and therefore counteract their cytotoxic function.
Interestingly, there is also an inhibitory effect of TGF-β1 on perforin and granzyme B mRNA expression as well as serine esterase activity in CD8+ T cells, and this serves to block cytotoxic function of perforin-dependent killers (43, 44). Whether this represents a similar requirement for c-Myc in the expression of these important components of cytotoxic granules is not known.
In contrast to its effects on FasL expression, we observed that TGF-β1 does not significantly affect activation-induced Fas expression in T cell hybridomas. T cell activation induces the appearance of Fas on naive T cells and an increased expression on some T cell hybridomas (15, 17). In the latter, CsA does not inhibit the activation-induced increase in Fas mRNA expression, although it does partially inhibit cell surface Fas (39). Similar observations have been made for retinoic acid analogues, which inhibit FasL but not Fas expression (20, 45). Thus, Fas is regulated in T cells in a fundamentally different way from its ligand, and this includes the effect of TGF-β1. However, in contrast to T cells, progenitors of dendritic cells show reduced Fas expression when cultured with TGF-β1 (6). Whether or not this is related to a requirement for c-Myc or some other TGF-β1–regulated factors is not known.
In our studies, the inhibition of FasL by TGF-β1 in T cell hybridomas correlated with a decrease in c-myc mRNA levels. This is in agreement with previous studies showing that TGF-β1 is a potent inhibitor of c-Myc expression in various cell types and in particular in mature T cells (27–29, 46). Our experiments using ectopic expression of the chimeric Myc-ER protein have now formally shown that the inhibition of FasL induced by TGF-β1 is a direct consequence of its downregulation of c-Myc expression.
The c-myc oncogene has been implicated in the control of cell proliferation and differentiation, as well as neoplastic transformation (47). Overexpression or inappropriate expression of the c-myc gene has been found to promote apoptosis in fibroblasts (48, 49). Recently, Hueber et al. (23) reported that c-myc–induced apoptosis of serum-starved fibroblasts requires the expression and function of Fas and FasL. Although they showed that this was associated with an increased susceptibility to Fas-induced apoptosis, they did not rule out a role for c-Myc in driving expression of FasL. We demonstrated previously that c-Myc function is required for AICD in T cells, using c-myc antisense oligonucleotides (21) and dominant negative mutants of Myc or Max, which antagonize the functional Myc/Max heterodimer (22). Our finding that TGF-β1 inhibits FasL expression via downregulation of c-myc mRNA expression strongly suggests that the requirement for c-Myc during AICD may be at the level of FasL expression.
There is evidence that proliferating T lymphocytes are more susceptible to AICD during the S phase of their cell cycle (50). Although TGF-β1 causes cell cycle arrest in G1 in some models (24–26) and can also block constitutive c-Myc expression in T cell hybridomas, surprisingly we did not find a significant difference in cell cycle distribution when we compared cells stimulated for 3–24 h in the presence or absence of TGF-β1. This observation could be explained by the fact that residual c-Myc expression may be sufficient for cell cycle progression but not for FasL expression. Nevertheless, our finding argues against the possibility that TGF-β1 regulates FasL expression and subsequent AICD via perturbation of the cell cycle. This result is consistent with the finding that TGF-β1 promotes effector T cell expansion in association with IL-2 in murine Th2 clones (14).
In contrast to T cells, TGF-β1 promotes apoptosis in immature B cell lines such as WEHI 231 or CH31 cells (51–54). Apoptosis induced by TGF-β1 in these cells is preceded by a decline in c-Myc expression, and stabilization or ectopic expression of c-Myc prevented this cell death (52, 55). More recently, Arsura et al. (51) demonstrated that inhibition of c-Myc expression and subsequent apoptosis in B cells after TGF-β1 treatment were a consequence of the transcriptional activation of IκBα, which prevented the direct regulation of c-Myc expression by NF-κB. Whether this plays a role in TGF-β1–induced downregulation of c-Myc in T cells is unknown.
The relevance of the antiapoptotic effect of TGF-β1 might be associated with the observation that addition of this cytokine together with IL-2 not only blocked AICD but also enhanced effector T cell expansion and long-term T cell survival (13, 14). These results strongly suggest that TGF-β1 may be one of the cytokines involved in T cell memory generation. The persistence of memory T cells has been suggested to be dependent on periodic restimulation by specific or cross-reacting antigens (56, 57). TGF-β1 might be necessary for inhibiting FasL expression, thus permitting the generation and survival of memory T cells (15).
The roles played in the development and function of the immune system by c-Myc and FasL, under the control of TGF-β1, are likely to be diverse. TGF-β1–deficient mice have smaller spleens with less distinct white pulp and fewer Peyer's patches with less distinct germinal centers than those of normal littermates (58), and it is thus possible that this is a consequence of deregulated FasL expression, normally controlled by this cytokine.
Acknowledgments
The authors thank G. Wong for providing reagents, P. Golstein for L1210 and L1210-Fas cell lines, G.I. Evan for the pBABE puroMyc-ER G525R, and W. Force for assistance with retrovirus production. We also thank T. Lin and H. Beere for critical reading of the manuscript.
This work was supported in part by the Association pour la Recherche sur le Cancer (ARC) and the Human Frontier Science Program (to L. Genestier), and by National Institutes of Health grant GM52735 (to D.R. Green). This is publication #227 from the La Jolla Institute for Allergy and Immunology.
Footnotes
1 Abbreviations used in this paper: AICD, activation-induced cell death; AS, antisense; CDK, cyclin-dependent kinase; CsA, cyclosporine A; NF, nuclear factor; NS, nonsense; 4-OHT, 4-hydroxytamoxifen; RT, reverse transcription.
T. Brunner's present address is Division of Immunopathology, Institute for Pathology, University of Bern, 3010 Bern, Switzerland.
References
- 1.Massague J. The transforming growth factor-beta family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 2.Kasid A, Bell GI, Director EP. Effects of transforming growth factor-beta on human lymphokine-activated killer cell precursors. Autocrine inhibition of cellular proliferation and differentiation to immune killer cells. J Immunol. 1988;141:690–698. [PubMed] [Google Scholar]
- 3.Fontana A, Frei K, Bodmer S, Hofer E, Schreier MH, Palladino MA, Jr, Zinkernagel RM. Transforming growth factor-beta inhibits the generation of cytotoxic T cells in virus-infected mice. J Immunol. 1989;143:3230–3244. [PubMed] [Google Scholar]
- 4.Bright JJ, Kerr LD, Sriram S. TGF-beta inhibits IL-2-induced tyrosine phosphorylation and activation of Jak-1 and Stat 5 in T lymphocytes. J Immunol. 1997;159:175–183. [PubMed] [Google Scholar]
- 5.Moses HL, Yang EY, Pietenpol JA. TGF-beta stimulation and inhibition of cell proliferation: new mechanistic insights. Cell. 1990;63:245–247. doi: 10.1016/0092-8674(90)90155-8. [DOI] [PubMed] [Google Scholar]
- 6.Riedl E, Strobl H, Majdic O, Knapp W. TGF-beta 1 promotes in vitro generation of dendritic cells by protecting progenitor cells from apoptosis. J Immunol. 1997;158:1591–1597. [PubMed] [Google Scholar]
- 7.Swain SL, Huston G, Tonkonogy S, Weinberg A. Transforming growth factor-beta and IL-4 cause helper T cell precursors to develop into distinct effector helper cells that differ in lymphokine secretion pattern and cell surface phenotype. J Immunol. 1991;147:2991–3000. [PubMed] [Google Scholar]
- 8.Johns LD, Flanders KC, Ranges GE, Sriram S. Successful treatment of experimental allergic encephalomyelitis with transforming growth factor-beta 1. J Immunol. 1991;147:1792–1796. [PubMed] [Google Scholar]
- 9.Kuruvilla AP, Shah R, Hochwald GM, Liggitt HD, Palladino MA, Thorbecke GJ. Protective effect of transforming growth factor beta 1 on experimental autoimmune diseases in mice. Proc Natl Acad Sci USA. 1991;88:2918–2921. doi: 10.1073/pnas.88.7.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santambrogio L, Hochwald GM, Saxena B, Leu CH, Martz JE, Carlino JA, Ruddle NH, Palladino MA, Gold LI, Thorbecke GJ. Studies on the mechanisms by which transforming growth factor-beta (TGF-beta) protects against allergic encephalomyelitis. Antagonism between TGF-beta and tumor necrosis factor. J Immunol. 1993;151:1116–1127. [PubMed] [Google Scholar]
- 11.Lee HM, Rich S. Co-stimulation of T cell proliferation by transforming growth factor-beta 1. J Immunol. 1991;147:1127–1133. [PubMed] [Google Scholar]
- 12.Rich S, Van Nood N, Lee HM. Role of alpha 5 beta 1 integrin in TGF-beta 1-costimulated CD8+ T cell growth and apoptosis. J Immunol. 1996;157:2916–2923. [PubMed] [Google Scholar]
- 13.Cerwenka A, Kovar H, Majdic O, Holter W. Fas- and activation-induced apoptosis are reduced in human T cells preactivated in the presence of TGF-beta 1. J Immunol. 1996;156:459–464. [PubMed] [Google Scholar]
- 14.Zhang X, Giangreco L, Broome HE, Dargan CM, Swain SL. Control of CD4 effector fate: transforming growth factor beta 1 and interleukin 2 synergize to prevent apoptosis and promote effector expansion. J Exp Med. 1995;182:699–709. doi: 10.1084/jem.182.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lynch DH, Ramsdell F, Alderson MR. Fas and FasL in the homeostatic regulation of immune responses. Immunol Today. 1995;16:569–574. doi: 10.1016/0167-5699(95)80079-4. [DOI] [PubMed] [Google Scholar]
- 16.Alderson MR, Tough TW, Davis-Smith T, Braddy S, Falk B, Schooley KA, Goodwin RG, Smith CA, Ramsdell F, Lynch DH. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995;181:71–77. doi: 10.1084/jem.181.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, Martin SJ, Force WR, Lynch DH, Ware CF, et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 18.Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 19.Ju ST, Panka DJ, Cui H, Ettinger R, el-Khatib M, Sherr DH, Stanger BZ, Marshak-Rothstein A. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Mercep M, Ware CF, Ashwell JD. Fas and activation-induced Fas ligand mediate apoptosis of T cell hybridomas: inhibition of Fas ligand expression by retinoic acid and glucocorticoids. J Exp Med. 1995;181:1673–1682. doi: 10.1084/jem.181.5.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Y, Glynn JM, Guilbert LJ, Cotter TG, Bissonnette RP, Green DR. Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science. 1992;257:212–214. doi: 10.1126/science.1378649. [DOI] [PubMed] [Google Scholar]
- 22.Bissonnette RP, McGahon A, Mahboubi A, Green DR. Functional Myc-Max heterodimer is required for activation-induced apoptosis in T cell hybridomas. J Exp Med. 1994;180:2413–2418. doi: 10.1084/jem.180.6.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hueber AO, Zornig M, Lyon D, Suda T, Nagata S, Evan GI. Requirement for the CD95 receptor-ligand pathway in c-Myc-induced apoptosis. Science. 1997;278:1305–1309. doi: 10.1126/science.278.5341.1305. [DOI] [PubMed] [Google Scholar]
- 24.Ewen ME, Sluss HK, Whitehouse LL, Livingston DM. TGF beta inhibition of Cdk4 synthesis is linked to cell cycle arrest. Cell. 1993;74:1009–1020. doi: 10.1016/0092-8674(93)90723-4. [DOI] [PubMed] [Google Scholar]
- 25.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 26.Iavarone A, Massague J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-beta in cells lacking the CDK inhibitor p15. Nature. 1997;387:417–422. doi: 10.1038/387417a0. [DOI] [PubMed] [Google Scholar]
- 27.Coffey RJ, Jr, Bascom CC, Sipes NJ, Graves-Deal R, Weissman BE, Moses HL. Selective inhibition of growth-related gene expression in murine keratinocytes by transforming growth factor beta. Mol Cell Biol. 1988;8:3088–3093. doi: 10.1128/mcb.8.8.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pietenpol JA, Stein RW, Moran E, Yaciuk P, Schlegel R, Lyons RM, Pittelkow MR, Munger K, Howley PM, Moses HL. TGF-beta 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell. 1990;61:777–785. doi: 10.1016/0092-8674(90)90188-k. [DOI] [PubMed] [Google Scholar]
- 29.Ruegemer JJ, Ho SN, Augustine JA, Schlager JW, Bell MP, McKean DJ, Abraham RT. Regulatory effects of transforming growth factor-beta on IL-2- and IL-4-dependent T cell-cycle progression. J Immunol. 1990;144:1767–1776. [PubMed] [Google Scholar]
- 30.Ashwell JD, Cunningham RE, Noguchi PD, Hernandez D. Cell growth cycle block of T cell hybridomas upon activation with antigen. J Exp Med. 1987;165:173–194. doi: 10.1084/jem.165.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, Fontana A, Tschopp J. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med. 1998;187:1205–1213. doi: 10.1084/jem.187.8.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benhamou LE, Watanabe T, Kitamura D, Cazenave PA, Sarthou P. Signaling properties of anti-immunoglobulin–resistant variants of WEHI-231 B lymphoma cells. Eur J Immunol. 1994;24:1993–1999. doi: 10.1002/eji.1830240909. [DOI] [PubMed] [Google Scholar]
- 33.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Force WR, Cheung TC, Ware CF. Dominant negative mutants of TRAF3 reveal an important role for the coiled coil domains in cell death signaling by the lymphotoxin-beta receptor. J Biol Chem. 1997;272:30835–30840. doi: 10.1074/jbc.272.49.30835. [DOI] [PubMed] [Google Scholar]
- 35.Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 37.Shi YF, Sahai BM, Green DR. Cyclosporin A inhibits activation-induced cell death in T-cell hybridomas and thymocytes. Nature. 1989;339:625–626. doi: 10.1038/339625a0. [DOI] [PubMed] [Google Scholar]
- 38.Eilers M, Picard D, Yamamoto KR, Bishop JM. Chimaeras of myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature. 1989;340:66–68. doi: 10.1038/340066a0. [DOI] [PubMed] [Google Scholar]
- 39.Brunner T, Yoo NJ, LaFace D, Ware CF, Green DR. Activation-induced cell death in murine T cell hybridomas. Differential regulation of Fas (CD95) versus Fas ligand expression by cyclosporin A and FK506. Int Immunol. 1996;8:1017–1026. doi: 10.1093/intimm/8.7.1017. [DOI] [PubMed] [Google Scholar]
- 40.Latinis KM, Norian LA, Eliason SL, Koretzky GA. Two NFAT transcription factor binding sites participate in the regulation of CD95 (Fas) ligand expression in activated human T cells. J Biol Chem. 1997;272:31427–31434. doi: 10.1074/jbc.272.50.31427. [DOI] [PubMed] [Google Scholar]
- 41.Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature. 1996;382:511–517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- 42.Bodmer S, Strommer K, Frei K, Siepl C, de Tribolet N, Heid I, Fontana A. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J Immunol. 1989;143:3222–3229. [PubMed] [Google Scholar]
- 43.Inge TH, McCoy KM, Susskind BM, Barrett SK, Zhao G, Bear HD. Immunomodulatory effects of transforming growth factor-beta on T lymphocytes. Induction of CD8 expression in the CTLL-2 cell line and in normal thymocytes. J Immunol. 1992;148:3847–3856. [PubMed] [Google Scholar]
- 44.Smyth MJ, Strobl SL, Young HA, Ortaldo JR, Ochoa AC. Regulation of lymphokine-activated killer activity and pore-forming protein gene expression in human peripheral blood CD8+ T lymphocytes. Inhibition by transforming growth factor-beta. J Immunol. 1991;146:3289–3297. [PubMed] [Google Scholar]
- 45.Bissonnette RP, Brunner T, Lazarchik SB, Yoo NJ, Boehm MF, Green DR, Heyman RA. 9-cis retinoic acid inhibition of activation-induced apoptosis is mediated via regulation of fas ligand and requires retinoic acid receptor and retinoid X receptor activation. Mol Cell Biol. 1995;15:5576–5585. doi: 10.1128/mcb.15.10.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Green DR, Mahboubi A, Nishioka W, Oja S, Echeverri F, Shi Y, Glynn J, Yang Y, Ashwell J, Bissonnette R. Promotion and inhibition of activation-induced apoptosis in T-cell hybridomas by oncogenes and related signals. Immunol Rev. 1994;142:321–342. doi: 10.1111/j.1600-065x.1994.tb00895.x. [DOI] [PubMed] [Google Scholar]
- 47.Luscher B, Eisenman RN. New light on Myc and Myb. Part I. Myc. Genes Dev. 1990;4:2025–2035. doi: 10.1101/gad.4.12a.2025. [DOI] [PubMed] [Google Scholar]
- 48.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 49.Klefstrom J, Arighi E, Littlewood T, Jäättel M, Saksela E, Evan GI, Alitalo K. Induction of TNF-sensitive cellular phenotype by c-Myc involves p53 and impaired NF-kappaB activation. EMBO (Eur Mol Biol Organ) J. 1997;16:7382–7392. doi: 10.1093/emboj/16.24.7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boehme SA, Lenardo MJ. Propriocidal apoptosis of mature T lymphocytes occurs at S phase of the cell cycle. Eur J Immunol. 1993;23:1552–1560. doi: 10.1002/eji.1830230724. [DOI] [PubMed] [Google Scholar]
- 51.Arsura M, Wu M, Sonenshein GE. TGF beta 1 inhibits NF-kappa B/Rel activity inducing apoptosis of B cells: transcriptional activation of I kappa B alpha. Immunity. 1996;5:31–40. doi: 10.1016/s1074-7613(00)80307-6. [DOI] [PubMed] [Google Scholar]
- 52.Fischer G, Kent SC, Joseph L, Green DR, Scott DW. Lymphoma models for B cell activation and tolerance. X. Anti-mu–mediated growth arrest and apoptosis of murine B cell lymphomas is prevented by the stabilization of myc. J Exp Med. 1994;179:221–228. doi: 10.1084/jem.179.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sonenshein GE. Down-modulation of c-myc expression induces apoptosis of B lymphocyte models of tolerance via clonal deletion. J Immunol. 1997;158:1994–1997. [PubMed] [Google Scholar]
- 54.Warner GL, Ludlow JW, Nelson DA, Gaur A, Scott DW. Anti-immunoglobulin treatment of murine B-cell lymphomas induces active transforming growth factor beta but pRB hypophosphorylation is transforming growth factor beta independent. Cell Growth Differ. 1992;3:175–181. [PubMed] [Google Scholar]
- 55.Wu M, Arsura M, Bellas RE, FitzGerald MJ, Lee H, Schauer SL, Sherr DH, Sonenshein GE. Inhibition of c-myc expression induces apoptosis of WEHI 231 murine B cells. Mol Cell Biol. 1996;16:5015–5025. doi: 10.1128/mcb.16.9.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beverley PC. Is T-cell memory maintained by crossreactive stimulation? . Immunol Today. 1990;11:203–205. doi: 10.1016/0167-5699(90)90083-l. [DOI] [PubMed] [Google Scholar]
- 57.Gray D, Matzinger P. T cell memory is short-lived in the absence of antigen. J Exp Med. 1991;174:969–974. doi: 10.1084/jem.174.5.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]