

Abstract
Pathogenic effector T cells in experimental autoimmune uveitis (EAU) are T helper type 1–like, and interleukin (IL)-12 is required for their generation and function. Therefore, we expected that IL-12 administration would have disease-enhancing effects. Mice were immunized with a uveitogenic regimen of the retinal antigen interphotoreceptor retinoid-binding protein, treated with IL-12 (100 ng/d for 5 d), and EAU was assessed by histopathology. Unexpectedly, IL-12 treatment failed to enhance EAU in resistant strains and downregulated disease in susceptible strains. Only treatment during the first, but not during the second, week after immunization was consistently protective. High levels of interferon γ (IFN-γ) were present in the serum during IL-12 treatment, but subsequent antigen-specific IFN-γ production in protected mice was diminished, as were IL-5 production, lymph node cell proliferation, and serum antibody levels. Treated mice had fewer cells and evidence of enhanced apoptosis in the draining lymph nodes. Unlike wild-type mice, IFN-γ–deficient, inducible nitric oxide synthase (iNOS)-deficient, and Bcl-2lck transgenic mice were poorly protected by IL-12, whereas IL-10–deficient mice were protected. We conclude that administration of IL-12 aborts disease by curtailing development of uveitogenic effector T cells. The data are compatible with the interpretation that IL-12 induces systemic hyperinduction of IFN-γ, causing activation of iNOS and production of NO, which mediates protection at least in part by triggering Bcl-2 regulated apoptotic deletion of the antigen-specific T cells as they are being primed.
Keywords: interleukin 12, apoptosis, autoimmune disease, uveitis, T helper type 1
Experimental autoimmune uveitis (EAU)1 is an organ-specific, T cell–mediated disease that is characterized by inflammation and subsequent destruction of the neural retina and related tissues, resulting in blindness. EAU can be induced by immunization with one of several retinal antigens in adjuvant in rodents and nonhuman primates, or by adoptive transfer of retinal-specific CD4+ T cells between syngeneic rodents (1–4). The pathology of EAU in the mouse model closely resembles that of human uveitic diseases of putative autoimmune etiology (2). Therefore, the study of immunological mechanisms affecting EAU can help to understand human uveitis as well as other organ-specific, T cell–mediated autoimmune diseases.
IL-12 is a heterodimeric protein composed of two disulfide-linked subunits of 40 kD (p40) and 35 kD (p35) (5, 6), and is secreted predominantly from APCs in response to T cell engagement of the MHC class II and CD40 molecules (7, 8). Once secreted, IL-12 induces the release of IFN-γ from NK cells and T cells and augments cell-mediated immune responses in vitro and in vivo (9–15). IL-12 also regulates T cell–dependent immune responses by inducing differentiation toward the Th1 pathway (16, 17) and by priming T cells for high IFN-γ production (18–20). Th1 cells characteristically produce IFN-γ, lymphotoxin, and IL-2, increase opsonizing antibody levels, and promote cell-mediated immune reactions such as delayed hypersensitivity. This is in contrast to Th2 cells, which produce IL-4, IL-5, IL-10, and IL-13 and are involved in allergic responses (21).
We wished to investigate the effects of IL-12 on EAU induction and expression. Results from our laboratory suggest that the pathogenesis of EAU is associated with a Th1 response. Susceptible strains of mice and rats characteristically mount a Th1-dominated response to the uveitogen and resistant strains do not (22, 23). Furthermore, IL-12 is necessary to generate the functional uveitogenic effector T cell (24). Because IL-12 promotes the differentiation of the antigen-specific lymphocytes toward the Th1 pathway, we hypothesized that potentiating the Th1 response might overcome resistance in some nonsusceptible strains of mice. Unexpectedly, IL-12 administration during the first week after uveitogenic immunization consistently suppressed, rather than exacerbated, EAU in all tested strains of mice. This was accompanied by an overall reduction in immune responsiveness to the immunizing uveitogenic antigen and upregulation of apoptosis in the lymph nodes. Experiments with gene knockout (KO) and transgenic mice supported the interpretation that IL-12, through IFN-γ–driven induction of nitric oxide (NO), inhibits priming of antigen-specific lymphocytes at least in part through Bcl-2–regulated apoptosis, thus curtailing generation of uveitogenic effector cells and resulting in protection from disease.
Materials and Methods
Mice.
C57Bl/6, BALB/c, A/J, AKR, DBA/1, and B10.BR female mice were purchased from The Jackson Laboratory. Gene-targeted stock was bred at the National Institutes of Health animal facility. Mice with a targeted disruption of the IFN-γ gene (GKO) were developed, screened, and back-crossed for eight generations onto the C57Bl/6 background by Dalton et al. (25), and were obtained from Genentech. Mice with a targeted disruption of the IL-10 gene (10KO) were developed by W. Müller (Köln, Germany), screened, and were back-crossed for eight generations onto the C57Bl/6 background by Renate Morawetz (National Institute of Allergy and Infectious Diseases). Inducible nitric oxide synthase– deficient (iNOS KO) mice were bred from the original stock developed by John MacMicking and Carl Nathan (Cornell University Medical College, New York) and John Mudgett (Merck Research Laboratories, Rahway, NJ) (26). These mice are hybrids of C57Bl/6 × 129. Wild-type C57Bl/6 × 129 control mice were purchased from The Jackson Laboratory. Bcl-2 transgenic mice expressing human Bcl-2 under control of the lck promoter were bred from the stock developed by the group of Korsmeyer and co-workers (Washington University School of Medicine, St. Louis, MO [27]) and were used as B6 × C3H hybrids. Control mice of the same genotype were purchased from The Jackson Laboratory. All animals were housed under specific pathogen–free conditions, were given water and chow ad libitum, and were used between 8 wk and 6 mo of age. The care and use of the animals was in compliance with institutional guidelines.
Antigen and Reagents.
Interphotoreceptor retinoid-binding protein (IRBP) was isolated from bovine retinas by Con A–Sepharose affinity chromatography and fast performance liquid chromatography as described previously (28). Pertussis toxin (PTX) and CFA were purchased from Sigma Chemical Co. Mycobacterium tuberculosis strain H37RA was purchased from Difco Laboratories, Inc. Murine recombinant IL-12 was generously provided by M.K. Gately of Hoffman-LaRoche (Nutley, NJ).
Immunization and IL-12 Administration.
Mice were immunized subcutaneously in the thighs and base of tail with 100–150 μg IRBP in 0.2 ml emulsion 1:1 vol/vol with CFA containing 2.5 mg/ml M. tuberculosis. At the same time, mice were injected intraperitoneally with 1 μg PTX in 0.1 ml as an additional adjuvant. In experiments with 10KO mice, the M. tuberculosis was decreased to 1 mg/ml, because higher concentrations of bacteria increased fatalities, and the concentration of PTX was raised to 1.5–2.0 μg/mouse. IL-12–treated mice were injected intraperitoneally with graded doses of murine recombinant IL-12, as specified, for five consecutive days either early (days 0–4) or late (days 7–11) after immunization (day 0).
Histopathology and Scoring of EAU.
Whole eyes were collected and prepared for histopathological evaluation at the termination of an experiment (days 17–20 after immunization for 10KO experiments or days 21–22 for all other experiments). The eyes were immersed for 1 h in 4% phosphate-buffered glutaraldehyde and then transferred into 10% phosphate-buffered formaldehyde until processing. Fixed and dehydrated tissue was embedded in methacrylate, and 4–6-μm sections were cut through the pupillary-optic nerve plane. Sections were stained by hematoxylin and eosin. Presence or absence of disease was evaluated in a masked fashion by examining six sections cut at different levels for each eye. Severity of EAU was scored on a scale of 0 (no disease) to 4 (maximum disease) in half-point increments, according to a semiquantitative system described previously (29), which takes into account lesion type, size, and number. In brief, the minimal criterion to score an eye as positive by histopathology was inflammatory cell infiltration of the ciliary body, choroid, or retina (EAU grade 0.5). Progressively higher grades were assigned for presence of discrete lesions in the tissue such as vasculitis, granuloma formation, retinal folding and/or detachment, photoreceptor damage, etc.
Delayed Type Hypersensitivity.
2 d before the termination of an experiment, mice received 10 μg of IRBP in 10 μl intradermally into the pinna of one ear. The other ear was injected similarly, but with PBS. Ear swelling was measured at the termination of the experiment 48 h later with a spring-loaded micrometer. Delayed type hypersensitivity (DTH) results are expressed as antigen-specific swelling, calculated as the difference between the thickness of the IRBP-injected ear and the PBS-injected ear.
Lymphocyte Proliferation.
Draining lymph nodes, the inguinals and iliacs, were collected and pooled within each group at the termination of an experiment (17–22 d after immunization). Triplicate cultures of 5 × 105 cells/well were stimulated with 30 μg/ml IRBP in 96-well round-bottomed plates in RPMI supplemented with 2-ME, glutamine, nonessential amino acids, sodium pyruvate, and antibiotics as described (1), 1% fresh-frozen normal mouse serum, and 20 mg/ml α-methyl mannopyranoside (to neutralize any possible traces of Con A, which is used in the initial stages of IRBP purification) (Sigma Chemical Co.). The cultures were incubated for 60 h and were pulsed with [3H]thymidine (1.0 μCi/10 μl per well) for the last 18 h.
Determination of Lymphokine Titers.
Draining lymph node cells harvested 21 d after immunization were cultured as for the proliferation assay above, except that double the number of cells per well (106) were stimulated with 50 μg/ml IRBP. Supernatants were collected for cytokine analysis after 48 h. Blood for determination of IFN-γ serum titers was collected from the tail vein. All samples were kept at −70°C until being assayed. IFN-γ, IL-4, IL-5, IL-6, and IL-10 were measured by ELISA using antibody pairs from PharMingen essentially as described previously (30). ELISA detection kits from Endogen, Inc. were used to measure TNF-α and IL-10 in some experiments.
Detection of Apoptosis.
IRBP-immunized and naive mice were treated with 100 ng/d of IL-12. Lymph nodes from treated and untreated mice were collected on the specified days 24 h after the last IL-12 injection, and were fixed for 18 h in 10% neutral buffered formalin. Fixed tissue was paraffin-embedded, sectioned, and stained for apoptosis using the in situ TUNEL staining kit from Oncor, Inc., as per the manufacturer's instructions. TUNEL-positive cells were counted under the microscope. Sections were scored in a blind fashion. For each lymph node section, the entire section was scanned in consecutive fields, and the average number of cells per field was calculated. Four to seven sections originating from two to three draining lymph nodes were evaluated per mouse.
Measurement of Antigen-specific IgG Antibody Isotypes.
Serum levels of anti-IRBP IgG2a and IgG1 subclasses were determined by ELISA in sera collected 21 d after immunization, as described previously for another antigen (31). In brief, 96-well microtiter plates (Costar Corp.) were coated with IRBP (1 μg/ml). After blocking the plates with BSA (Sigma Chemical Co.) and an overnight incubation with serum samples, the plates were developed using horseradish peroxidase–conjugated goat anti-IgG subclass– specific antibodies (PharMingen). The concentration of anti-IRBP antibody was estimated using standard curves constructed by coating wells with anti-Ig antibody and by adding polyclonal Ig standards of the pertinent isotype.
Reproducibility and Statistical Analysis.
Experiments were repeated at least twice, and usually three or more times. Figures show data compiled from several experiments, or from a representative experiment, as specified. Statistical analysis of EAU scores was by Snedecor and Cochran's test for linear trend in proportions (nonparametric, frequency-based) (32). Each mouse (average of both eyes) was treated as one statistical event. Antibody titers and TUNEL staining data were analyzed using the independent t test. Probability values of ≤0.05 were considered significant.
Results
IL-12 Does Not Enhance EAU in Resistant Mouse Strains, and Prevents EAU in Susceptible Strains.
We have shown previously that susceptibility to EAU in rodents is associated with a Th1-dominant response, whereas resistance is associated with Th1-low response to the uveitogenic antigen (22, 23). Therefore, we assumed that administration of IL-12 would enhance susceptibility in at least some EAU-resistant mouse strains. Several mouse strains of different genetic background and MHC, whose susceptibility to EAU was characterized previously (23, 33), were injected with 100 ng/d of IL-12 for the first 5 d after immunization with IRBP, as described in Materials and Methods. After IL-12 treatment, the resistant BALB/c strain remained resistant to EAU. The moderately susceptible C57Bl/6 and minimally susceptible DBA.1 were completely protected. The highly susceptible B10.BR had strongly reduced disease scores (Table I). In four subsequent experiments using the C57Bl/6 strain, it was determined that only early treatment (days 0–4 relative to immunization), but not late treatment (days 7–11), significantly decreased the incidence and severity of EAU (P < 0.006) (Fig. 1 A). Both incidence and severity of disease in controls were typical of the C57Bl/6 strain, as seen by us previously (33). In a series of experiments designed to determine the dose–response of the protective effect, graded doses of IL-12 (100, 10, and 1 ng/d) were administered on days 0–4 (Fig. 1 B). A clear dose–response was apparent, in which the protective effect titered out over two orders of magnitude of IL-12 concentrations. None of the tested doses resulted in an enhancement of disease scores.
Table I.
IL-12 Administration Protects from EAU
| Animal | BALB/c (H-2d) | C57Bl/6 (H-2b) | DBA/1 (H-2q) | B10.BR (H-2k) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IL-12 | None | IL-12 | None | IL-12 | None | IL-12 | None | |||||||||
| 1 | 0* | 0 | 0 | 0.5 | 0 | 0 | 0.5 | 1.25 | ||||||||
| 2 | 0 | 0 | 0 | 0.9 | 0 | 0.75 | 0.25 | 0.5 | ||||||||
| 3 | 0 | 0 | 0 | 0.5 | 0 | 0.25 | 0.25 | 3 | ||||||||
| 4 | 0 | 0 | 0 | 0.6 | 0 | 2 | 0 | 3 | ||||||||
| 5 | 0 | 0 | 0 | 0.25 | 0 | 0 | 0.5 | 2.5 | ||||||||
| Average score | 0 | 0 | 0 | 0.6 | 0 | 0.6 | 0.3 | 2.1 | ||||||||
| Incidence | 0/5 | 0/5 | 0/5 | 5/5 | 0/5 | 3/5 | 4/5 | 5/5 | ||||||||
EAU score (average of both eyes).
Figure 1.


C57Bl/6 mice are protected from EAU by IL-12 administered early, but not late, after immunization. C57Bl/6 mice were immunized with IRBP on day 0 and were treated (or not) with recombinant murine IL-12, as indicated. (A) Timing of treatment: after immunization, mice were given daily injections of 100 ng of IL-12 early (days 0–4), late (days 7–11), or were left untreated (None). (B) Dose–response. On days 0–4 after immunization mice were given daily injections of graded doses of IL-12; high dose (100 ng), intermediate (10 ng), or low dose (1 ng), or were left untreated (None). Eyes were harvested for histopathology on days 21–22 and were graded on a scale of 0 (no disease) to 4 (maximal disease) in half-point increments. Each point is one mouse (average of both eyes). The average of each group is denoted by a horizontal bar. The data are compiled from six experiments (A), and three experiments (B).
IL-12 Administration Induces High Levels of Systemic IFN-γ.
In a previous study, we noted that neutralization of systemic IFN-γ in mice had an EAU-exacerbating effect, and augmentation of systemic IFN-γ had a suppressive effect (34). Because IL-12 induces IFN-γ, we hypothesized that the protective effect of IL-12 might be connected to a systemic upregulation of IFN-γ. Analysis of sera from IL-12– treated mice showed that a single injection of 100 ng of IL-12 was followed by an increase in serum IFN-γ that was first detected at 9 h, and still remained elevated at 24 h (Fig. 2 A). Because IL-12 treatment was administered in the EAU experiments every 24 h, IFN-γ serum titers of mice treated with IL-12 would be expected to remain continuously elevated throughout the 5-d treatment. This was supported by another series of experiments, where mice given a uveitogenic immunization of IRBP were bled 12 h after the first and the last IL-12 injections. IL-12–treated groups, irrespective of the timing of the treatment (early versus late), had nanogram quantities of IFN-γ in the serum, whereas no IFN-γ could be detected in the serum of untreated controls (Fig. 2 B). A dose–response was apparent, with mice treated with the highest dose of IL-12 showing the highest serum IFN-γ titers (Fig. 2 C).
Figure 2.

Systemic production of IFN-γ under IL-12 treatment. (A) After a single injection of IL-12. Naive C57Bl/6 mice were injected with 100 ng of IL-12 at 0 h. Serum was collected at the indicated times after IL-12 injection and serum IFN-γ was measured by specific ELISA. (B) Timing. Mice received 100 ng IL-12 injections early (days 0–4) or late (days 7–10) or were left untreated (None). (C) Dose– response. On days 0–4 after immunization mice received graded doses of IL-12; high dose (100 ng/d), intermediate dose (10 ng/ d), low dose (1 ng/d), or were left untreated (None). Blood was collected from the tail vein 12 h after their first and last IL-12 injections. The sera were pooled within each group (three to six mice) and were analyzed for IFN-γ by ELISA.
IL-12 Administration Does Not Protect IFN-γ–deficient Mice From EAU.
To test the hypothesis that elevated IFN-γ serum titers were in fact connected to the protective effect of IL-12, we used GKO mice on the C57Bl/6 background. GKO mice and their wild-type littermates were given a uveitogenic immunization of IRBP and were treated with 100 ng/d of IL-12 on days 0–4. IL-12 treatment did not protect GKO mice from developing EAU, although it did protect the wild-type littermates (P < 0.009) (Fig. 3). The incidence and severity of EAU in the GKO mice were similar to our previous observations (35).
Figure 3.

IL-12 treatment fails to protect IFN-γ–deficient mice from EAU. Mice were immunized with IRBP on day 0 and received daily injections of 100 ng of IL-12 (days 0–4) or were left untreated (None). Eyes for EAU evaluation were harvested for histopathology on day 21 and were graded on a scale of 0 (no disease) to 4 (maximal disease) in half-point increments. Each point is one mouse (average of both eyes). The average of each group is denoted by a horizontal bar. Data are compiled from three experiments.
In addition to upregulating IFN-γ, IL-12 has been reported to upregulate IL-10 (20, 36). Because IL-10 has a suppressive effect on EAU (30), it was necessary to address the possibility that protection from EAU could at least in part be due to upregulation of IL-10. Therefore, we treated IRBP-immunized 10KO mice and their littermates with 100 ng/d of IL-12 on days 0–4 after immunization. Histopathology of eyes collected 21 d later showed that IL-10– deficient mice were protected equally to their wild-type littermates, indicating that the protective effect of IL-12 is independent of IL-10 (data not shown). Interestingly, the 10KO mice treated with IL-12 had four times as much circulating IFN-γ as their wild-type littermates during treatment (14.3 versus 3.6 ng/ml, respectively). This was in line with the previously described suppressive effect of IL-10 on IFN-γ, an effect which is, of course, absent in 10KO mice (37, 38).
Antigen-specific Production of Both IFN-γ and IL-5 Is Reduced in Mice Treated Early with IL-12.
Antigen-specific IFN-γ production is considered indicative of the Th1 response, whereas antigen-specific IL-4 and IL-5 production can be used to assess the Th2 response. Mice were immunized for EAU induction and were treated with the protective, or the nonprotective, IL-12 regimen. Draining lymph node cells were collected on day 21 and were stimulated in culture with IRBP. Supernatants were assayed for lymphokine content by ELISA as described in Materials and Methods. The protected early treatment group, but not the unprotected late treatment group, consistently had strongly decreased production of IFN-γ to IRBP in culture, which showed a clear dose–response (Fig. 4, A and B). The same mice also showed reduced IRBP-specific production of IL-5, albeit to a somewhat lesser extent than of IFN-γ. Again, a dose–response was apparent (Fig. 4, C and D). Antigen-specific TNF-α and IL-10 production did not differ significantly between the two IL-12 treatment groups and untreated controls, and IL-4 was not detectable by ELISA in any of the supernatants (data not shown).
Figure 4.

Antigen-specific production of IFN-γ (A and B) and IL-5 (C and D) in mice treated with IL-12. (A) Timing. IFN-γ production after injections of IL-12 early (days 0–4) or late (days 7–11) or untreated (None). (B) Dose–response. IFN-γ production after administration of graded doses of IL-12 on days 0–4. (C) Timing. IL-5 production by the mice described in A. (D) Dose–response. IL-5 production by the mice described in B. Draining lymph node cells were collected on day 21 and were pooled within each group. Cultures were stimulated with IRBP (50 μg/ml) and supernatants collected at 48 h were assayed by ELISA. Data represent an average of four experiments, each normalized to its control group to compensate for interexperiment variation. Shown is the percentage of response relative to control ± standard deviation.
IL-12 Treatment Decreases Antigen-specific DTH and Antigen-specific Proliferation.
19 d after uveitogenic immunization IL-12–treated and control C57Bl/6 mice were challenged for DTH response by ear assay. 48 h later the ear swelling was measured and specific DTH responses were calculated. IL-12–treated mice had decreased DTH responses, with the early treatment group having the lowest scores (P vs. untreated < 0.003) (Fig. 5). The reduction of scores in the late treatment group was not significantly different from control (P < 0.13). Statistical significance notwithstanding, the strong reduction of disease scores contrasts with the milder reduction of DTH. This is observed consistently in our experiments with whole bovine IRBP as immunogen. While disease is, by definition, a manifestation of the response to the conserved autologous epitopes, DTH represents the sum of the response to autologous and to immunodominant foreign epitopes. The more restricted self-reactive repertoire may be easier to inhibit than the stronger response to the xenogeneic epitopes, which can explain the more dramatic effect on disease than on DTH.
Figure 5.

DTH is reduced in IL-12–treated mice. Mice received intradermal injections of 10 μg IRBP in one ear and PBS in the other ear 2 d before the termination of an experiment. After 48 h, ear swelling was measured. DTH results are expressed as antigen-specific swelling, calculated as the difference between the thickness of the IRBP-injected ear and the PBS-injected ear. Each point of the graph represents an individual mouse. The data are compiled from four experiments. The means are shown as horizontal bars.
Draining lymph nodes of IRBP-immunized, IL-12– treated mice were cultured with IRBP. Lymph node cells of mice that received early IL-12 treatment exhibited a suppression of IRBP-specific proliferation, which was clearly dose–dependent (Fig. 6, A and B). In contrast, lymph node cell proliferation of GKO mice was much less amenable to inhibition by early IL-12 treatment, in keeping with their lack of protection from EAU. In a representative experiment, proliferation of wild-type lymph node cells was reduced by half (from 122,000 to 63,000 cpm), whereas proliferation of GKO cells was reduced by only 12% (from 207,000 to 181,000 cpm).
Figure 6.
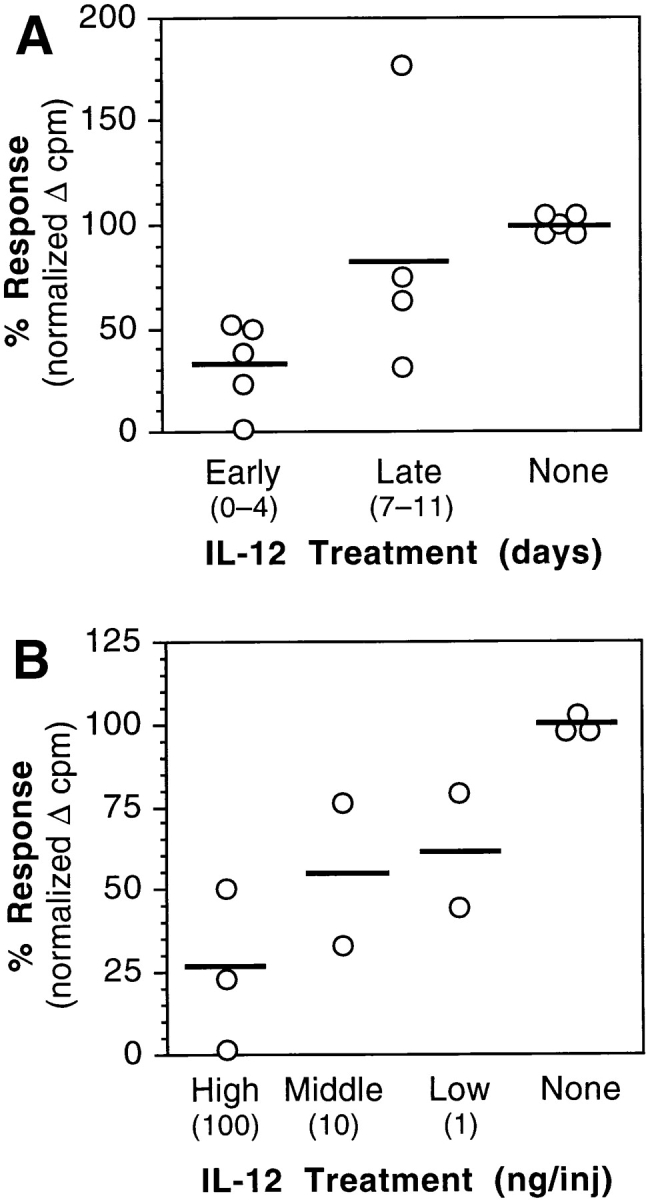
IRBP-specific in vitro proliferation is reduced in lymph node cultures of IL-12– treated mice. (A) Timing. C57Bl/6 mice received a high dose of IL-12 (100 ng/d) either early (days 0–4) or late (days 7– 11) after immunization with IRBP. (B) Dose–response. C57Bl/6 mice received a high dose (100 ng/d), intermediate dose (10 ng/d), or low dose (1 ng/d) of IL-12 on days 0–4 after immunization with IRBP. Draining lymph node cells were collected 21 d after immunization and were pooled within each group. Triplicate cultures were stimulated with IRBP (30 μg/ml). Proliferation of each group is shown as percent response relative to the control group, after background subtraction. Each point represents one experiment. The means are shown as horizontal bars.
Anti-IRBP Antibody Response of IL-12–treated Mice.
C57Bl/6 mice were immunized with 100 μg of IRBP, and were given either timed treatment of 100 ng IL-12, or an early treatment with graded doses of IL-12. Sera were collected from individual mice 21 d after immunization and were analyzed for IRBP-specific antibody production as described in Materials and Methods. In mice treated with an early regimen of high-dose IL-12 the IgG2a/IgG1 ratio was reversed, reminiscent of a Th1-dependent isotype switch (Table II). However, at the same time, these mice showed evidence of immune suppression, in that they had the lowest combined IgG antibody level (IgG1 + IgG2a) (P versus untreated < 0.003). The extent of suppression in IgG Ab level was dependent on IL-12 timing and dose, and generally correlated with protection from disease.
Table II.
Protective Administration of IL-12 Reverses the Isotype Ratios But Suppresses Total Antibody Levels
| IL-12 treatment | Amount of IL-12 | IgG1 | IgG2a | IgG1/IgG2a ratio | Number of samples | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| (ng) | (μg/ml)* | (μg/ml)* | ||||||||
| Early | 100 | 8,476 | 19,747 | 2.33 | 20 | |||||
| 10 | 166,958 | 14,753 | 0.09 | 8 | ||||||
| 1 | 275,749 | 2,436 | 0.01 | 12 | ||||||
| Late | 100 | 43,081 | 6,317 | 0.15 | 11 | |||||
| None | 210,483 | 4,089 | 0.02 | 18 |
Calculated by averaging the titers of individually assayed serum samples.
IL-12–treated Mice Have a Reduced Yield of Cells, and an Enhanced Number of TUNEL-positive Cells, in the Draining Lymph Nodes.
After observing that IL-12–treated mice had decreased antigen-specific in vitro proliferation, decreased DTH, and reduced lymphokine production, we investigated the possibility that IRBP-specific cells were being deleted in the lymph node during the priming of the immune response. Total cell counts from the draining lymph nodes were obtained at the termination of an experiment, and mice that had received IL-12 early had up to a 50% reduction in cell number compared to untreated controls (Fig. 7). To test the hypothesis that the decreased cell number was a result of IL-12–induced programmed cell death, lymph nodes obtained from IL-12–treated and untreated mice were extracted 24 h after a consecutive IL-12 treatment, and were sectioned and stained for apoptosis by the TUNEL method. The count and the distribution of TUNEL-positive cells in individual tissue sections were highly variable, necessitating evaluation of the entire section area in up to seven sections for each mouse. The number of TUNEL-positive cells in draining lymph nodes of IRBP-immunized mice showed a modest increase as a result of immunization alone, and was upregulated by the IL-12 treatment in a time-dependent fashion (Fig. 8). Unimmunized mice treated with IL-12 also had evidence of increased apoptosis in peripheral lymph nodes, as did immunized mice treated with IL-12 during the second week after immunization, that were not protected. In contrast, GKO mice appeared to develop fewer TUNEL-positive cells in the draining lymph nodes than did the wild-type mice as a result of IL-12 treatment. Furthermore, even without IL-12 treatment, their background levels of TUNEL-positive cells were low compared to wild-type mice (data not shown).
Figure 7.

Mice treated early with IL-12 have decreased numbers of cells from the draining lymph nodes. C57Bl/6 mice received a high dose of IL-12 (100 ng/d) either early (days 0–4) or late (days 7–11) after immunization with IRBP. Draining lymph node cells (iliacs and inguinals) were collected, pooled, and counted within each group at the termination of an experiment. Each point of the graph is a separate experiment (average number of lymph node cells per mouse in a group of five mice). The means are shown as horizontal bars.
Figure 8.
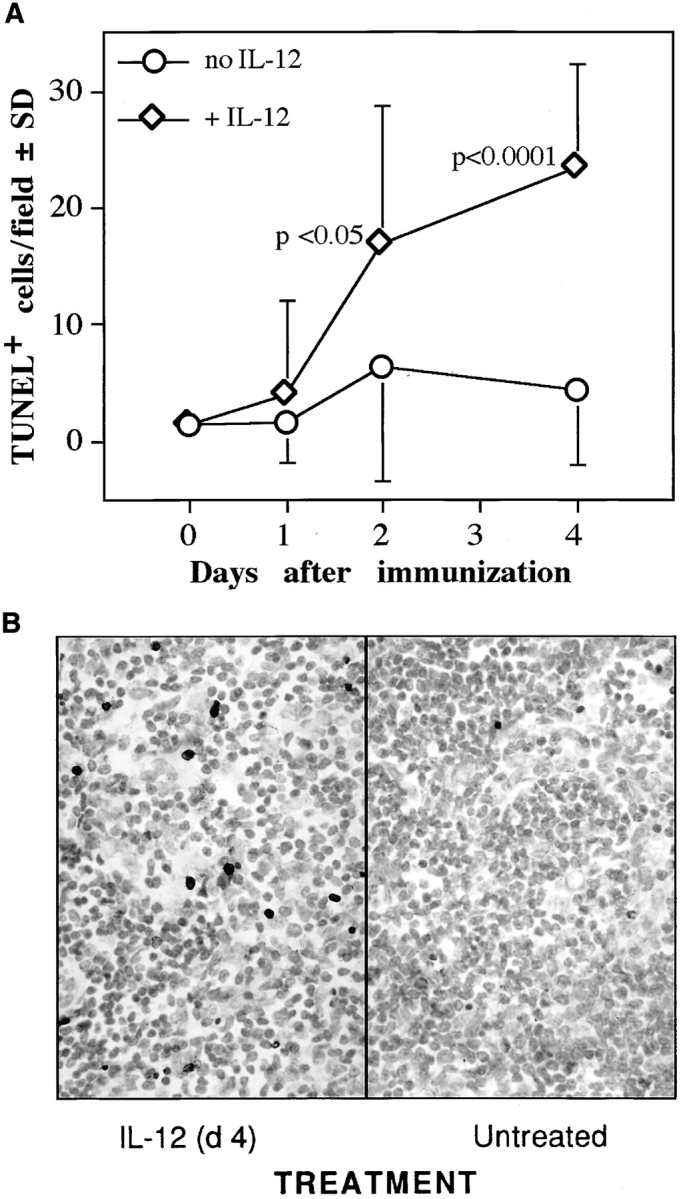
IL-12 treatment induces apoptosis in the draining lymph nodes. C57Bl/6 mice immunized with IRBP received 100 ng IL-12/d for 5 d after immunization. Lymph nodes were collected, fixed, and paraffin-embedded 1, 2, or 4 d after immunization, 24 h after the preceding IL-12 injection (one, two, or four treatments, respectively). TUNEL staining of tissue sections was performed as described in Materials and Methods. (A) Mean number of TUNEL-positive cells per field at each time point ± standard deviation. The value at time = 0 is naive mice. The data are a composite of two experiments, and represent a total of 66 lymph node sections derived from 15 mice (2–4 mice per time point). (B) Photomicrograph of draining lymph nodes from an IL-12–treated (day 4) or untreated mouse. Note numerous TUNEL-positive cells in the treated mouse (×400).
iNOS KO Mice Have a Reduced Ability to Be Protected from EAU by IL-12 Treatment.
IFN-γ is a major inducer of iNOS, and consequently of NO, which can trigger programmed cell death. Therefore, it was of interest to examine a possible connection between induction of iNOS and protection from EAU. To address this question, iNOS KO mice were immunized for induction of EAU and were treated with an early regimen of IL-12. The results showed that while EAU in wild-type mice was strongly suppressed, iNOS KO mice were not well protected by IL-12 treatment (Fig. 9 A). Whereas there was no statistically significant difference in disease scores between iNOS KO and wild-type mice that did not receive IL-12 (P = 0.74), scores of IL-12–treated iNOS KO and IL-12–treated wild-type mice differed at the highly significant probability value of P < 0.005. In keeping with the reduced protection, antigen-specific proliferation of lymph node cells from iNOS KO mice was not as effectively suppressed by IL-12 treatment as in the wild-type mouse (Fig. 9 B). TUNEL staining of lymph nodes from these mice confirmed that after 4 d of treatment with IL-12 the wild-type animals had more evidence of apoptosis, although the difference did not achieve statistical significance (P < 0.1). This may stem from the incomplete effect of IL-12 treatment in this strain, combined with the inherent limitations of TUNEL in tissue sections as a quantitative assay.
Figure 9.
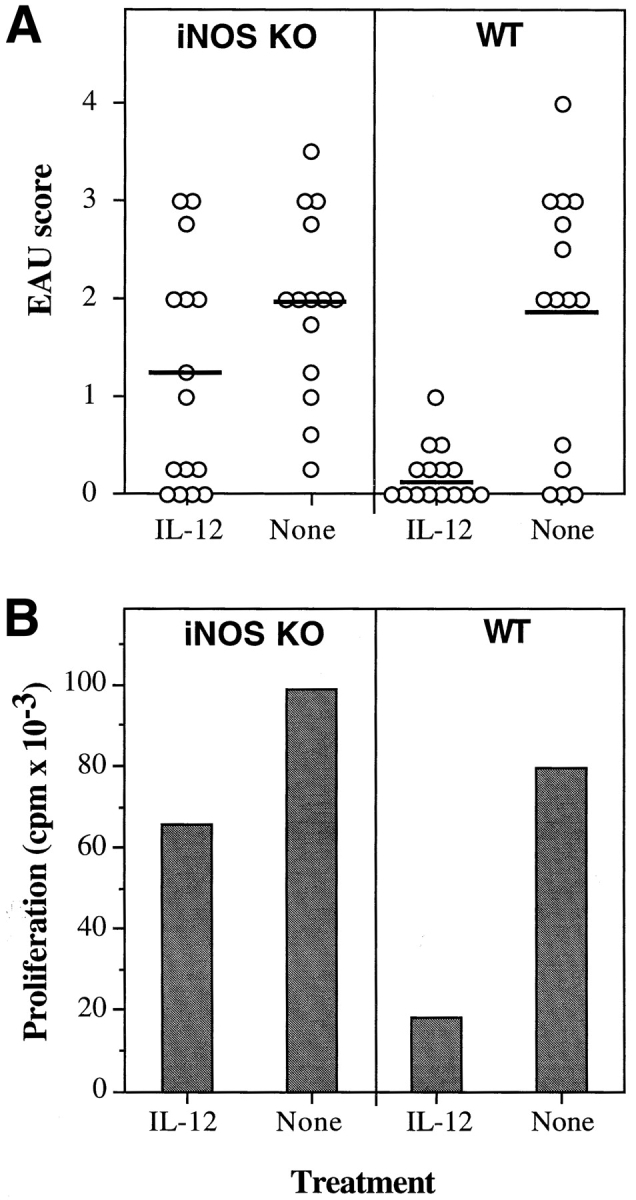
iNOS KO mice have reduced protection from disease and reduced inhibition of lymphocyte proliferation in response to IL-12 treatment. C57Bl/6 × 129 iNOS KO mice and matched wild-type (WT) controls were immunized with IRBP on day 0 and received daily injections of 100 ng of IL-12 (days 0–4) or were left untreated (None). (A) Eyes were harvested for histopathology on day 21 and were graded on a scale of 0 (no disease) to 4 (maximal disease) in half-point increments. Each point is one mouse (average of both eyes). The average of each group is denoted by a horizontal bar. The data are a composite of three experiments. (B) Proliferation of lymph node cells to IRBP in culture in counts per min (cpm) was assayed as described in Materials and Methods. Background counts ranged from 1.5 × 103 to 4 × 103 cpm.
Bcl-2 Transgenic Mice Have a Reduced Ability to be Protected by IL-12 Treatment.
NO-induced apoptosis is known to involve downregulation of the antiapoptotic protein Bcl-2, and forced expression of Bcl-2 counteracts NO-induced apoptosis (39–42). To test whether Bcl-2 overexpression could counteract IL-12–induced protection from EAU, we used mice transgenic for human Bcl-2 on the lck promoter, which overexpress Bcl-2 in their T lymphocytes (27). Bcl-2 transgenic and wild-type mice were treated with an early regimen of IL-12 after uveitogenic immunization. As with other strains, disease in the wild-type mice was essentially completely prevented by IL-12: only 2 mice of 11 developed minimal disease, and 1 of the 2 was affected in only one eye. In contrast, Bcl-2 transgenic mice were deficient in developing protection; 9 of 11 Bcl-2 transgenic mice treated with IL-12 developed bilateral EAU, albeit with lower scores than did untreated Bcl-2 transgenic animals (Fig. 10 A). The difference in disease scores between IL-12– treated wild-type and IL-12–treated Bcl-2 transgenic mice was highly statistically significant at P < 0.008. In keeping with their reduced protection from EAU, lymphocyte proliferation to IRBP of Bcl-2 transgenic mice was considerably less suppressed by IL-12 than that of wild-type mice (Fig. 10 B). DTH responses exhibited the same pattern (not shown). TUNEL staining of lymph nodes from IL-12– treated Bcl-2 transgenic mice revealed the presence of some apoptotic cells, although in reduced numbers compared with wild-type mice.
Figure 10.
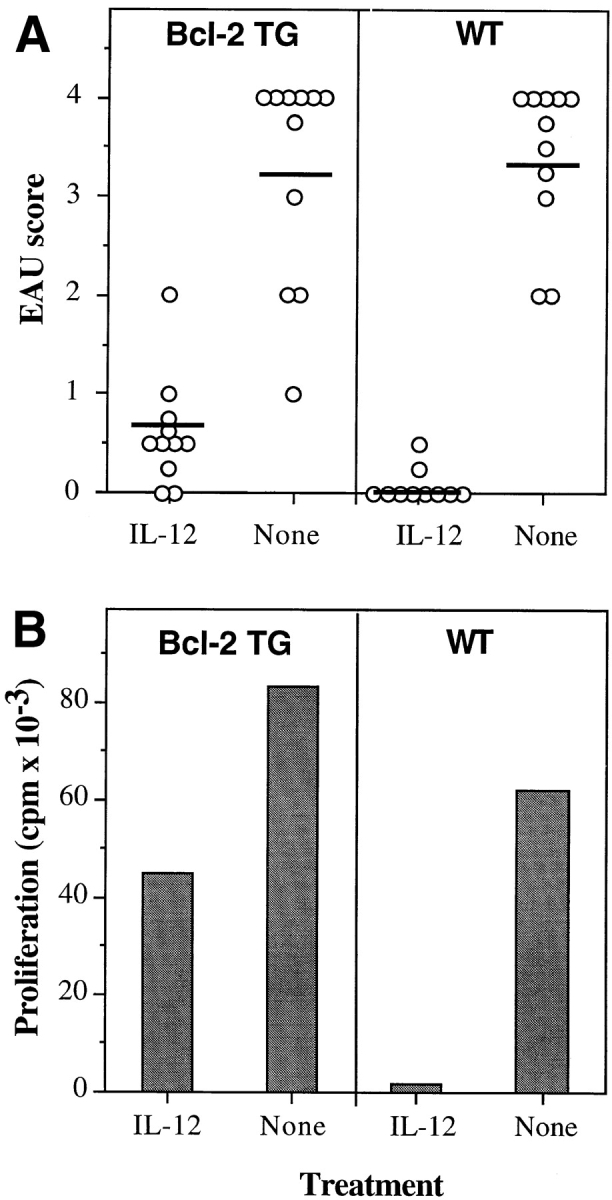
Bcl-2 transgenic mice have reduced protection from disease and reduced inhibition of lymphocyte proliferation in response to IL-12 treatment. C57BL/6 × C3H Bcl-2 transgenic (TG) mice and matched wild-type (WT) controls were immunized with IRBP on day 0 and received daily injections of 100 ng of IL-12 (days 0–4) or were left untreated (None). (A) Eyes were harvested for histopathology on day 21 and were graded on a scale of 0 (no disease) to 4 (maximal disease) in half-point increments. Each point is one mouse (average of both eyes). The data are a composite of two experiments. The mean score of each group is denoted by a horizontal bar. (B) Proliferation of lymph node cells to IRBP in culture in counts per min (cpm) was assayed as described in Materials and Methods. Background counts ranged from 0.5 × 103 to 1.5 × 103 cpm.
Discussion
The assumption that led to the present study was that if a Th1-low response underlies resistance to EAU, then treatment with IL-12 should upregulate disease. In view of the numerous published reports showing exacerbating effects of IL-12 treatment on cell-mediated autoimmunity (43– 46), the unequivocal protection seen in the present study was surprising. However, it was reminiscent of previously reported protective effects of systemic IFN-γ in both EAU and in experimental autoimmune encephalomyelitis (EAE) (34, 47). Because IL-12 induces a strong systemic IFN-γ response (16), we hypothesized that suppression of EAU might be related to an IL-12–induced elevation of systemic IFN-γ. Indeed, IL-12–treated mice exhibited nanogram amounts of circulating IFN-γ in the serum, whereas IFN-γ was undetectable in sera of untreated controls. The hypothesis that IFN-γ induction underlies the protective effect of IL-12 in the EAU model was borne out by the finding that IFN-γ–deficient mice could not be protected from EAU by treatment with the same regimen of IL-12 that was protective in wild-type mice. Although our experiments do not identify the cellular source of this IFN-γ, data published by others suggest that it is likely to be derived largely from NK cells and constitutes an antigen- independent effect (16).
While the protective early IL-12 administration elevated IFN-γ in the serum during treatment, it strongly inhibited the subsequent antigen-specific IFN-γ production by lymph node cells, indicating a suppressed Th1 response. This was not only contrary to expectation, but also in apparent contradiction to the observed shift towards IgG2a antibody isotype in sera of the protected mice. However, in view of the depressed overall IgG antibody response in these animals, we believe that this isotype shift was simply due to the presence of excessive systemic levels of IFN-γ (a switch factor to IgG2a) at the appropriate time during evolution of the antibody response, and not to enhanced Th1 help.
Because IL-12 in the human system has been shown to induce IL-10, a cytokine that suppresses IFN-γ production and inhibits the Th1 response (20, 36), and because treatment with IL-10 inhibits EAU (48), it was necessary to address the possibility that the protective effect of IL-12 may at least in part be due to induction of IL-10. The finding that antigen-driven IL-10 production in the protected mice was not affected did not tend to support this possibility. More importantly, IL-10–deficient mice were protected from disease equally to the wild-type mice, indicating that the protective effect of IL-12 in this system is independent of IL-10.
We next attempted to elucidate the mechanism by which systemic IFN-γ upregulation might mediate protection. The data showed that reduction of antigen-specific responses in the protected mice was not restricted to Th1 (IFN-γ). IL-5, a lymphokine produced by Th2 cells, as well as lymphocyte proliferation and DTH were also suppressed. This, together with a consistently reduced number of cells in the draining lymph nodes, raised the possibility that a deletion of IRBP-specific cells may be occurring. In keeping with this, TUNEL staining of lymph nodes from IL-12–treated mice showed increased numbers of apoptotic cells and led us to postulate that the systemic hyperinduction of IFN-γ caused by treatment with IL-12 protects from EAU by causing programmed death of IRBP-specific effector cells. This interpretation was further supported by the finding that upregulation of TUNEL-positive cells in IL-12–treated GKO mice, which were not protected, was lower and not as consistent as in wild-type mice. An observation unconnected to the IL-12 treatment was that, while in wild-type mice immunization alone increased the number of TUNEL-positive cells, that increase was minimal in GKO mice. This last observation suggests that programmed (activation-induced?) cell death in the draining lymph node is at least in part dependent on IFN-γ, and as a corollary, that the enhanced proliferative responses that we and others have noted in IFN-γ–deficient and IFN-γ receptor–deficient mice (35, 49) may be related to a failure of IFN-γ– driven elimination of antigen-specific cells.
We next asked the question, what might be the mechanism(s) downstream of IFN-γ that could result in apoptosis? IFN-γ is known to have antiproliferative effects on many cell types, and at least some of those effects might be connected to induction of cell death. IFN-γ strongly upregulates iNOS, and consequently NO, which can cause apoptosis, inhibition of lymphocyte growth, and downregulation of Th1 and Th2 cytokines (50–52). Other apoptosis-triggering molecules induced by IFN-γ include TNF-α and Fas (53–56). Finally, IFN-γ has been implicated in some studies as being able to directly induce apoptosis (57– 60). Because our previous work showed that GKO mice immunized with a uveitogenic regimen of IRBP do not upregulate iNOS (35), involvement of NO in protection from EAU was immediately suspected. In keeping with this hypothesis, IL-12–treated iNOS KO mice proved to be deficient in their ability to be protected from EAU and showed less suppression of their proliferative response to IRBP than the wild-type mice. The present results are in line with recent reports showing that inhibition of iNOS, or disruption of its gene, exacerbate EAE and enhance lymphocyte proliferation and IFN-γ production (61, 62). Taken together with these reports, our data support the interpretation that induction of NO is a major pathway through which upregulation of IFN-γ protects from autoimmune disease by curtailing the priming of autoaggressive cells. Our data also point to NO-induced apoptosis as a likely mechanism underlying the protective effects of systemic IFN-γ, that were repeatedly documented in Th1-dependent autoimmune diseases such as EAU and EAE, but were not adequately understood (34, 47, 49, 63).
NO-driven apoptosis is known to involve downregulation of the Bcl-2 gene product, and forced expression of Bcl-2 protects from NO-induced apoptosis (39–42). The finding that mice transgenic for Bcl-2 under control of the lck promoter were significantly deficient in developing protection from EAU after IL-12 treatment provides strong evidence that protection is due at least in part to apoptotic death of uveitogenic lymphocytes through the NO/Bcl-2 pathway. It should be noted, however, that the defect in developing protection, which was very clear in GKO mice, was not as complete in the iNOS knockouts and was partial in the Bcl-2 transgenic mice. Since TUNEL-positive cells were still evident in lymph nodes of Bcl-2 transgenic mice, overexpression of the Bcl-2 transgene either did not completely prevent NO-induced T cell apoptosis, or alternatively may indicate participation of other IFN-γ–driven apoptotic effects. Although experiments not shown here have so far failed to implicate in a major way either Fas/FasL or TNF-α–mediated apoptosis in the protection, they did not exclude them. The present data also do not exclude participation of nonapoptotic effects, such as inhibition of proliferation or induction of anergy. In addition, the somewhat more moderate suppression of antigen-specific production of IL-5 than of IFN-γ after IL-12 treatment could be indicative of a shift in the residual response towards a less pathogenic phenotype. Lastly, although our results strongly implicate apoptosis as a major mechanism in the protection, a T cell receptor–transgenic system amenable to clonotypic analysis is needed to demonstrate directly that antigen-specific lymphocytes are among the cells undergoing apoptosis. These questions will be the subject of a separate study.
The protective effect of IL-12 was a time-limited phenomenon, in that delayed IL-12 treatment was not protective despite the fact that it did elevate systemic IFN-γ. This suggested that the uveitogenic T cells are sensitive to elimination only during the initial phase of their differentiation, as they are being primed, but not later, when they have already become mature effector cells. Two observations showing a dissociation between protection and apoptosis are in apparent contradiction to this interpretation, and need to be reconciled: (a) lymph nodes of naive IL-12– treated animals had increased numbers of apoptotic cells. The answer to this is that naive mice are in fact constantly being primed by environmental antigens; and (b) mice receiving the delayed (nonprotective) IL-12 treatment had enhanced apoptosis in the draining lymph nodes. This is explained by the fact that during the second week new cells are continuing to be primed in the lymph node. While IL-12 treatment may be eliminating those, enough mature effectors have already been generated to induce disease. Finally, cells other than lymphocytes are likely to be undergoing apoptosis as well, resulting in a background of TUNEL-positive cells that is unrelated to an antigen-specific response occurring in the same vicinity.
The data reported here also shed new light on our previous observations concerning the antiinflammatory effect of IL-12 in endotoxin-induced uveitis, an acute model of anterior uveitis that is immune mediated, but not antigen specific (64). In that study, direct intraocular injection of IL-12 was able to reduce the number of inflammatory cells infiltrating the anterior chamber, an effect that was accompanied by elevated titers of IFN-γ in the aqueous humor. In view of the data reported here, we propose that this reduction in inflammatory cells could have been caused by their elimination through apoptosis.
The present results appear to differ from those reported in several other models of autoimmunity. Whether administered in vitro or in vivo, IL-12 accelerated the onset, and increased the incidence and severity of disease in the EAE and in the collagen-induced arthritis (CIA) models, as well as in the nonobese diabetic (NOD) mouse (43–46). However, other investigators reported amelioration of CIA in mice by continuous administration of high-dose IL-12, and some IL-12 treatment regimens ameliorated diabetes in NOD mice (65–67). Our results offer to reconcile the apparent contradiction between enhancing and protective effects of IL-12 in the same model. Although the protective effect of high-dose IL-12 in the CIA model (which is strongly dependent on humoral immunity) was felt by the authors to be due to an effect on antibody isotypes rather than inhibition of cellular responses (65), it is interesting to note that this study used CFA, itself a strong IL-12 inducer, in the immunization protocol. In contrast, none of the studies that documented enhancement of disease used an induction regimen incorporating CFA. Similarly, induction of EAU in the present study was achieved by immunization using both CFA and PTX as adjuvants. However, under conditions of adoptive transfer IL-12 increases the pathogenicity of retinal antigen–specific T cells (24, 68). Therefore it could tbe hypothesized that the immunization protocol for induction of EAU already generates a maximum Th1 response, which cannot be further upregulated by the exogenous IL-12 treatment, resulting in an effective IL-12 overdose. This interpretation is also in line with the finding that we did not obtain exacerbation of EAU at any of the IL-12 doses. Our data thus argue that an excess of IL-12 during priming can abort a Th1-mediated autoimmune disease instead of bringing about its exacerbation, by triggering a negative feedback loop.
In summary, the present study shows that administration of the Th1-inducing cytokine IL-12 protects from a Th1-dependent autoimmune disease, EAU, and inhibits across the board the cellular and humoral immune responses to the uveitogenic antigen. The mechanism of this phenomenon involves hyperinduction of systemic IFN-γ, causing upregulation of iNOS and production of NO, which protects at least in part by triggering Bcl-2 controlled apoptotic deletion of antigen-specific cells at a critical time point during antigen priming. We conclude that protection from EAU by IL-12 is secondary to a curtailment in generation of uveitogenic effector T cells. The present study also proposes a likely explanation for the well-documented protective effects of systemic IFN-γ in Th1-dependent autoimmunity.
Acknowledgments
The authors thank Dr. Maurice Gately of Hoffman La-Roche for providing the IL-12 used in these experiments and to Ms. Dawn Matteson for Fas/FasL immunostaining. We are grateful to Drs. Mark Doherty and Alan Sher for providing the iNOS KO mice; Dr. Renate Morawetz for providing the B6-backcrossed IL-10 KO breeding stock; Dr. Rajeev Agarwal for the IFN-γ–deficient mice; and Dr. Stanley Korsmeyer and Barbara Klocke for the Bcl-2 transgenic breeding stock. The help of Ms. Heather Gitchell in the experiments with Bcl-2 transgenic mice is gratefully acknowledged.
Footnotes
Some of the material in this manuscript was presented at the AAAAI/ AAI/CIS joint meeting in San Francisco, February 1997. Portions of this work have appeared in abstract form (1997. J. Allergy Clin. Immunol. 99[No. 5, Pt. 2]).
Abbreviations used in this paper: CIA, collagen-induced arthritis; DTH, delayed type hypersensitivity; EAE, experimental autoimmune encephalomyelitis; EAU, experimental autoimmune uveitis; GKO, IFN-γ knockout; iNOS, inducible nitric oxide synthase; IRBP, interphotoreceptor retinoid-binding protein; KO, knockout; NO, nitric oxide; NOD, nonobese diabetic; PTX, pertussis toxin.
References
- 1.Caspi RR, Roberge FG, McAllister CG, el-Saied M, Kuwabara T, Gery I, Hanna E, Nussenblatt RB. T cell lines mediating experimental autoimmune uveoretinitis (EAU) in the rat. J Immunol. 1986;136:928–933. [PubMed] [Google Scholar]
- 2.Gery, I., M. Mochizuki, and R.B. Nussenblatt. 1986. Retinal specific antigens and immunopathogenic processes they provoke. In Progress in Retinal Research. N.N. Osborne and G.J. Chader, editors. Pergamon Press, Oxford. 75–109.
- 3.Sanui H, Redmond TM, Kotake S, Wiggert B, Hu LH, Margalit H, Berzofsky JA, Chader GJ, Gery I. Identification of an immunodominant and highly immunopathogenic determinant in the retinal interphotoreceptor retinoid–binding protein (IRBP) J Exp Med. 1989;169:1947–1960. doi: 10.1084/jem.169.6.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizzo LV, Silver P, Wiggert B, Hakim F, Gazzinelli RT, Chan CC, Caspi RR. Establishment and characterization of a murine CD4+T cell line and clone that induce experimental autoimmune uveoretinitis in B10.A mice. J Immunol. 1996;156:1654–1660. [PubMed] [Google Scholar]
- 5.Wolf SF, Temple PA, Kobayashi M, Young D, Dicig M, Lowe L, Dzialo R, Fitz L, Ferenz C, Hewick RM, et al. Cloning of cDNA for natural killer cell stimulatory factor, a heterodimeric cytokine with multiple biologic effects on T and natural killer cells. J Immunol. 1991;146:3074–3081. [PubMed] [Google Scholar]
- 6.Schoenhaut DS, Chua AO, Wolitzky AG, Quinn PM, Dwyer CM, McComas W, Familletti PC, Gately MK, Gubler U. Cloning and expression of murine IL-12. J Immunol. 1992;148:3433–3440. [PubMed] [Google Scholar]
- 7.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T–T help via APC activation. J Exp Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koch F, Stanzl U, Jennewein P, Janke K, Heufler C, Kampgen E, Romani N, Schuler G. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J Exp Med. 1996;184:741–746. doi: 10.1084/jem.184.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 10.Germann T, Gately MK, Schoenhaut DS, Lohoff M, Mattner F, Fischer S, Jin SC, Schmitt E, Rude E. Interleukin-12/T cell stimulating factor, a cytokine with multiple effects on T helper type 1 (Th1) but not on Th2 cells. Eur J Immunol. 1993;23:1762–1770. doi: 10.1002/eji.1830230805. [DOI] [PubMed] [Google Scholar]
- 11.Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, Wysocka M, Trinchieri G, Murphy KM, O'Garra A. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+T cells. J Immunol. 1995;154:5071–5079. [PubMed] [Google Scholar]
- 12.Schmitt E, Hoehn P, Germann T, Rude E. Differential effects of interleukin-12 on the development of naive mouse CD4+T cells. Eur J Immunol. 1994;24:343–347. doi: 10.1002/eji.1830240211. [DOI] [PubMed] [Google Scholar]
- 13.Afonso LC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P. The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. . Science. 1994;263:235–237. doi: 10.1126/science.7904381. [DOI] [PubMed] [Google Scholar]
- 14.Heinzel FP, Schoenhaut DS, Rerko RM, Rosser LE, Gately MK. Recombinant interleukin 12 cures mice infected with Leishmania major. . J Exp Med. 1993;177:1505–1509. doi: 10.1084/jem.177.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schijns VE, Haagmans BL, Horzinek MC. IL-12 stimulates an antiviral type 1 cytokine response but lacks adjuvant activity in IFN-gamma-receptor–deficient mice. J Immunol. 1995;155:2525–2532. [PubMed] [Google Scholar]
- 16.McKnight AJ, Zimmer GJ, Fogelman I, Wolf SF, Abbas AK. Effects of IL-12 on helper T cell–dependent immune responses in vivo. J Immunol. 1994;152:2172–2179. [PubMed] [Google Scholar]
- 17.Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri G, Romagnani S. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)–specific immune responses and inhibits the development of IL-4–producing Th cells. J Exp Med. 1993;177:1199–1204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manetti R, Gerosa F, Giudizi MG, Biagiotti R, Parronchi P, Piccinni MP, Sampognaro S, Maggi E, Romagnani S, Trinchieri G, et al. Interleukin 12 induces stable priming for interferon gamma (IFN-gamma) production during differentiation of human T helper (Th) cells and transient IFN-gamma production in established Th2 cell clones. J Exp Med. 1994;179:1273–1283. doi: 10.1084/jem.179.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paganin C, Frank I, Trinchieri G. Priming for high interferon-gamma production induced by interleukin-12 in both CD4+ and CD8+T cell clones from HIV-infected patients. J Clin Invest. 1995;96:1677–1682. doi: 10.1172/JCI118209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerosa F, Paganin C, Peritt D, Paiola F, Scupoli MT, Aste-Amezaga M, Frank I, Trinchieri G. Interleukin-12 primes human CD4 and CD8 T cell clones for high production of both interferon-gamma and interleukin 10. J Exp Med. 1996;183:2559–2569. doi: 10.1084/jem.183.6.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 22.Caspi RR, Silver PB, Chan CC, Sun B, Agarwal RK, Wells J, Oddo S, Fujino Y, Najafian F, Wilder RL. Genetic susceptibility to experimental autoimmune uveoretinitis in the rat is associated with an elevated Th1 response. J Immunol. 1996;157:2668–2675. [PubMed] [Google Scholar]
- 23.Sun B, Rizzo LV, Sun S-H, Chan C-C, Wiggert B, Wilder RL, Caspi RR. Genetic susceptibility to experimental autoimmune uveitis involves more than a predisposition to generate a T helper-1-like or a T helper-2-like response. J Immunol. 1997;159:1004–1011. [PubMed] [Google Scholar]
- 24.Tarrant TK, Silver PB, Chan CC, Wiggert B, Caspi RR. Endogenous IL-12 is required for induction and expression of experimental autoimmune uveitis. J Immunol. 1998;161:122–127. [PubMed] [Google Scholar]
- 25.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 26.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase [published erratum appears in Cell.1995. 81: following 1170] Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 27.Sentman CL, Shutter JR, Hockenbery D, Kanagawa O, Korsmeyer SJ. bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell. 1991;67:879–888. doi: 10.1016/0092-8674(91)90361-2. [DOI] [PubMed] [Google Scholar]
- 28.Pepperberg DR, Okajima TL, Ripps H, Chader GJ, Wiggert B. Functional properties of interphotoreceptor retinoid–binding protein. Photochem Photobiol. 1991;54:1057–1060. doi: 10.1111/j.1751-1097.1991.tb02129.x. [DOI] [PubMed] [Google Scholar]
- 29.Chan CC, Caspi RR, Ni M, Leake WC, Wiggert B, Chader GJ, Nussenblatt RB. Pathology of experimental autoimmune uveoretinitis in mice. J Autoimmun. 1990;3:247–255. doi: 10.1016/0896-8411(90)90144-h. [DOI] [PubMed] [Google Scholar]
- 30.Rizzo LV, Miller-Rivero NE, Chan CC, Wiggert B, Nussenblatt RB, Caspi RR. Interleukin 2 treatment potentiates induction of oral tolerance in a murine model of autoimmunity. J Clin Invest. 1994;94:1668–1672. doi: 10.1172/JCI117511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rizzo LV, DeKruyff RH, Umetsu DT, Caspi RR. Regulation of the interaction between Th1 and Th2 T cell clones to provide help for antibody production in vivo. Eur J Immunol. 1995;25:708–716. doi: 10.1002/eji.1830250312. [DOI] [PubMed] [Google Scholar]
- 32.Snedecor, G.W., and W.G. Cochran. 1967. Statistical Methods. Iowa State University Press, Ames, IA. p. 248.
- 33.Caspi RR, Grubbs BG, Chan CC, Chader GJ, Wiggert B. Genetic control of susceptibility to experimental autoimmune uveoretinitis in the mouse model: concomitant regulation by MHC and non-MHC genes. J Immunol. 1992;148:2384–2389. [PubMed] [Google Scholar]
- 34.Caspi RR, Chan CC, Grubbs BG, Silver PB, Wiggert B, Parsa CF, Bahmanyar S, Billiau A, Heremans H. Endogenous systemic IFN-gamma has a protective role against ocular autoimmunity in mice. J Immunol. 1994;152:890–899. [PubMed] [Google Scholar]
- 35.Jones LS, Rizzo LV, Agarwal RK, Tarrant TK, Chan CC, Wiggerrt B, Caspi RR. Interferon gamma–deficient mice develop experimental autoimmune uveitis in the context of a deviant effector response. J Immunol. 1997;158:5997–6005. [PubMed] [Google Scholar]
- 36.Windhagen A, Anderson DE, Carrizosa A, Williams RE, Hafler DA. IL-12 induces human T cells secreting IL-10 with IFN-gamma. J Immunol. 1996;157:1127–1131. [PubMed] [Google Scholar]
- 37.Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, Muller W, Trinchieri G, Sher A. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF- alpha. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 38.Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis and colon cancer in interleukin-10– deficient mice are associated with aberrant cytokine production and CD4(+) TH1–like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie K, Wang Y, Huang S, Xu L, Bielenberg D, Salas T, McConkey DJ, Jiang W, Fidler IJ. Nitric oxide–mediated apoptosis of K-1735 melanoma cells is associated with downregulation of Bcl-2. Oncogene. 1997;15:771–779. doi: 10.1038/sj.onc.1201239. [DOI] [PubMed] [Google Scholar]
- 40.Xie K, Huang S, Wang Y, Beltran PJ, Juang SH, Dong Z, Reed JC, McDonnell TJ, McConkey DJ, Fidler IJ. Bcl-2 protects cells from cytokine-induced nitric-oxide–dependent apoptosis. Cancer Immunol Immunother. 1996;43:109–115. doi: 10.1007/s002620050310. [DOI] [PubMed] [Google Scholar]
- 41.Messmer UK, Reed UK, Brune B. Bcl-2 protects macrophages from nitric oxide–induced apoptosis. J Biol Chem. 1996;271:20192–20197. doi: 10.1074/jbc.271.33.20192. [DOI] [PubMed] [Google Scholar]
- 42.Melkova Z, Lee SB, Rodriguez D, Esteban M. Bcl-2 prevents nitric oxide–mediated apoptosis and poly (ADP-ribose) polymerase cleavage. FEBS (Fed Eur Biochem Soc) Lett. 1997;403:273–278. doi: 10.1016/s0014-5793(97)00065-3. [DOI] [PubMed] [Google Scholar]
- 43.Trembleau S, Germann T, Gately MK, Adorini L. The role of IL-12 in the induction of organ-specific autoimmune diseases. Immunol Today. 1995;16:383–386. doi: 10.1016/0167-5699(95)80006-9. [DOI] [PubMed] [Google Scholar]
- 44.Leonard JP, Waldburger KE, Goldman SJ. Prevention of experimental autoimmune encephalomyelitis by antibodies against interleukin 12. J Exp Med. 1995;181:381–386. doi: 10.1084/jem.181.1.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waldburger KE, Hastings RC, Schaub RG, Goldman SJ, Leonard JP. Adoptive transfer of experimental allergic encephalomyelitis after in vitro treatment with recombinant murine interleukin-12. Preferential expansion of interferon-gamma–producing cells and increased expression of macrophage-associated inducible nitric oxide synthase as immunomodulatory mechanisms. Am J Pathol. 1996;148:375–382. [PMC free article] [PubMed] [Google Scholar]
- 46.Germann T, Szeliga J, Hess H, Storkel S, Podlaski FJ, Gately MK, Schmitt E, Rude E. Administration of interleukin 12 in combination with type II collagen induces severe arthritis in DBA/1 mice. Proc Natl Acad Sci USA. 1995;92:4823–4827. doi: 10.1073/pnas.92.11.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, Carton H. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J Immunol. 1988;140:1506–1510. [PubMed] [Google Scholar]
- 48.Rizzo LV, Xu H, Chan CC, Wiggert B, Caspi RR. IL-10 has a protective role in experimental autoimmune uveoretinitis. Int Immunol. 1998;10:807–814. doi: 10.1093/intimm/10.6.807. [DOI] [PubMed] [Google Scholar]
- 49.Willenborg DO, Fordham S, Bernard CCA, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein–induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- 50.Sveinbjornsson B, Olsen R, Seternes OM, Seljelid R. Macrophage cytotoxicity against murine meth A sarcoma involves nitric oxide–mediated apoptosis. Biochem Biophys Res Commun. 1996;223:643–649. doi: 10.1006/bbrc.1996.0948. [DOI] [PubMed] [Google Scholar]
- 51.Geng YJ, Wu Q, Muszynski M, Hansson GK, Libby P. Apoptosis of vascular smooth muscle cells induced by in vitro stimulation with interferon-gamma, tumor necrosis factor–alpha, and interleukin-1 beta. Arterioscler Thromb Vasc Biol. 1996;16:19–27. doi: 10.1161/01.atv.16.1.19. [DOI] [PubMed] [Google Scholar]
- 52.Bauer H, Jung T, Tsikas D, Stichtenoth DO, Frolich JC, Neumann C. Nitric oxide inhibits the secretion of T-helper 1– and T-helper 2–associated cytokines in activated human T cells. Immunology. 1997;90:205–211. doi: 10.1046/j.1365-2567.1997.00161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geng YJ, Hellstrand K, Wennmalm A, Hansson GK. Apoptotic death of human leukemic cells induced by vascular cells expressing nitric oxide synthase in response to gamma-interferon and tumor necrosis factor–alpha. Cancer Res. 1996;56:866–874. [PubMed] [Google Scholar]
- 54.Han X, Becker K, Degen HJ, Jablonowski H, Strohmeyer G. Synergistic stimulatory effects of tumour necrosis factor alpha and interferon gamma on replication of human immunodeficiency virus type 1 and on apoptosis of HIV-1–infected host cells. Eur J Clin Invest. 1996;26:286–292. doi: 10.1046/j.1365-2362.1996.116271.x. [DOI] [PubMed] [Google Scholar]
- 55.Iwahashi H, Hanafusa T, Eguchi Y, Nakajima H, Miyagawa J, Itoh N, Tomita K, Namba M, Kuwajima M, Noguchi T, et al. Cytokine-induced apoptotic cell death in a mouse pancreatic beta-cell line: inhibition by Bcl-2. Diabetologia. 1996;39:530–536. doi: 10.1007/BF00403299. [DOI] [PubMed] [Google Scholar]
- 56.Jo T, Tomiyama T, Ohashi K, Saji F, Tanizawa O, Ozaki M, Yamamoto R, Yamamoto T, Nishizawa Y, Terada N. Apoptosis of cultured mouse luteal cells induced by tumor necrosis factor–alpha and interferon-gamma. Anat Rec. 1995;241:70–76. doi: 10.1002/ar.1092410110. [DOI] [PubMed] [Google Scholar]
- 57.Levy-Strumpf N, Deiss LP, Berissi H, Kimchi A. DAP-5, a novel homolog of eukaryotic translation initiation factor 4G isolated as a putative modulator of gamma interferon–induced programmed cell death. Mol Cell Biol. 1997;17:1615–1625. doi: 10.1128/mcb.17.3.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brysk MM, Selvanayagam P, Arany I, Brysk H, Tyring SK, Rajaraman S. Induction of apoptotic nuclei by interferon-gamma and by predesquamin in cultured keratinocytes. J Interferon Cytokine Res. 1995;15:1029–1035. doi: 10.1089/jir.1995.15.1029. [DOI] [PubMed] [Google Scholar]
- 59.Maier JA, Morelli D, Balsari A. The differential response to interferon gamma by normal and transformed endothelial cells. Biochem Biophys Res Commun. 1995;214:582–588. doi: 10.1006/bbrc.1995.2325. [DOI] [PubMed] [Google Scholar]
- 60.Trubiani O, Bosco D, Di Primio R. Interferon-gamma (IFN-gamma) induces programmed cell death in differentiated human leukemic B cell lines. Exp Cell Res. 1994;215:23–27. doi: 10.1006/excr.1994.1309. [DOI] [PubMed] [Google Scholar]
- 61.Gold DP, Schroder K, Powell HC, Kelly CJ. Nitric oxide and the immunomodulation of experimental allergic encephalomyelitis. Eur J Immunol. 1997;27:1–7. doi: 10.1002/eji.1830271118. [DOI] [PubMed] [Google Scholar]
- 62.Fenyk-Melody JE, Garrison AE, Brunnert SR, Weidner JR, Shen F, Shelton BA, Mudgett JS. Experimental autoimmune encephalomyelitis is exacerbated in mice lacking the NOS2 gene. J Immunol. 1998;160:2940–2946. [PubMed] [Google Scholar]
- 63.Krakowski M, Owens T. Interferon-γ confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26:1641–1646. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- 64.Whitcup SM, Rizzo LV, Lai JC, Hayashi S, Gazzinelli R, Chan CC. IL-12 inhibits endotoxin-induced inflammation in the eye. Eur J Immunol. 1996;26:995–999. doi: 10.1002/eji.1830260506. [DOI] [PubMed] [Google Scholar]
- 65.Hess H, Gately MK, Rude E, Schmitt E, Szeliga J, Germann T. High doses of interleukin-12 inhibit the development of joint disease in DBA/1 mice immunized with type II collagen in complete Freund's adjuvant. Eur J Immunol. 1996;26:187–191. doi: 10.1002/eji.1830260129. [DOI] [PubMed] [Google Scholar]
- 66.Szeliga J, Hess H, Rude E, Schmitt E, Germann T. IL-12 promotes cellular but not humoral type II collagen–specific Th 1–type responses in C57BL/6 and B10.Q mice and fails to induce arthritis. Int Immunol. 1996;8:1221–1227. doi: 10.1093/intimm/8.8.1221. [DOI] [PubMed] [Google Scholar]
- 67.O'Hara RM, Jr, Henderson SL, Nagelin A. Prevention of a Th1 disease by a Th1 cytokine: IL-12 and diabetes in NOD mice. Ann NY Acad Sci. 1996;795:241–249. doi: 10.1111/j.1749-6632.1996.tb52673.x. [DOI] [PubMed] [Google Scholar]
- 68.Xu H, Rizzo LV, Silver PB, Caspi RR. Uveitogenicity is associated with a Th1-like lymphokine profile: cytokine-dependent modulation of primary and committed T cells in EAU. Cell Immunol. 1997;178:69–78. doi: 10.1006/cimm.1997.1121. [DOI] [PubMed] [Google Scholar]